
Role of MicroRNAs in Signaling Pathways Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis: A Focus on Epithelial-Mesenchymal Transition

 ,
,  and
and
Abstract
1. Idiopathic Pulmonary Fibrosis (IPF)
2. MicroRNAs (miRNAs)
3. MicroRNAs and IPF
4. Importance of Epithelial Mesenchymal Transition (EMT) in the Pathology of IPF
5. Signaling Pathways Involved in the EMT Process
5.1. Smads Intracellular Effectors
5.2. Smad-Dependent Signaling Pathways in EMT Induced by TGFβ1
6. Non-Smad-Dependent Signaling Pathways in EMT Induced by TGFβ
6.1. Erk, JNK, and P38 MAPK Pathways and Their Effects on the Development of EMT
6.2. PI3K/Akt/mTOR Pathway
6.3. PI3K/Akt Signaling Pathway
6.4. Rho-Like GTPases Regulated by TGFβ
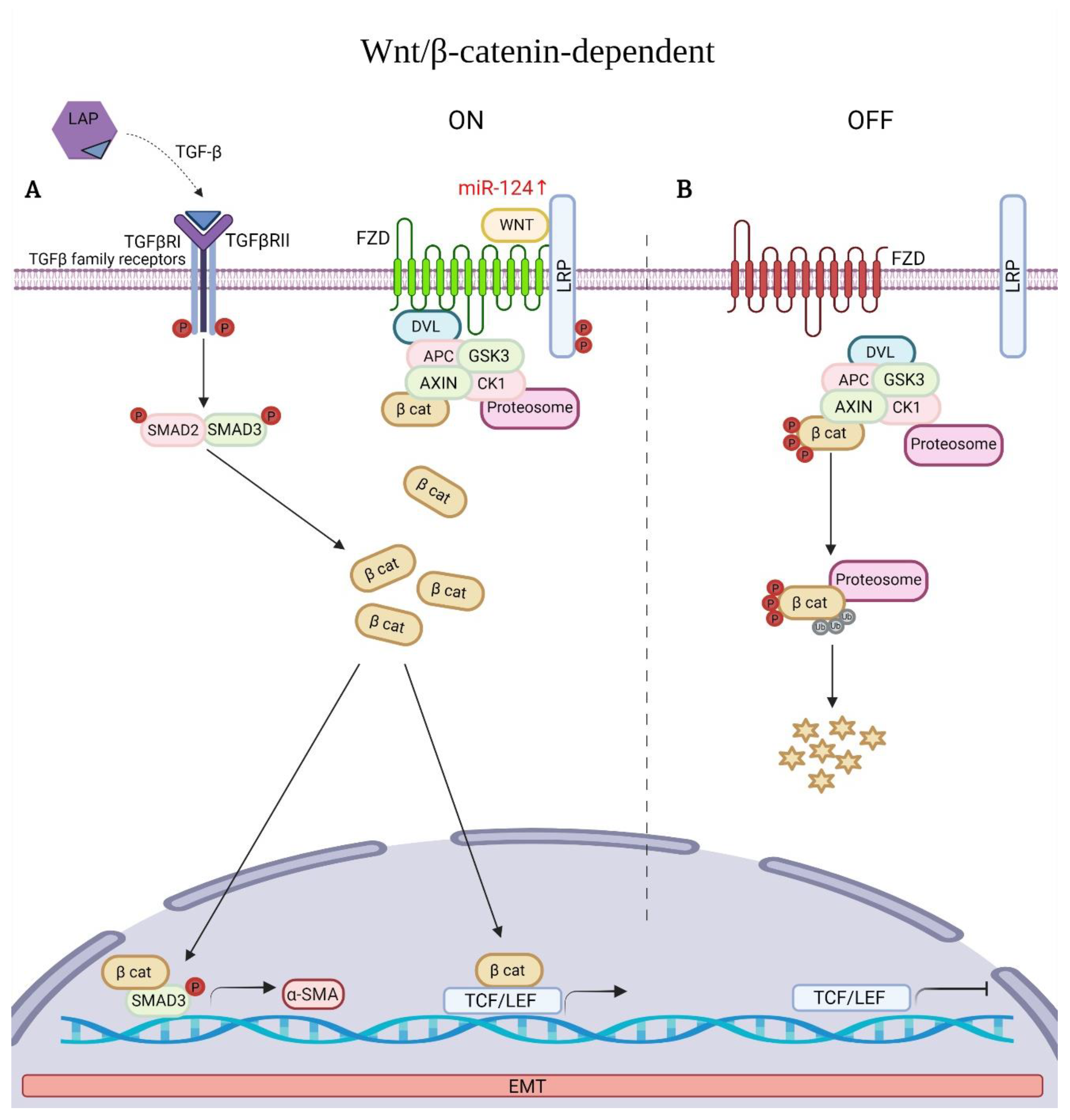
6.5. Wnt Signaling Pathway Associated with EMT
7. MicroRNAs with Pro-Fibrotic Properties and with Regulatory Activity of Signaling Pathways Linked with IPF
MicroRNAs with Pro-Fibrotic Properties but Negative Regulators of Smad6/Smad7 Expression
8. MicroRNAs with Anti-Fibrotic Properties and with Regulatory Activity of Signaling Pathways Linked with IPF
MicroRNAs Down-Regulated and Associated with Apoptosis
9. Discussion and Conclusions
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Thoracic Society. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment. International Consensus Statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative Stress in Pulmonary Fibrosis: A Possible Role for Redox Modulatory Therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging Cellular and Molecular Determinants of Idiopathic Pulmonary Fibrosis. Cell. Mol. Life Sci. CMLS 2021, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and Cellular Mechanisms of Pulmonary Fibrosis. Fibrogenesis Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH Oxidase Expression Is Increased in Pulmonary Fibroblasts from Patients with Idiopathic Pulmonary Fibrosis and Mediates TGFbeta1-Induced Fibroblast Differentiation into Myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef]
- Fierro-Fernández, M.; Busnadiego, Ó.; Sandoval, P.; Espinosa-Díez, C.; Blanco-Ruiz, E.; Rodríguez, M.; Pian, H.; Ramos, R.; López-Cabrera, M.; García-Bermejo, M.L.; et al. MiR-9-5p Suppresses pro-Fibrogenic Transformation of Fibroblasts and Prevents Organ Fibrosis by Targeting NOX4 and TGFBR2. EMBO Rep. 2015, 16, 1358–1377. [Google Scholar] [CrossRef]
- Malsin, E.S.; Kamp, D.W. The Mitochondria in Lung Fibrosis: Friend or Foe? Transl. Res. J. Lab. Clin. Med. 2018, 202, 1–23. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 Deficiency Impairs Mitochondrial Homeostasis and Promotes Lung Fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial Dysfunction in Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388. [Google Scholar] [CrossRef]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant Protein A2 Mutations Associated with Pulmonary Fibrosis Lead to Protein Instability and Endoplasmic Reticulum Stress. J. Biol. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic Reticulum Stress: Cell Life and Death Decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A Key Regulatory Factor in Tumour Growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia Signaling in Human Diseases and Therapeutic Targets. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Bodempudi, V.; Hergert, P.; Smith, K.; Xia, H.; Herrera, J.; Peterson, M.; Khalil, W.; Kahm, J.; Bitterman, P.B.; Henke, C.A. MiR-210 Promotes IPF Fibroblast Proliferation in Response to Hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L283–L294. [Google Scholar] [CrossRef]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-Mesenchymal Transition in Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. From Pulmonary Fibrosis to Progressive Pulmonary Fibrosis: A Lethal Pathobiological Jump. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L600–L607. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, X.; Zhang, Y.; Pan, Z.; Sun, Z.; Huang, Z.; Zheng, S.; Pan, H.; Zou, X.; Huang, P. Heterogeneous Microenvironment Analysis to Explore the Potential Regulatory Role of Endothelial-Mesenchymal Transition in Idiopathic Pulmonary Fibrosis. Ann. Transl. Med. 2022, 10, 486. [Google Scholar] [CrossRef]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; Ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef]
- Posadino, A.M.; Erre, G.L.; Cossu, A.; Emanueli, C.; Eid, A.H.; Zinellu, A.; Pintus, G.; Giordo, R. NADPH-Derived ROS Generation Drives Fibrosis and Endothelial-to-Mesenchymal Transition in Systemic Sclerosis: Potential Cross Talk with Circulating MiRNAs. Biomol. Concepts 2022, 13, 11–24. [Google Scholar] [CrossRef]
- Coultas, D.B.; Zumwalt, R.E.; Black, W.C.; Sobonya, R.E. The Epidemiology of Interstitial Lung Diseases. Am. J. Respir. Crit. Care Med. 1994, 150, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Keogh, B.A.; Crystal, R.G. Alveolitis: The Key to the Interstitial Lung Disorders. Thorax 1982, 37, 1–10. [Google Scholar] [CrossRef][Green Version]
