
Large Benefit from Simple Things: High-Dose Vitamin A Improves RBP4-Related Retinal Dystrophy

 ,
,  and
and 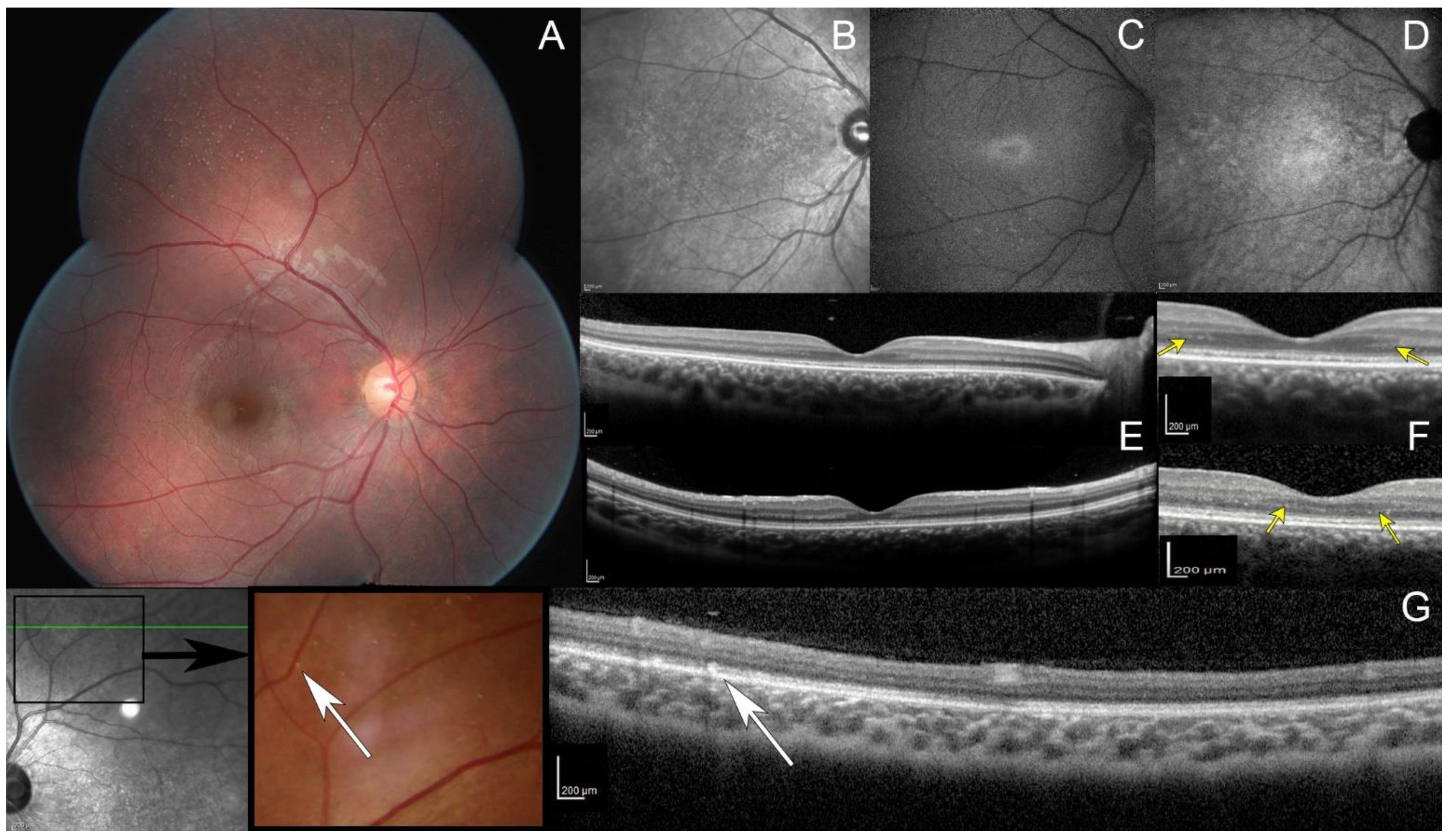{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Detailed Case Description
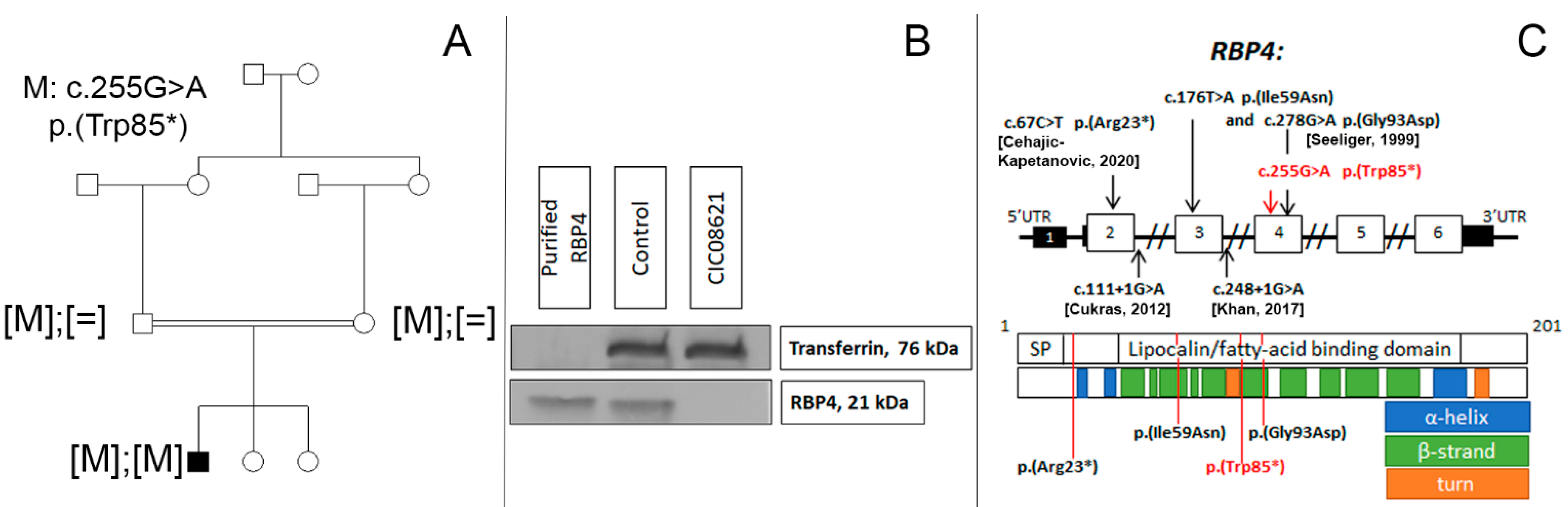
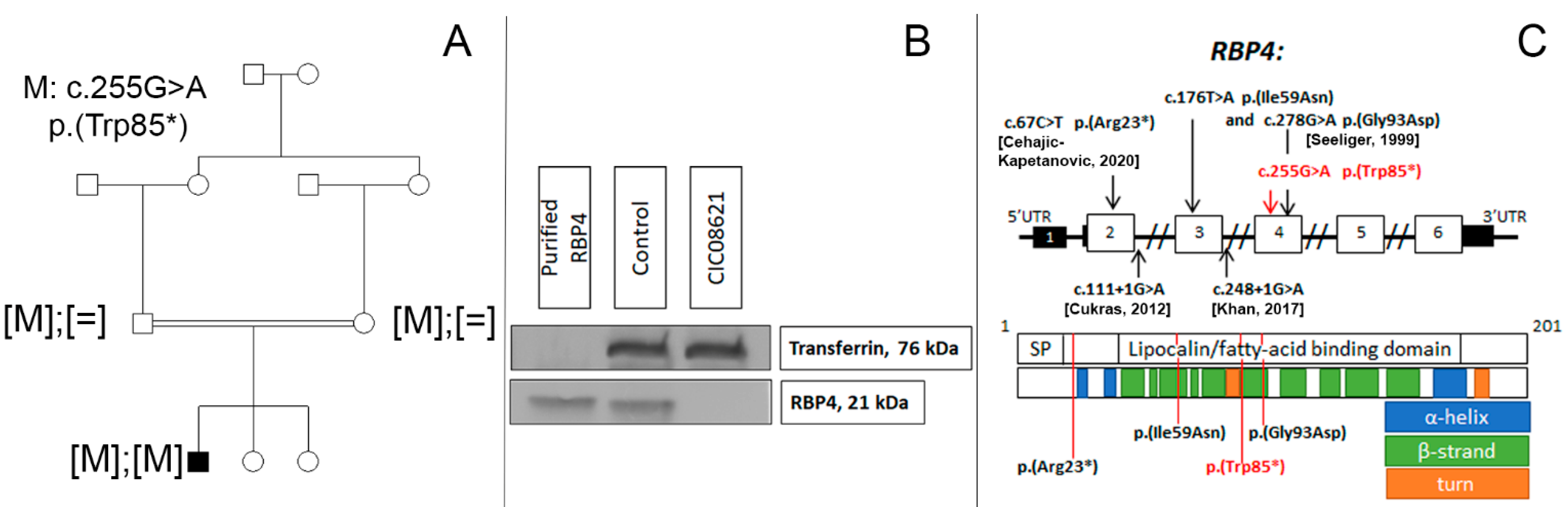
2.2. Genetic and Functional Studies
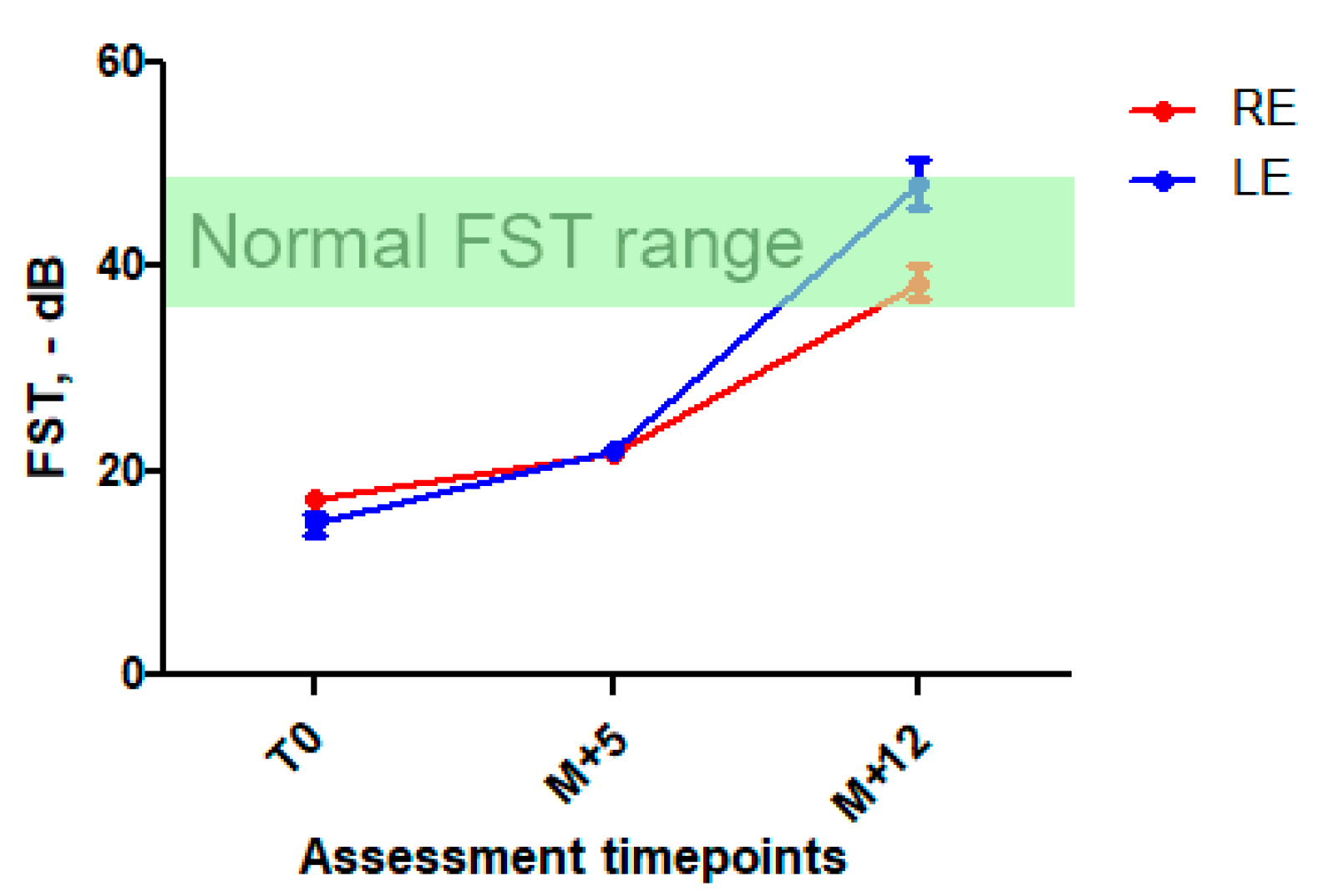
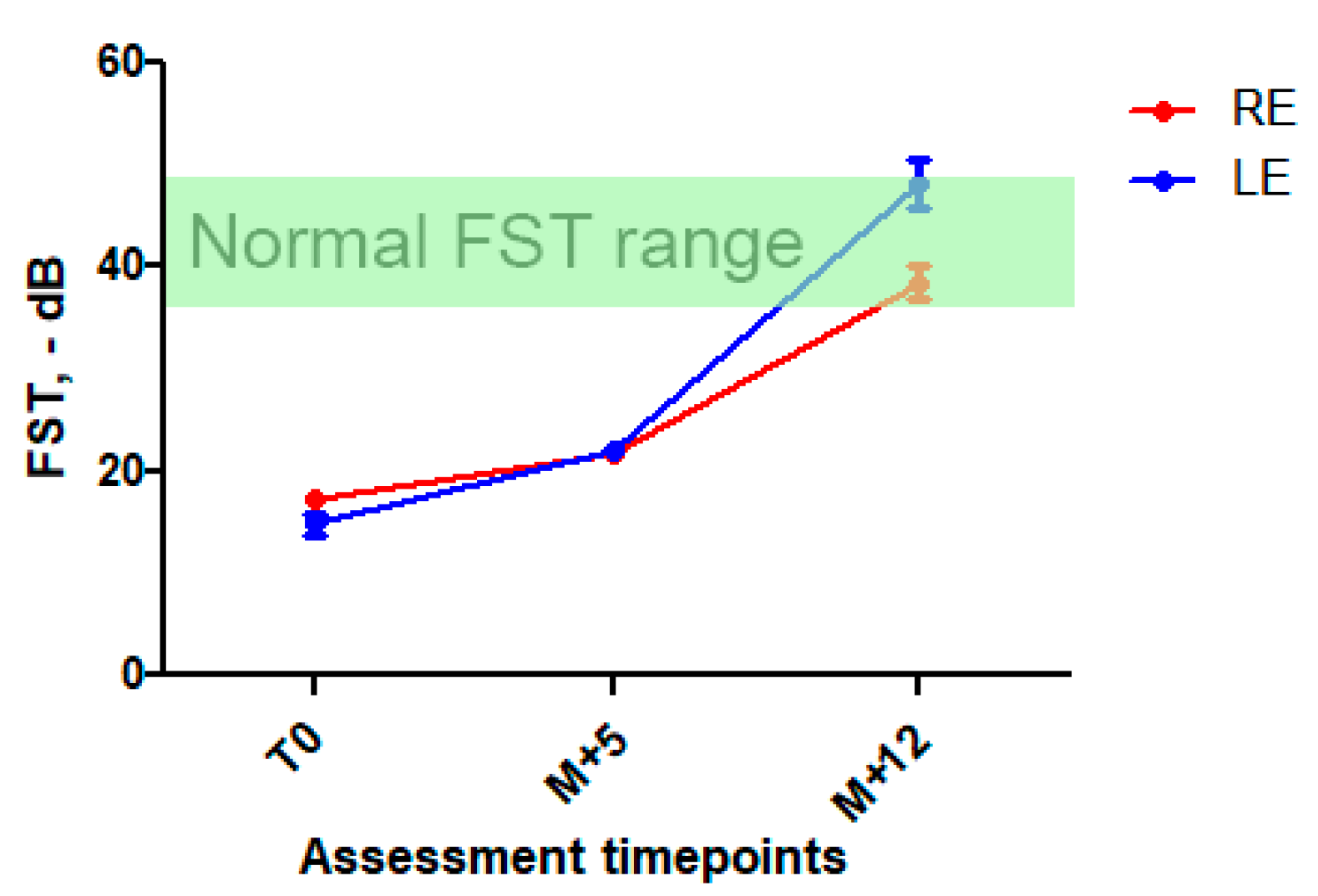
2.3. Treatment and Follow-Up
3. Discussion
4. Materials and Methods
4.1. Clinical Studies
4.2. Genetic Analysis
4.3. Western Blot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grant, C.A.; Berson, E.L. Treatable Forms of Retinitis Pigmentosa Associated with Systemic Neurological Disorders. Int Ophthalmol. Clin. 2001, 41, 103–110. [Google Scholar] [CrossRef]
- Weleber, R.G.; Kurz, D.E.; Trzupek, K.M. Treatment of Retinal and Choroidal Degenerations and Dystrophies: Current Status and Prospects for Gene-Based Therapy. Ophthalmol. Clin. N. Am. 2003, 16, 583–593. [Google Scholar] [CrossRef]
- Chowers, I.; Banin, E.; Merin, S.; Cooper, M.; Granot, E. Long-Term Assessment of Combined Vitamin A and E Treatment for the Prevention of Retinal Degeneration in Abetalipoproteinaemia and Hypobetalipoproteinaemia Patients. Eye 2001, 15, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balfoort, B.M.; Buijs, M.J.N.; Ten Asbroek, A.L.M.A.; Bergen, A.A.B.; Boon, C.J.F.; Ferreira, E.A.; Houtkooper, R.H.; Wagenmakers, M.A.E.M.; Wanders, R.J.A.; Waterham, H.R.; et al. A Review of Treatment Modalities in Gyrate Atrophy of the Choroid and Retina (GACR). Mol. Genet. Metab. 2021, 134, 96–116. [Google Scholar] [CrossRef] [PubMed]
