
Patient-Derived Tumor Organoids for Guidance of Personalized Drug Therapies in Recurrent Glioblastoma

 , , , ,
, , , ,
Abstract

1. Introduction
2. Results
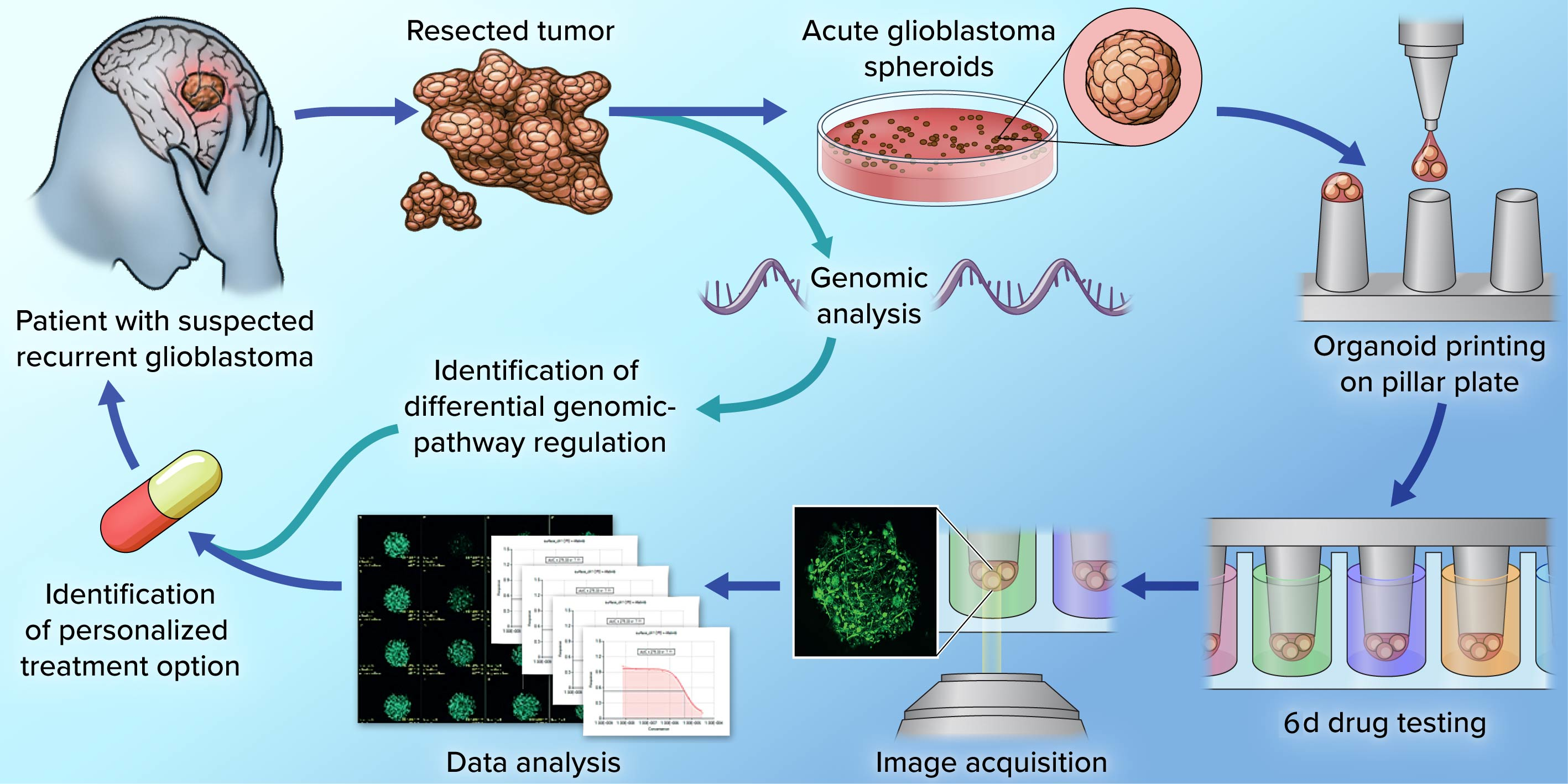
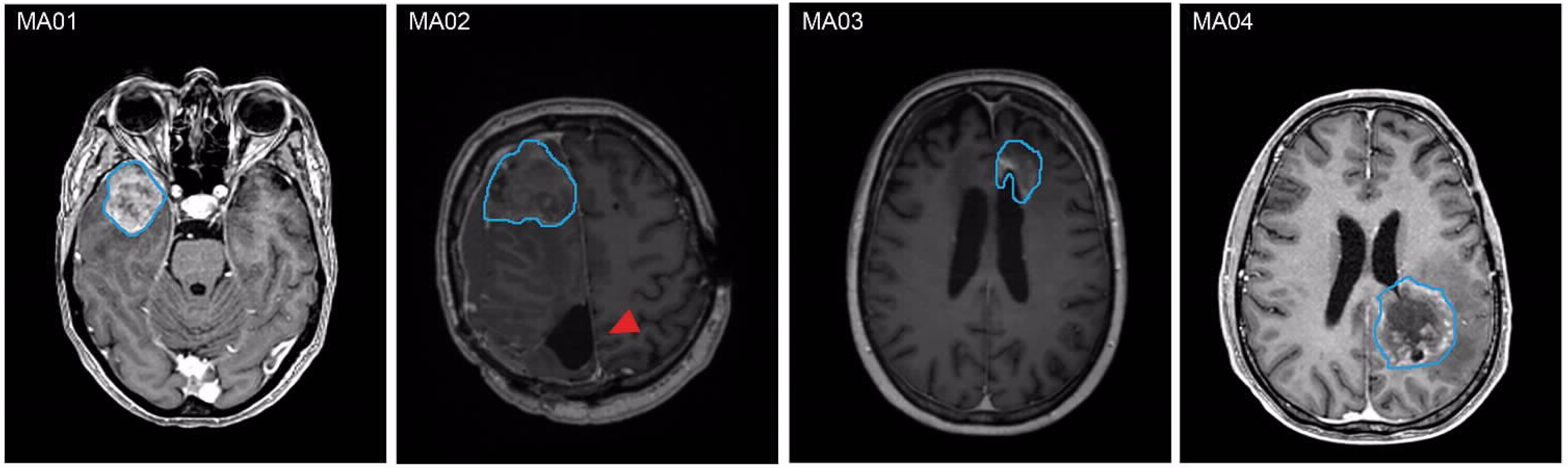
2.1. PD-GBOs Can Be Generated from Resected Tumor Tissue
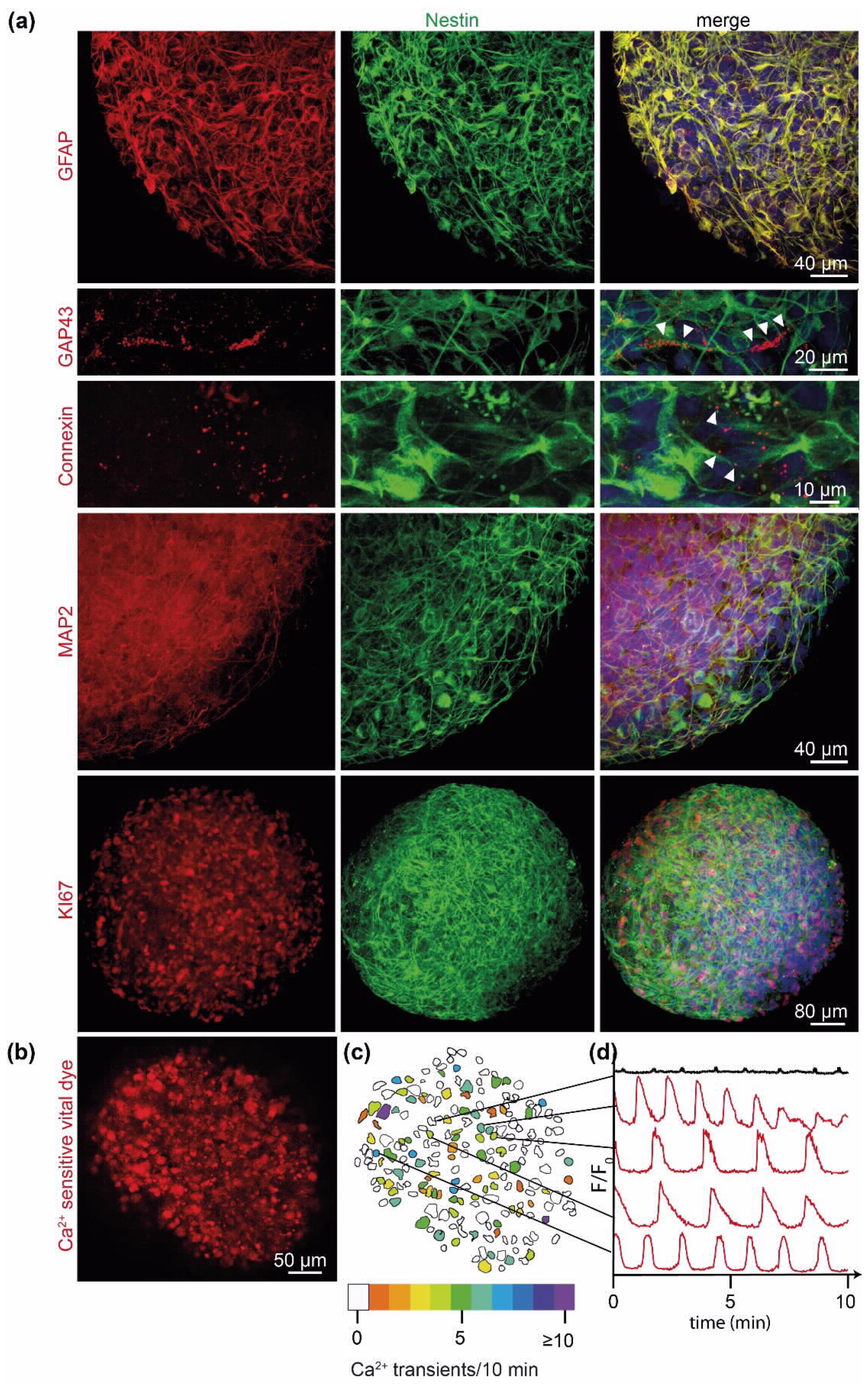