- Selman, M.; King, T.E.; Pardo, A.; American Thoracic Society; European Respiratory Society. American College of Chest Physicians Idiopathic Pulmonary Fibrosis: Prevailing and Evolving Hypotheses about Its Pathogenesis and Implications for Therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. Idiopathic Pulmonary Fibrosis: New Insights in Its Pathogenesis. Int. J. Biochem. Cell Biol. 2002, 34, 1534–1538. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Role of Epithelial Cells in Idiopathic Pulmonary Fibrosis: From Innocent Targets to Serial Killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Thannickal, V.J. Mechanistic Links between Aging and Lung Fibrosis. Biogerontology 2013, 14, 609–615. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Revealing the Pathogenic and Aging-Related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis. an Integral Model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; Kaminski, N. Idiopathic Pulmonary Fibrosis: Aberrant Recapitulation of Developmental Programs? PLoS Med. 2008, 5, e62. [Google Scholar] [CrossRef]
- Zhang, H.; Song, M.; Guo, J.; Ma, J.; Qiu, M.; Yang, Z. The Function of Non-Coding RNAs in Idiopathic Pulmonary Fibrosis. Open Med. Wars. Pol. 2021, 16, 481–490. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, H.; Zhao, F.; Li, H.; Gao, R.; Yan, B.; Ren, J.; Yang, J. Decrypting the Crosstalk of Noncoding RNAs in the Progression of IPF. Mol. Biol. Rep. 2020, 47, 3169–3179. [Google Scholar] [CrossRef]
- Li, Q.; Li, M.; Zheng, K.; Li, H.; Yang, H.; Ma, S.; Zhong, M. Detection of MicroRNA Expression Levels Based on Microarray Analysis for Classification of Idiopathic Pulmonary Fibrosis. Exp. Ther. Med. 2020, 20, 3096–3103. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Zamore, P.D. MicroPrimer: The Biogenesis and Function of MicroRNA. Dev. Camb. Engl. 2005, 132, 4645–4652. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of Primary MicroRNAs by the Microprocessor Complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lee, R.C.; Ambros, V. An Extensive Class of Small RNAs in Caenorhabditis Elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer Is Essential for Mouse Development. Nat. Genet. 2003, 35, 215–217. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most Mammalian MRNAs Are Conserved Targets of MicroRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Plasterk, R.H.A. The Diverse Functions of MicroRNAs in Animal Development and Disease. Dev. Cell 2006, 11, 441–450. [Google Scholar] [CrossRef]
- Londin, E.; Loher, P.; Telonis, A.G.; Quann, K.; Clark, P.; Jing, Y.; Hatzimichael, E.; Kirino, Y.; Honda, S.; Lally, M.; et al. Analysis of 13 Cell Types Reveals Evidence for the Expression of Numerous Novel Primate- and Tissue-Specific MicroRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E1106–E1115. [Google Scholar] [CrossRef]
- Ambros, V. The Functions of Animal MicroRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Thomson, J.M.; Wong, H.Y.F.; Hammond, S.M.; Hogan, B.L.M. Transgenic Over-Expression of the MicroRNA MiR-17-92 Cluster Promotes Proliferation and Inhibits Differentiation of Lung Epithelial Progenitor Cells. Dev. Biol. 2007, 310, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.J.; Pérez de Castro, I.; Malumbres, M. Control of Cell Proliferation Pathways by MicroRNAs. Cell Cycle Georget. Tex 2008, 7, 3143–3148. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Tsitsiou, E.; Herrick, S.E.; Lindsay, M.A. MicroRNAs and the Regulation of Fibrosis. FEBS J. 2010, 277, 2015–2021. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes MiR15 and MiR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of MicroRNAs in Serum: A Novel Class of Biomarkers for Diagnosis of Cancer and Other Diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and Role of Let-7d in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. MiR-21 Mediates Fibrogenic Activation of Pulmonary Fibroblasts and Lung Fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Pandit, K.V.; Milosevic, J.; Kaminski, N. MicroRNAs in Idiopathic Pulmonary Fibrosis. Transl. Res. 2011, 157, 191–199. [Google Scholar] [CrossRef]
- Fan, L.; Yu, X.; Huang, Z.; Zheng, S.; Zhou, Y.; Lv, H.; Zeng, Y.; Xu, J.-F.; Zhu, X.; Yi, X. Analysis of Microarray-Identified Genes and MicroRNAs Associated with Idiopathic Pulmonary Fibrosis. Mediators Inflamm. 2017, 2017, 1804240. [Google Scholar] [CrossRef]
- Wang, L.; Huang, W.; Zhang, L.; Chen, Q.; Zhao, H. Molecular Pathogenesis Involved in Human Idiopathic Pulmonary Fibrosis Based on an Integrated MicroRNA-MRNA Interaction Network. Mol. Med. Rep. 2018, 18, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.E.; Ahangari, F.; Li, Q.; Jain, S.; Verleden, S.E.; Herazo-Maya, J.; Vukmirovic, M.; DeIuliis, G.; Tzouvelekis, A.; Tanabe, N.; et al. Transcriptional Regulatory Model of Fibrosis Progression in the Human Lung. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Qian, W.; Cai, X.; Qian, Q.; Zhang, X. Identification and Validation of Potential Biomarkers and Pathways for Idiopathic Pulmonary Fibrosis by Comprehensive Bioinformatics Analysis. BioMed Res. Int. 2021, 2021, 5545312. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Xiong, Y.; Zhang, G.; Chang, J. MicroRNAs in Idiopathic Pulmonary Fibrosis, New Research Progress and Their Pathophysiological Implication. Exp. Lung Res. 2018, 44, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Stolzenburg, L.R.; Harris, A. The Role of MicroRNAs in Chronic Respiratory Disease: Recent Insights. Biol. Chem. 2018, 399, 219–234. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray Analysis Shows That Some MicroRNAs Downregulate Large Numbers of Target MRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Radisky, D.C. Epithelial-Mesenchymal Transition. J. Cell Sci. 2005, 118, 4325–4326. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex Networks Orchestrate Epithelial-Mesenchymal Transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The Epithelial-Mesenchymal Transition: New Insights in Signaling, Development, and Disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Thannickal, V.J.; Pardo, A.; Zisman, D.A.; Martinez, F.J.; Lynch, J.P. Idiopathic Pulmonary Fibrosis: Pathogenesis and Therapeutic Approaches. Drugs 2004, 64, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells by Transforming Growth Factor-Β1. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic Pulmonary Fibrosis. Lancet Lond. Engl. 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-Beta-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Königshoff, M.; Balsara, N.; Pfaff, E.-M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt Signaling Is Increased in Idiopathic Pulmonary Fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef]
- Yan, Z.; Kui, Z.; Ping, Z. Reviews and Prospectives of Signaling Pathway Analysis in Idiopathic Pulmonary Fibrosis. Autoimmun. Rev. 2014, 13, 1020–1025. [Google Scholar] [CrossRef]
- Chanda, D.; Otoupalova, E.; Hough, K.P.; Locy, M.L.; Bernard, K.; Deshane, J.S.; Sanderson, R.D.; Mobley, J.A.; Thannickal, V.J. Fibronectin on the Surface of Extracellular Vesicles Mediates Fibroblast Invasion. Am. J. Respir. Cell Mol. Biol. 2019, 60, 279–288. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.-Q.; Kalluri, R.; Müller, G.A.; Neilson, E.G. Role of Basic Fibroblast Growth Factor-2 in Epithelial-Mesenchymal Transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef]
- Uttamsingh, S.; Bao, X.; Nguyen, K.T.; Bhanot, M.; Gong, J.; Chan, J.L.-K.; Liu, F.; Chu, T.T.; Wang, L.-H. Synergistic Effect between EGF and TGF-Beta1 in Inducing Oncogenic Properties of Intestinal Epithelial Cells. Oncogene 2008, 27, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Morali, O.G.; Delmas, V.; Moore, R.; Jeanney, C.; Thiery, J.P.; Larue, L. IGF-II Induces Rapid Beta-Catenin Relocation to the Nucleus during Epithelium to Mesenchyme Transition. Oncogene 2001, 20, 4942–4950. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-Beta Signaling and the Fibrotic Response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Niehrs, C. The Complex World of WNT Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Lynch, J.P.; Martinez, F.J. Mechanisms of Pulmonary Fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-Beta1 Induces Human Alveolar Epithelial to Mesenchymal Cell Transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis: From Targeting to Biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad Regulation in TGF-Beta Signal Transduction. J. Cell Sci. 2001, 114, 4359–4369. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-Dependent and Smad-Independent Pathways in TGF-Beta Family Signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Massague, J. Integration of Smad and MAPK Pathways: A Link and a Linker Revisited. Genes Dev. 2003, 17, 2993–2997. [Google Scholar] [CrossRef]
- Miyazono, K.; ten Dijke, P.; Heldin, C.H. TGF-Beta Signaling by Smad Proteins. Adv. Immunol. 2000, 75, 115–157. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. How Cells Read TGF-Beta Signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Bartram, U.; Speer, C.P. The Role of Transforming Growth Factor Beta in Lung Development and Disease. Chest 2004, 125, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Xaubet, A.; Marin-Arguedas, A.; Lario, S.; Ancochea, J.; Morell, F.; Ruiz-Manzano, J.; Rodriguez-Becerra, E.; Rodriguez-Arias, J.M.; Inigo, P.; Sanz, S.; et al. Transforming Growth Factor-Beta1 Gene Polymorphisms Are Associated with Disease Progression in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 431–435. [Google Scholar] [CrossRef]
- Flanders, K.C. Smad3 as a Mediator of the Fibrotic Response. Int. J. Exp. Pathol. 2004, 85, 47–64. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. TGF-Beta-Induced EMT: Mechanisms and Implications for Fibrotic Lung Disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. New Cellular and Molecular Mechanisms of Lung Injury and Fibrosis in Idiopathic Pulmonary Fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Xu, L.; Chen, Y.G.; Massagué, J. The Nuclear Import Function of Smad2 Is Masked by SARA and Unmasked by TGFbeta-Dependent Phosphorylation. Nat. Cell Biol. 2000, 2, 559–562. [Google Scholar] [CrossRef]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of Transforming Growth Factor Beta in Human Disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial-Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 Binds to Smurf2 to Form an E3 Ubiquitin Ligase That Targets the TGF Beta Receptor for Degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- He, W.; Dai, C. Key Fibrogenic Signaling. Curr. Pathobiol. Rep. 2015, 3, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-Beta1-Induced EMT in Vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Böttinger, E.P. TGF-Beta and Epithelial-to-Mesenchymal Transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Kang, H. Role of MicroRNAs in TGF-β Signaling Pathway-Mediated Pulmonary Fibrosis. Int. J. Mol. Sci. 2017, 18, 2527. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 Family and MiR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.-Y.; et al. An Autocrine TGF-Beta/ZEB/MiR-200 Signaling Network Regulates Establishment and Maintenance of Epithelial-Mesenchymal Transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef]