- Casalino, G.; Pierro, L.; Manitto, M.P.; Michaelides, M.; Bandello, F. Resolution of Cystoid Macular Edema Following Arginine-Restricted Diet and Vitamin B6 Supplementation in a Case of Gyrate Atrophy. J. AAPOS 2018, 22, 321–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüether, K.; Baldwin, E.; Casteels, M.; Feher, M.D.; Horn, M.; Kuranoff, S.; Leroy, B.P.; Wanders, R.J.; Wierzbicki, A.S. Adult Refsum Disease: A Form of Tapetoretinal Dystrophy Accessible to Therapy. Surv. Ophthalmol. 2010, 55, 531–538. [Google Scholar] [CrossRef]
- Benson, M.D.; MacDonald, I.M.; Sheehan, M.; Jain, S. Improved Electroretinographic Responses Following Dietary Intervention in a Patient with Refsum Disease. JIMD Rep. 2020, 55, 32–37. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, M.W.; Biesalski, H.K.; Wissinger, B.; Gollnick, H.; Gielen, S.; Frank, J.; Beck, S.; Zrenner, E. Phenotype in Retinol Deficiency Due to a Hereditary Defect in Retinol Binding Protein Synthesis. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3–11. [Google Scholar]
- Cehajic-Kapetanovic, J.; Jasani, K.M.; Shanks, M.; Clouston, P.; MacLaren, R.E. A Novel Homozygous c.67C>T Variant in Retinol Binding Protein 4 (RBP4) Associated with Retinitis Pigmentosa and Childhood Acne Vulgaris. Ophthalmic. Genet. 2020, 41, 288–292. [Google Scholar] [CrossRef]
- Roman, A.J.; Cideciyan, A.V.; Aleman, T.S.; Jacobson, S.G. Full-Field Stimulus Testing (FST) to Quantify Visual Perception in Severely Blind Candidates for Treatment Trials. Physiol. Meas. 2007, 28, N51–N56. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Birch, D.G. Psychophysical Assessment of Low Visual Function in Patients with Retinal Degenerative Diseases (RDDs) with the Diagnosys Full-Field Stimulus Threshold (D-FST). Doc. Ophthalmol. 2009, 119, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiser, P.D.; Palczewski, K. Retinoids and Retinal Diseases. Annu. Rev. Vis. Sci. 2016, 2, 197–234. [Google Scholar] [CrossRef] [Green Version]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a Complex of Two Plasma Proteins: Transthyretin and Retinol-Binding Protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A Membrane Receptor for Retinol Binding Protein Mediates Cellular Uptake of Vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Quadro, L.; Hamberger, L.; Colantuoni, V.; Gottesman, M.E.; Blaner, W.S. Understanding the Physiological Role of Retinol-Binding Protein in Vitamin A Metabolism Using Transgenic and Knockout Mouse Models. Mol. Asp. Med. 2003, 24, 421–430. [Google Scholar] [CrossRef]
- Quadro, L. Impaired Retinal Function and Vitamin A Availability in Mice Lacking Retinol-Binding Protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Phelps, E.; Balangue, M.J.; Dockins, A.; Moiseyev, G.; Shin, Y.; Kane, S.; Otalora, L.; Ma, J.-X.; Farjo, R.; et al. Transgenic Mice Over-Expressing RBP4 Have RBP4-Dependent and Light-Independent Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4375–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukras, C.; Gaasterland, T.; Lee, P.; Gudiseva, H.V.; Chavali, V.R.M.; Pullakhandam, R.; Maranhao, B.; Edsall, L.; Soares, S.; Reddy, G.B.; et al. Exome Analysis Identified a Novel Mutation in the RBP4 Gene in a Consanguineous Pedigree with Retinal Dystrophy and Developmental Abnormalities. PLoS ONE 2012, 7, e50205. [Google Scholar] [CrossRef]