- Yu, L.; Hébert, M.C.; Zhang, Y.E. TGF-Beta Receptor-Activated P38 MAP Kinase Mediates Smad-Independent TGF-Beta Responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 Mediates Smad-Independent Activation of JNK and P38 by TGF-Beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-Induced Activation of MTOR Complex 2 Drives Epithelial-Mesenchymal Transition and Cell Invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Horowitz, J.C. Evolving Concepts of Apoptosis in Idiopathic Pulmonary Fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Hergert, P.; Kahm, J.; Jessurun, J.; Henke, C. Pathological Alteration of FoxO3a Activity Promotes Idiopathic Pulmonary Fibrosis Fibroblast Proliferation on Type i Collagen Matrix. Am. J. Pathol. 2011, 179, 2420–2430. [Google Scholar] [CrossRef]
- Nho, R.S.; Xia, H.; Diebold, D.; Kahm, J.; Kleidon, J.; White, E.; Henke, C.A. PTEN Regulates Fibroblast Elimination during Collagen Matrix Contraction. J. Biol. Chem. 2006, 281, 33291–33301. [Google Scholar] [CrossRef]
- Yamada, K.M.; Araki, M. Tumor Suppressor PTEN: Modulator of Cell Signaling, Growth, Migration and Apoptosis. J. Cell Sci. 2001, 114, 2375–2382. [Google Scholar] [CrossRef]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.-R.; Zhang, Y.; Wrana, J.L. Regulation of the Polarity Protein Par6 by TGFbeta Receptors Controls Epithelial Cell Plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef]
- Pellegrin, S.; Mellor, H. Actin Stress Fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/Beta-Catenin Pathway Activation in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between β-Catenin and Transforming Growth Factor-β Signaling Pathways Mediate Epithelial-Mesenchymal Transition and Are Dependent on the Transcriptional Co-Activator CAMP-Response Element-Binding Protein (CREB)-Binding Protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef] [PubMed]
- Pongracz, J.E.; Stockley, R.A. Wnt Signalling in Lung Development and Diseases. Respir. Res. 2006, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL Receptor-Related Proteins 5 and 6 in Wnt/Beta-Catenin Signaling: Arrows Point the Way. Dev. Camb. Engl. 2004, 131, 1663–1677. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Habas, R.; Dawid, I.B. Dishevelled and Wnt Signaling: Is the Nucleus the Final Frontier? J. Biol. 2005, 4, 2. [Google Scholar] [CrossRef][Green Version]
- Tao, Q.; Yokota, C.; Puck, H.; Kofron, M.; Birsoy, B.; Yan, D.; Asashima, M.; Wylie, C.C.; Lin, X.; Heasman, J. Maternal Wnt11 Activates the Canonical Wnt Signaling Pathway Required for Axis Formation in Xenopus Embryos. Cell 2005, 120, 857–871. [Google Scholar] [CrossRef]
- Steinhart, Z.; Angers, S. Wnt Signaling in Development and Tissue Homeostasis. Dev. Camb. Engl. 2018, 145, dev146589. [Google Scholar] [CrossRef]
- Vuga, L.J.; Ben-Yehudah, A.; Kovkarova-Naumovski, E.; Oriss, T.; Gibson, K.F.; Feghali-Bostwick, C.; Kaminski, N. WNT5A Is a Regulator of Fibroblast Proliferation and Resistance to Apoptosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 583–589. [Google Scholar] [CrossRef]
- Burgy, O.; Medina, A.M.; Mayr, C.H.; Behr, J.; Schiller, H.B.; Königshoff, M. Extracellular Vesicles from Broncho-Alveolar Lavage Act on Cellular Behavior during Lung Fibrosis. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, T.; Shan, S.; Wang, S.; Bian, W.; Ren, T.; Yang, D. MiR-124 Regulates Transforming Growth Factor-Β1 Induced Differentiation of Lung Resident Mesenchymal Stem Cells to Myofibroblast by Repressing Wnt/β-Catenin Signaling. Dev. Biol. 2019, 449, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Teague, T.T.; Payne, S.R.; Kelly, B.T.; Dempsey, T.M.; McCoy, R.G.; Sangaralingham, L.R.; Limper, A.H. Evaluation for Clinical Benefit of Metformin in Patients with Idiopathic Pulmonary Fibrosis and Type 2 Diabetes Mellitus: A National Claims-Based Cohort Analysis. Respir. Res. 2022, 23, 91. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Yu, L.; Deng, Y.; Li, D.; Yu, X.; Chen, D.; Lu, Y.; Liu, S.; Chen, R. Pemafibrate Attenuates Pulmonary Fibrosis by Inhibiting Myofibroblast Differentiation. Int. Immunopharmacol. 2022, 108, 108728. [Google Scholar] [CrossRef] [PubMed]
- Simonson, B.; Das, S. MicroRNA Therapeutics: The Next Magic Bullet? Mini Rev. Med. Chem. 2015, 15, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Usman, K.; Hsieh, A.; Hackett, T.-L. The Role of MiRNAs in Extracellular Matrix Repair and Chronic Fibrotic Lung Diseases. Cells 2021, 10, 1706. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, Q.; Zhang, Q.; Wang, Z.; Guan, S. A Novel Molecular Mechanism of MicroRNA-21 Inducing Pulmonary Fibrosis and Human Pulmonary Fibroblast Extracellular Matrix through Transforming Growth Factor Β1-Mediated SMADs Activation. J. Cell. Biochem. 2018, 119, 7834–7843. [Google Scholar] [CrossRef]