- Khan, K.N.; Carss, K.; Raymond, F.L.; Islam, F.; Nihr BioResource-Rare Diseases Consortium, null; Moore, A.T.; Michaelides, M.; Arno, G. Vitamin A Deficiency Due to Bi-Allelic Mutation of RBP4: There’s More to It than Meets the Eye. Ophthalmic. Genet. 2017, 38, 465–466. [Google Scholar] [CrossRef]
- Riggs, L.A. Electroretinography in Cases of Night Blindness. Am. J. Ophthalmol. 1954, 38, 70–78. [Google Scholar] [CrossRef]
- Driessen, C.A.; Winkens, H.J.; Hoffmann, K.; Kuhlmann, L.D.; Janssen, B.P.; Van Vugt, A.H.; Van Hooser, J.P.; Wieringa, B.E.; Deutman, A.F.; Palczewski, K.; et al. Disruption of the 11-Cis-Retinol Dehydrogenase Gene Leads to Accumulation of Cis-Retinols and Cis-Retinyl Esters. Mol. Cell. Biol. 2000, 20, 4275–4287. [Google Scholar] [CrossRef] [Green Version]
- Jang, G.F.; Van Hooser, J.P.; Kuksa, V.; McBee, J.K.; He, Y.G.; Janssen, J.J.; Driessen, C.A.; Palczewski, K. Characterization of a Dehydrogenase Activity Responsible for Oxidation of 11-Cis-Retinol in the Retinal Pigment Epithelium of Mice with a Disrupted RDH5 Gene. A Model for the Human Hereditary Disease Fundus Albipunctatus. J. Biol. Chem. 2001, 276, 32456–32465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstedt, M.; Jonsson, F.; Köhn, L.; Burstedt, M.; Kivitalo, M.; Golovleva, I. Genotype-Phenotype Correlations in Bothnia Dystrophy Caused by RLBP1 Gene Sequence Variations. Acta Ophthalmol. 2013, 91, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Dessalces, E.; Bocquet, B.; Bourien, J.; Zanlonghi, X.; Verdet, R.; Meunier, I.; Hamel, C.P. Early-Onset Foveal Involvement in Retinitis Punctata Albescens With Mutations in RLBP1. JAMA Ophthalmol. 2013, 131, 1314. [Google Scholar] [CrossRef] [Green Version]
- Burstedt, M.S.I.; Sandgren, O.; Golovleva, I.; Wachtmeister, L. Effects of Prolonged Dark Adaptation in Patients with Retinitis Pigmentosa of Bothnia Type: An Electrophysiological Study. Doc. Ophthalmol. 2008, 116, 193–205. [Google Scholar] [CrossRef]
- Souied, E.; Soubrane, G.; Benlian, P.; Coscas, G.J.; Gerber, S.; Munnich, A.; Kaplan, J. Retinitis Punctata Albescens Associated With the Arg135Trp Mutation in the Rhodopsin Gene. Am. J. Ophthalmol. 1996, 121, 19–25. [Google Scholar] [CrossRef]
- Kajiwara, K.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. A Null Mutation in the Human Peripherin/RDS Gene in a Family with Autosomal Dominant Retinitis Punctata Albescens. Nat. Genet. 1993, 3, 208–212. [Google Scholar] [CrossRef]
- Littink, K.W.; van Genderen, M.M.; van Schooneveld, M.J.; Visser, L.; Riemslag, F.C.C.; Keunen, J.E.E.; Bakker, B.; Zonneveld, M.N.; den Hollander, A.I.; Cremers, F.P.M.; et al. A Homozygous Frameshift Mutation in LRAT Causes Retinitis Punctata Albescens. Ophthalmology 2012, 119, 1899–1906. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; van Duuren, R.J.G.; Van Cauwenbergh, C.; Ten Brink, J.B.; De Baere, E.; Florijn, R.J.; Schalij-Delfos, N.E.; Leroy, B.P.; Bergen, A.A.; et al. Long-Term Follow-Up of Retinal Degenerations Associated With LRAT Mutations and Their Comparability to Phenotypes Associated With RPE65 Mutations. Transl. Vis. Sci. Technol. 