- Liu, H.; He, Y.; Jiang, Z.; Shen, S.; Mei, J.; Tang, M. Prodigiosin Alleviates Pulmonary Fibrosis Through Inhibiting MiRNA-410 and TGF-Β1/ADAMTS-1 Signaling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 501–511. [Google Scholar] [CrossRef]
- Dai, W.-J.; Qiu, J.; Sun, J.; Ma, C.-L.; Huang, N.; Jiang, Y.; Zeng, J.; Ren, B.-C.; Li, W.-C.; Li, Y.-H. Downregulation of MicroRNA-9 Reduces Inflammatory Response and Fibroblast Proliferation in Mice with Idiopathic Pulmonary Fibrosis through the ANO1-Mediated TGF-β-Smad3 Pathway. J. Cell. Physiol. 2019, 234, 2552–2565. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Zhong, M.; Wang, Y.; Zou, L.; Wang, M.; Gong, X.; Wang, X.; Zhou, C.; Ma, X.; et al. MiR-301a Suppression within Fibroblasts Limits the Progression of Fibrosis through the TSC1/MTOR Pathway. Mol. Ther. Nucleic Acids 2020, 21, 217–228. [Google Scholar] [CrossRef]
- Huang, Y.; Xie, Y.; Abel, P.W.; Wei, P.; Plowman, J.; Toews, M.L.; Strah, H.; Siddique, A.; Bailey, K.L.; Tu, Y. TGF-Β1-Induced MiR-424 Promotes Pulmonary Myofibroblast Differentiation by Targeting Slit2 Protein Expression. Biochem. Pharmacol. 2020, 180, 114172. [Google Scholar] [CrossRef]
- Wang, C.; Cao, H.; Gu, S.; Shi, C.; Chen, X.; Han, X. Expression Analysis of MicroRNAs and MRNAs in Myofibroblast Differentiation of Lung Resident Mesenchymal Stem Cells. Differ. Res. Biol. Divers. 2020, 112, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gu, S.; Cao, H.; Li, Z.; Xiang, Z.; Hu, K.; Han, X. MiR-877-3p Targets Smad7 and Is Associated with Myofibroblast Differentiation and Bleomycin-Induced Lung Fibrosis. Sci. Rep. 2016, 6, 30122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Q.; Zhou, Y.; Yang, Z.; Tan, M. Inhibition of MiR-182-5p Attenuates Pulmonary Fibrosis via TGF-β/Smad Pathway. Hum. Exp. Toxicol. 2020, 39, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Li, B.-B.; Tan, Y.-J.; Zhang, G.-M.; Cheng, G.-L.; Ren, Y.-S. MicroRNA-31/184 Is Involved in Transforming Growth Factor-β-Induced Apoptosis in A549 Human Alveolar Adenocarcinoma Cells. Life Sci. 2020, 242, 117205. [Google Scholar] [CrossRef]
- Ge, L.; Habiel, D.M.; Hansbro, P.M.; Kim, R.Y.; Gharib, S.A.; Edelman, J.D.; Königshoff, M.; Parimon, T.; Brauer, R.; Huang, Y.; et al. MiR-323a-3p Regulates Lung Fibrosis by Targeting Multiple Profibrotic Pathways. JCI Insight 2016, 1, e90301. [Google Scholar] [CrossRef]
- Yamada, Y.; Takanashi, M.; Sudo, K.; Ueda, S.; Ohno, S.-I.; Kuroda, M. Novel Form of MiR-29b Suppresses Bleomycin-Induced Pulmonary Fibrosis. PLoS ONE 2017, 12, e0171957. [Google Scholar] [CrossRef]
- Tsitoura, E.; Wells, A.U.; Karagiannis, K.; Lasithiotaki, I.; Vasarmidi, E.; Bibaki, E.; Koutoulaki, C.; Sato, H.; Spandidos, D.A.; Siafakas, N.M.; et al. MiR-185/AKT and MiR-29a/Collagen 1a Pathways Are Activated in IPF BAL Cells. Oncotarget 2016, 7, 74569–74581. [Google Scholar] [CrossRef]
- Lei, G.-S.; Kline, H.L.; Lee, C.-H.; Wilkes, D.S.; Zhang, C. Regulation of Collagen V Expression and Epithelial-Mesenchymal Transition by MiR-185 and MiR-186 during Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2016, 186, 2310–2316. [Google Scholar] [CrossRef]
- Li, S.; Geng, J.; Xu, X.; Huang, X.; Leng, D.; Jiang, D.; Liang, J.; Wang, C.; Jiang, D.; Dai, H. MiR-130b-3p Modulates Epithelial-Mesenchymal Crosstalk in Lung Fibrosis by Targeting IGF-1. PLoS ONE 2016, 11, e0150418. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Liu, J.-S.; Tang, H.-K.; Nie, J.; Zhu, J.-X.; Wen, L.-L.; Guo, Q.-L. MiR-221 Targets HMGA2 to Inhibit Bleomycin-induced Pulmonary Fibrosis by Regulating TGF-β1/Smad3-Induced EMT. Int. J. Mol. Med. 2016, 38, 1208–1216. [Google Scholar] [CrossRef]
- Stolzenburg, L.R.; Wachtel, S.; Dang, H.; Harris, A. MiR-1343 Attenuates Pathways of Fibrosis by Targeting the TGF-β Receptors. Biochem. J. 2016, 473, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Banerjee, S.; Xie, N.; Ge, J.; Liu, R.-M.; Matalon, S.; Thannickal, V.J.; Liu, G. MicroRNA-27a-3p Is a Negative Regulator of Lung Fibrosis by Targeting Myofibroblast Differentiation. Am. J. Respir. Cell Mol. Biol. 2016, 54, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Huang, C.; Senavirathna, L.; Wang, P.; Liu, L. MiR-27b Inhibits Fibroblast Activation via Targeting TGFβ Signaling Pathway. BMC Cell Biol. 2017, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ye, H.; Xiang, F.; Song, L.-J.; Zhou, L.-L.; Cai, P.-C.; Zhang, J.-C.; Yu, F.; Shi, H.-Z.; Su, Y.; et al. MiR-18a-5p Inhibits Sub-Pleural Pulmonary Fibrosis by Targeting TGF-β Receptor II. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 728–738. [Google Scholar] [CrossRef]
- Huang, C.; Xiao, X.; Yang, Y.; Mishra, A.; Liang, Y.; Zeng, X.; Yang, X.; Xu, D.; Blackburn, M.R.; Henke, C.A.; et al. MicroRNA-101 Attenuates Pulmonary Fibrosis by Inhibiting Fibroblast Proliferation and Activation. J. Biol. Chem. 2017, 292, 16420–16439. [Google Scholar] [CrossRef]