2019, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Schatz, P.; Preising, M.; Lorenz, B.; Sander, B.; Larsen, M.; Rosenberg, T. Fundus Albipunctatus Associated with Compound Heterozygous Mutations in RPE65. Ophthalmology 2011, 118, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Ramtohul, P.; Denis, D. RPE65-Mutation Associated Fundus Albipunctatus with Cone Dystrophy. Ophthalmol. Retina 2019, 3, 535. [Google Scholar] [CrossRef]
- Apushkin, M.A.; Fishman, G.A. Improvement in Visual Function and Fundus Findings for a Patient with Vitamin A-Deficient Retinopathy. Retina 2005, 25, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Genead, M.A.; Fishman, G.A.; Lindeman, M. Fundus White Spots and Acquired Night Blindness Due to Vitamin A Deficiency. Doc. Ophthalmol. 2009, 119, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Garrity, S.T.; Brucker, A.J. Retinal Structure in Vitamin A Deficiency as Explored with Multimodal Imaging. Doc. Ophthalmol. 2013, 127, 239–243. [Google Scholar] [CrossRef]
- Cvekl, A.; Wang, W.-L. Retinoic Acid Signaling in Mammalian Eye Development. Exp. Eye Res. 2009, 89, 280–291. [Google Scholar] [CrossRef] [Green Version]
- MacGilchrist, A.J.; Mills, P.R.; Noble, M.; Foulds, W.S.; Simpson, J.A.; Watkinson, G. Abetalipoproteinaemia in Adults: Role of Vitamin Therapy. J. Inherit. Metab. Dis. 1988, 11, 184–190. [Google Scholar] [CrossRef]
- Bishara, S.; Merin, S.; Cooper, M.; Azizi, E.; Delpre, G.; Deckelbaum, R.J. Combined Vitamin A and E Therapy Prevents Retinal Electrophysiological Deterioration in Abetalipoproteinaemia. Br. J. Ophthalmol. 