- Chi, L.; Xiao, Y.; Zhu, L.; Zhang, M.; Xu, B.; Xia, H.; Jiang, Z.; Wu, W. MicroRNA-155 Attenuates Profibrotic Effects of Transforming Growth Factor-Beta on Human Lung Fibroblasts. J. Biol. Regul. Homeost. Agents 2019, 33, 1415–1424. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, Y.; Ping, F.; Yi, L.; Zeng, Z.; Li, Y. MiR-200b/c Attenuates Lipopolysaccharide-Induced Early Pulmonary Fibrosis by Targeting ZEB1/2 via P38 MAPK and TGF-β/Smad3 Signaling Pathways. Lab. Investig. J. Tech. Methods Pathol. 2018, 98, 339–359. [Google Scholar] [CrossRef]
- Souma, K.; Shichino, S.; Hashimoto, S.; Ueha, S.; Tsukui, T.; Nakajima, T.; Suzuki, H.I.; Shand, F.H.W.; Inagaki, Y.; Nagase, T.; et al. Lung Fibroblasts Express a MiR-19a-19b-20a Sub-Cluster to Suppress TGF-β-Associated Fibroblast Activation in Murine Pulmonary Fibrosis. Sci. Rep. 2018, 8, 16642. [Google Scholar] [CrossRef]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming Growth Factor (TGF)-Β1-Induced MiR-133a Inhibits Myofibroblast Differentiation and Pulmonary Fibrosis. Cell Death Dis. 2019, 10, 670. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, T.; Hu, X.; Liu, Z.; Zhao, L.; Liu, H.; Liu, Z.; Ma, L. Downregulation of MicroRNA-30a in Bronchoalveolar Lavage Fluid from Idiopathic Pulmonary Fibrosis Patients. Mol. Med. Rep. 2018, 18, 5799–5806. [Google Scholar] [CrossRef]
- Wu, G.; Xie, B.; Lu, C.; Chen, C.; Zhou, J.; Deng, Z. MicroRNA-30a Attenuates TGF-Β1-Induced Activation of Pulmonary Fibroblast Cell by Targeting FAP-α. J. Cell. Mol. Med. 2020, 24, 3745–3750. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.-Q.; Guo, Y.-F.; Yang, S.-M.; Ma, H.-H.; Li, J. MiR-340-5p Mitigates the Proliferation and Activation of Fibroblast in Lung Fibrosis by Targeting TGF-β/P38/ATF1 Signaling Pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6252–6261. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Ishiyama, J. MicroRNA-29c Regulates Apoptosis Sensitivity via Modulation of the Cell-Surface Death Receptor, Fas, in Lung Fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1050–L1061. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Liang, J.; Geng, Y.; Liu, N.; Kurkciyan, A.; Kulur, V.; Leng, D.; Deng, N.; Liu, Z.; Song, J.; et al. MicroRNA-29c Prevents Pulmonary Fibrosis by Regulating Epithelial Cell Renewal and Apoptosis. Am. J. Respir. Cell Mol. Biol. 2017, 57, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Lin, S.C.; Zhao, M.S.; Yu, B.; Li, X.Y.; Gao, Q.; Lin, D.J. MicroRNA-142-3p Inhibits Apoptosis and Inflammation Induced by Bleomycin through down-Regulation of Cox-2 in MLE-12 Cells. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Medicas E Biol. 2017, 50, e5974. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; An, Y.; Zhang, X.; Wang, Z.; Duan, H. Experimental Pulmonary Fibrosis Was Suppressed by MicroRNA-506 through NF-Kappa-Mediated Apoptosis and Inflammation. Cell Tissue Res. 2019, 378, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xu, F.; Xie, S.; Zuo, W.; Wen, G.; Zhao, T.; Wan, X. MicroRNA-448 Overexpression Inhibits Fibroblast Proliferation and Collagen Synthesis and Promotes Cell Apoptosis via Targeting ABCC3 through the JNK Signaling Pathway. J. Cell. Physiol. 2020, 235, 1374–1385. [Google Scholar] [CrossRef]
- Ma, X.; Yan, F.; Deng, Q.; Li, F.; Lu, Z.; Liu, M.; Wang, L.; Conklin, D.J.; McCracken, J.; Srivastava, S.; et al. Modulation of Tumorigenesis by the Pro-Inflammatory MicroRNA MiR-301a in Mouse Models of Lung Cancer and Colorectal Cancer. Cell Discov. 2015, 1, 15005. [Google Scholar] [CrossRef]
- Pandit, K.V.; Milosevic, J. MicroRNA Regulatory Networks in Idiopathic Pulmonary Fibrosis. Biochem. Cell Biol. Biochim. Biol. Cell. 2015, 93, 129–137. [Google Scholar] [CrossRef]
- Oak, S.R.; Murray, L.; Herath, A.; Sleeman, M.; Anderson, I.; Joshi, A.D.; Coelho, A.L.; Flaherty, K.R.; Toews, G.B.; Knight, D.; et al. A Micro RNA Processing Defect in Rapidly Progressing Idiopathic Pulmonary Fibrosis. PLoS ONE 2011, 6, e21253. [Google Scholar] [CrossRef]
- Vittal, R.; Mickler, E.A.; Fisher, A.J.; Zhang, C.; Rothhaar, K.; Gu, H.; Brown, K.M.; Emtiazjoo, A.; Lott, J.M.; Frye, S.B.; et al. Type V Collagen Induced Tolerance Suppresses Collagen Deposition, TGF-β and Associated Transcripts in Pulmonary Fibrosis. PLoS ONE 2013, 8, e76451. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Enomoto, M.; Fujii, H.; Sekiya, Y.; Yoshizato, K.; Ikeda, K.; Kawada, N. MicroRNA-221/222 Upregulation Indicates the Activation of Stellate Cells and the Progression of Liver Fibrosis. Gut 2012, 61, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 Is Induced during the Macrophage Inflammatory Response. Proc. Natl. Acad. Sci. USA. 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, M.; Pereira, P.M.; Dunand-Sauthier, I.; Barras, E.; Reith, W.; Santos, M.A.; Pierre, P. MicroRNA-155 Modulates the Interleukin-1 Signaling Pathway in Activated Human Monocyte-Derived Dendritic Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Pottier, N.; Maurin, T.; Chevalier, B.; Puisségur, M.-P.; Lebrigand, K.; Robbe-Sermesant, K.; Bertero, T.; Lino Cardenas, C.L.; Courcot, E.; Rios, G.; et al. Identification of Keratinocyte Growth Factor as a Target of MicroRNA-155 in Lung Fibroblasts: Implication in Epithelial-Mesenchymal Interactions. PLoS ONE 2009, 4, e6718. [Google Scholar] [CrossRef]
- Yang, S.; Banerjee, S.; de Freitas, A.; Sanders, Y.Y.; Ding, Q.; Matalon, S.; Thannickal, V.J.; Abraham, E.; Liu, G. Participation of MiR-200 in Pulmonary Fibrosis. Am. J. Pathol. 2012, 180, 484–493. [Google Scholar] [CrossRef]
- Yang, G.; Yang, C.; She, Y.; Shen, Z.; Gao, P. LINC01354 Enhances the Proliferation and Invasion of Lung Cancer Cells by Regulating MiR-340-5p/ATF1 Signaling Pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3737–3744. [Google Scholar] [CrossRef]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. Mir-29 Regulates Mcl-1 Protein Expression and Apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. MiR-29 Is a Major Regulator of Genes Associated with Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- He, Y.; Huang, C.; Lin, X.; Li, J. MicroRNA-29 Family, a Crucial Therapeutic Target for Fibrosis Diseases. Biochimie 2013, 95, 1355–1359. [Google Scholar] [CrossRef]
- Wang, Y.; Ouyang, M.; Wang, Q.; Jian, Z. MicroRNA-142-3p Inhibits Hypoxia/Reoxygenation-induced Apoptosis and Fibrosis of Cardiomyocytes by Targeting High Mobility Group Box 1. Int. J. Mol. Med. 2016, 38, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Mezzanzanica, D.; Zhang, W. MiR-506: A Multitasker in Suppression of the Epithelial-to-Mesenchymal Transition. RNA Dis. Houst. Tex 2014, 1, e447. [Google Scholar] [CrossRef]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the Complexities of the NF-KappaB Signalling Pathway Using Mouse Knockout and Transgenic Models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Fei, F.; Li, F.; Zhuang, B.; Zheng, Y.; Wan, Y.; Chen, J. MiR-448 Is a Novel Prognostic Factor of Lung Squamous Cell Carcinoma and Regulates Cells Growth and Metastasis by Targeting DCLK1. Biomed. Pharmacother. 2017, 89, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Ladewig, E.; Okamura, K.; Flynt, A.S.; Westholm, J.O.; Lai, E.C. Discovery of Hundreds of Mirtrons in Mouse and Human Small RNA Data. Genome Res. 2012, 22, 1634–1645. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. MiRBase: MicroRNA Sequences, Targets and Gene Nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L.M. Multiple Stromal Populations Contribute to Pulmonary Fibrosis without Evidence for Epithelial to Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, X.; Long, Y.; Chen, Y. Emerging Therapeutic Potential of Mesenchymal Stem Cell-Derived Extracellular Vesicles in Chronic Respiratory Diseases: An Overview of Recent Progress. Front. Bioeng. Biotechnol. 2022, 10, 845042. [Google Scholar] [CrossRef]
- Santos-Álvarez, J.C.; Velázquez-Enríquez, J.M.; García-Carrillo, R.; Rodríguez-Beas, C.; Ramírez-Hernández, A.A.; Reyes-Jiménez, E.; González-García, K.; López-Martínez, A.; Pérez-Campos Mayoral, L.; Aguilar-Ruiz, S.R.; et al. MiRNAs Contained in Extracellular Vesicles Cargo Contribute to the Progression of Idiopathic Pulmonary Fibrosis: An In Vitro Aproach. Cells 2022, 11, 1112. [Google Scholar] [CrossRef]
- Zhou, J.; Lin, Y.; Kang, X.; Liu, Z.; Zhang, W.; Xu, F. MicroRNA-186 in Extracellular Vesicles from Bone Marrow Mesenchymal Stem Cells Alleviates Idiopathic Pulmonary Fibrosis via Interaction with SOX4 and DKK1. Stem Cell Res. Ther. 2021, 12, 96. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Experimental Model | Mi-RNA | Function | Target of Action | Status in IPF | Effect on IPF and Pathway Associated | Reference |
|---|---|---|---|---|---|---|
| BLM fibrosis mice model and HELF-TGFβind | miR-21 | Pro-fibrotic | Smad 2 Smad 3 Smad 4 | Up-regulated | TGFβ/Smad pathway activation | [126] |
| BLM fibrosis rat model | miR-410 | Pro-fibrotic | ADAMTS1 | Up regulated | ↑ECM proteins deposition and fibroblasts proliferation | [127] |
| LR-MSCs-TGFβind | miR-124 | Pro fibrotic | AXIN1 | Up regulated | ↑Fibrogenic differentiation by Wnt signaling pathway activation | [121] |
| BLM fibrosis mice model | miR-9 | Pro-fibrotic | ANO1 | Up regulated | TGFβ-Smad-3 pathway activation and apoptosis suppression | [128] |
| HFLT, HFLF-TGFβind and LF of BLM fibrosis mice model | miR-301a | Pro-fibrotic | TSC1 | Up regulated | Activation of TSC1/mTOR pathway | [129] |
| NHLF-TGFβind and HFLF | miR-424 | Pro-fibrotic | SLIT2 | Up regulated | ↑myofibroblast differentiation ↑CTGF and α-SMA by TGFβ1/Smad3 pathway | [130] |
| Mice LR-MSC-TGFβind | miR-152–3p, 140–3p, 148b-3p, 7a-5p _______ miR-34a-5p, 27b-3p, 323-3p, 27a-3p, 34c-5p, 128–3p, 224–5p | Pro-fibrotic _______ Pro-fibrotic | KLF4 ________ ING5 | Up regulated _______ Up regulated | Activation of TGFβ and Wnt/β- catenin pathways ____________ Activation of PI3K/AKT and Wnt/β- catenin pathways | [131] |
| 1.2 MiRNAs with pro-fibrotic properties but negative regulators of Smad6/Smad7 expression. | ||||||
| LR-MSCs-TGFβ and bleomycin mice model | miR-877-3p | Pro-fibrotic | Smad7 | Up regulated | ↑fibrotic markers and myofibroblasts differentiation induced by TGFβ pathway | [132] |
| BLM fibrosis mice model and HELF-TGFβind | miR-182-5p | Pro-fibrotic | Smad7 | Up regulated | ↑pro-fibrotic markers by pSmad2, pSmad3 and TGFβ pathway activation | [133] |
| A549-TGFβind | miR-31 | Pro-fibrotic | Smad6 | Up-regulated | TGFβ Smad2 activation | [134] |
| 1.3 MiRNAs with anti-fibrotic properties and with regulatory activity of signaling pathways linked with IPF. | ||||||