1982, 66, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; Frank, J.; Beck, S.C.; Heinrich, F.; Illek, B.; Reifen, R.; Gollnick, H.; Seeliger, M.W.; Wissinger, B.; Zrenner, E. Biochemical but Not Clinical Vitamin A Deficiency Results from Mutations in the Gene for Retinol Binding Protein. Am. J. Clin. Nutr. 1999, 69, 931–936. [Google Scholar] [CrossRef]
- Hathcock, J.N.; Hattan, D.G.; Jenkins, M.Y.; McDonald, J.T.; Sundaresan, P.R.; Wilkening, V.L. Evaluation of Vitamin A Toxicity. Am. J. Clin. Nutr. 1990, 52, 183–202. [Google Scholar] [CrossRef]
- During, A.; Doraiswamy, S.; Harrison, E.H. Xanthophylls Are Preferentially Taken up Compared with β-Carotene by Retinal Cells via a SRBI-Dependent Mechanism. J. Lipid Res. 2008, 49, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Audo, I. An Unusual Retinal Phenotype Associated With a Novel Mutation in RHO. Arch. Ophthalmol. 2010, 128, 1036. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for Full-Field Clinical Electroretinography (2015 Update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Lancelot, M.; Mohand-Saïd, S.; Antonio, A.; Germain, A.; Sahel, J.; Bhattacharya, S.S.; Zeitz, C. Novel C2orf71 Mutations Account for ∼1% of Cases in a Large French ArRP Cohort. Hum. Mutat. 2011, 32, E2091–E2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audo, I.; Bujakowska, K.M.; Léveillard, T.; Mohand-Saïd, S.; Lancelot, M.-E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.-P.; Letexier, M.; et al. Development and Application of a Next-Generation-Sequencing (NGS) Approach to Detect Known and Novel Gene Defects Underlying Retinal Diseases. Orphanet. J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.E.; Wason, C.J.; Blüher, M.; Kahn, B.B. Shortcomings in Methodology Complicate Measurements of Serum Retinol Binding Protein (RBP4) in Insulin-Resistant Human Subjects. Diabetologia 2007, 50, 814–823. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnov, V.M.; Wilmet, B.; Nassisi, M.; Condroyer, C.; Antonio, A.; Andrieu, C.; Devisme, C.; Sancho, S.; Sahel, J.-A.; Zeitz, C.; et al. Large Benefit from Simple Things: High-Dose Vitamin A Improves RBP4-Related Retinal Dystrophy. Int. J. Mol. Sci. 2022, 23, 6590. https://doi.org/10.3390/ijms23126590
Smirnov VM, Wilmet B, Nassisi M, Condroyer C, Antonio A, Andrieu C, Devisme C, Sancho S, Sahel J-A, Zeitz C, et al. Large Benefit from Simple Things: High-Dose Vitamin A Improves RBP4-Related Retinal Dystrophy. International Journal of Molecular Sciences. 2022; 23(12):6590. https://doi.org/10.3390/ijms23126590
Chicago/Turabian StyleSmirnov, Vasily M., Baptiste Wilmet, Marco Nassisi, Christel Condroyer, Aline Antonio, Camille Andrieu, Céline Devisme, Serge Sancho, José-Alain Sahel, Christina Zeitz, and et al. 2022. "Large Benefit from Simple Things: High-Dose Vitamin A Improves RBP4-Related Retinal Dystrophy" International Journal of Molecular Sciences 23, no. 12: 6590. https://doi.org/10.3390/ijms23126590
APA StyleSmirnov, V. M., Wilmet, B., Nassisi, M., Condroyer, C., Antonio, A., Andrieu, C., Devisme, C., Sancho, S., Sahel, J.-A., Zeitz, C., & Audo, I. (2022). Large Benefit from Simple Things: High-Dose Vitamin A Improves RBP4-Related Retinal Dystrophy. International Journal of Molecular Sciences, 23(12), 6590. https://doi.org/10.3390/ijms23126590