| A549-TGFβind | miR-184 | Anti- fibrotic | Smad2/Akt | Down-regulated | TGFβ-P13k-AKt pathway activation | [134] |
| HFLT and BLM fibrosis mice model | miR-323a-3p | Anti-fibrotic | TGFα Smad2 | Down regulated | Activation of both TGFα and TGFβ signaling pathways | [135] |
| BLM fibrosis mice model | miR-29b | Anti-fibrotic | COL1A1 and COL3A1 | Down-regulated | ↑collagen expression | [136] |
| Human BAL cells and THP-1 cells | miR-185 miR-29a | Anti-fibrotic Anti- fibrotic | AKT COL1A1 | Down regulated Down Regulated | ↑collagen expression and ↑of EMC proteins deposition | [137] |
| Adenocarcinoma epithelial cell lines and HFLT | miR-185 and miR-186 | Anti-fibrotic | COL5A1 | Down-regulated | ↑EMT process | [138] |
| HFLT | miR-130b-3p | Anti-fibrotic | IGF-1 | Down regulated | ↑Collagen1A1 expression and ↑proliferation and migration of fibroblasts | [139] |
| HFLT, A549, HBEC and BLM fibrosis mice model | miR-221 | Anti-fibrotic | HMGA2 | Down regulated | ↑EMT and fibrotic markers by activation of TGFβ1/Smad3 pathway | [140] |
| A549-TGFβind NHLF-TGFβind | miR-1343 | Anti-fibrotic | TGFβRI and TGFβRII | Down regulated | Activation of TGFβ pathway and ↑expression of fibrotic markers and EMT | [141] |
| HFLF and NHLF | miR-27a-3p | Anti-fibrotic | Smad2 Smad4 and α-SMA | Down Regulated | ↑ myofibroblasts differentiation by TGFβ pathway activation | [142] |
| Lung and LF of BLM fibrosis mice model | miR-27b | Anti-fibrotic | TGFβR1 and Smad2 | Down Regulated | ↑fibrotic markers by TGFβ pathway activation | [143] |
| PMCs | miR-18a-5p | Anti-fibrotic | TGFβRII | Down regulated | ↑EMT through TGFβ1 Smad2/3 complex | [144] |
| LL29, NHLF and BLM mice model | MiR-101 | Anti-fibrotic | Col1A1 Col3A1 α-SMA | Down regulated | ↑fibroblast proliferation and activation via TGFBR1 | [145] |
| NHLF-TGFβind | MiR-155 | Anti-fibrotic | Smad1 | Down regulated | ↑fibroblast proliferation, migration, and collagen synthesis by TGFβ activity | [146] |
| LPS early pulmonary fibrosis mouse model | miR-200b/c | Anti-fibrotic | ZEB1/2 | Down regulated | Activation of EMT via p38 MAPK and TGF-β/Smad3 pathway | [147] |
| LF of BLM and silica fibrotic mice model | miR-19a- 19b-20a sub-cluster | Anti-fibrotic | TGFβRII | Down regulated | Activation of TGFβ pathway ↑expression of pro-fibrotic genes | [148] |
| NHLF-TGFβind | miR-133a | Anti-fibrotic | COL1A1 CTGF α-SMA TGFβR1 | Up-regulated | Functions as a feed-back negative regulator of TGFβ pathway | [149] |
| 293T-TGFβind A549-TGFβind | miR-30a | Anti-fibrotic | TAB3 αSMA FNT | Down regulated | Activation of TGFB pathway | [150] |
| MRC-5 cells | miR-30a | Anti-fibrotic | FAP-α | Down regulated | ↑FAP-α, col1a and α-α-SMA synthesis | [151] |
| NHLF-TGFβind | miR-340-5p | Anti-fibrotic | FNTATF1 | Down- regulated | ↑TGFβ/P38/ATF1 pathway | [152] |
| 1.4 MicroRNAs down-regulated and associated with Apoptosis. | ||||||
| LF cell lines and LT of BLM-fibrotic mice model | miR-29c | Anti-fibrotic | PARP-1 LOXL2 COL3A1 SPARC | Down-regulated | ↑Resistance to Fas-mediated apoptosis and ↓activity of caspase-8/caspase-3/PARP-1 pathway | [153] |
| AEC2s of IPF, healthy LT and MLECs | miR-29c | Anti-fibrotic | Foxo3a | Down regulated | ↑apoptosis associated with reduced epithelial cell renewal | [154] |
| MLE-12 cells plusBleomycin | miR-142-3p | Anti-fibrotic | Cox-2 | Down regulated | ↑apoptosisP13K/AKT/mTOR pathway inactivation | [155] |
| LPS-lung fibrosis mice model | MiR-506 | Anti-fibrotic | p65 (NFκβ subunit) | Down regulated | Apoptosis resistance | [156] |
| LF of BLM fibrosis mice model | miR-448 | Anti-fibrotic | ABCC3 | Down regulated | Apoptosis resistance by JNK signaling pathway ↑proliferation and collagen synthesis | [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadena-Suárez, A.R.; Hernández-Hernández, H.A.; Alvarado-Vásquez, N.; Rangel-Escareño, C.; Sommer, B.; Negrete-García, M.C. Role of MicroRNAs in Signaling Pathways Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis: A Focus on Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2022, 23, 6613. https://doi.org/10.3390/ijms23126613
Cadena-Suárez AR, Hernández-Hernández HA, Alvarado-Vásquez N, Rangel-Escareño C, Sommer B, Negrete-García MC. Role of MicroRNAs in Signaling Pathways Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis: A Focus on Epithelial-Mesenchymal Transition. International Journal of Molecular Sciences. 2022; 23(12):6613. https://doi.org/10.3390/ijms23126613
Chicago/Turabian StyleCadena-Suárez, Ana Ruth, Hilda Arely Hernández-Hernández, Noé Alvarado-Vásquez, Claudia Rangel-Escareño, Bettina Sommer, and María Cristina Negrete-García. 2022. "Role of MicroRNAs in Signaling Pathways Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis: A Focus on Epithelial-Mesenchymal Transition" International Journal of Molecular Sciences 23, no. 12: 6613. https://doi.org/10.3390/ijms23126613
APA StyleCadena-Suárez, A. R., Hernández-Hernández, H. A., Alvarado-Vásquez, N., Rangel-Escareño, C., Sommer, B., & Negrete-García, M. C. (2022). Role of MicroRNAs in Signaling Pathways Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis: A Focus on Epithelial-Mesenchymal Transition. International Journal of Molecular Sciences, 23(12), 6613. https://doi.org/10.3390/ijms23126613