2.2. PD-GBOs Recapitulate Actual Glioblastoma Properties
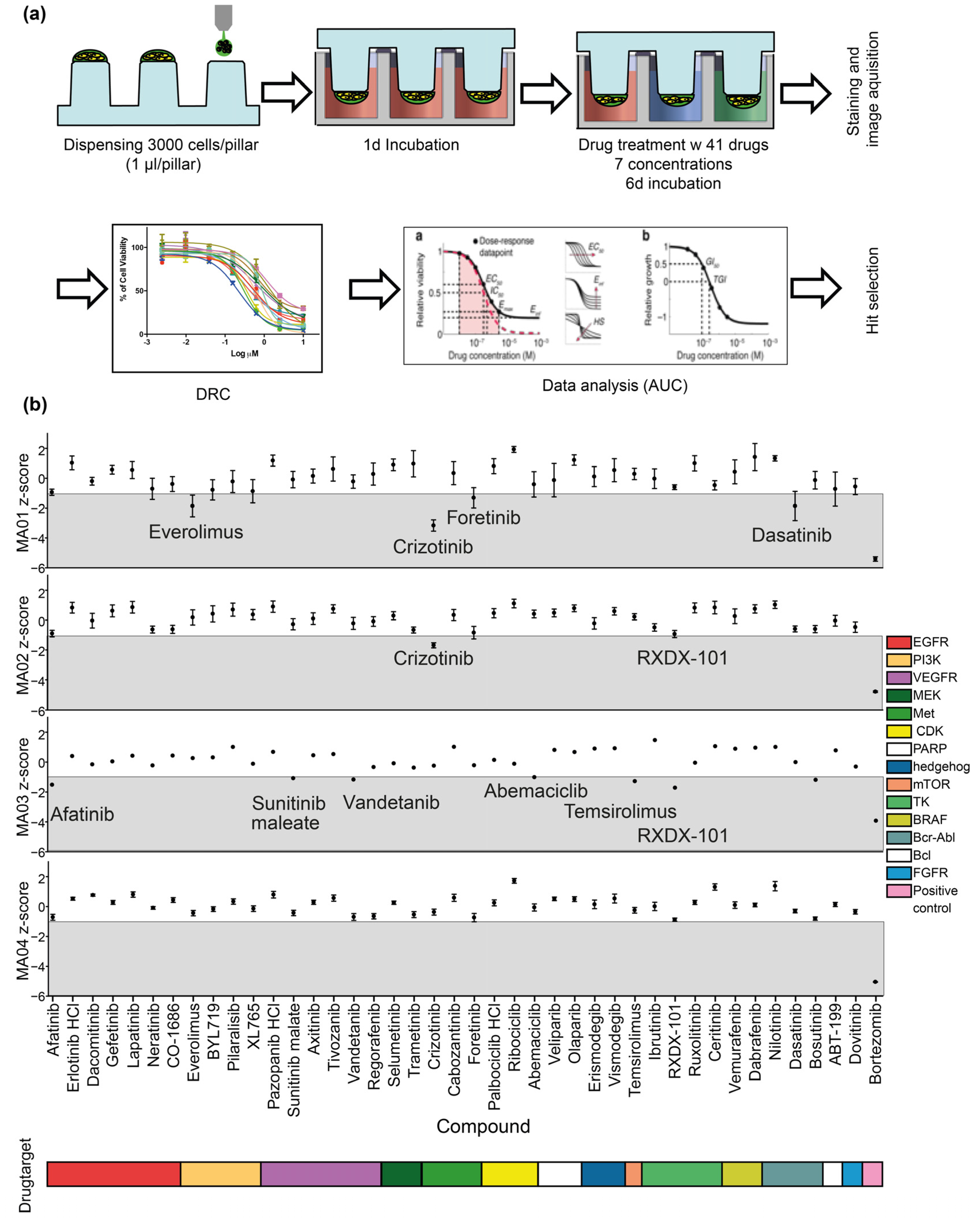
2.3. Personalized 3D Drug Screens Using PD-GBOs
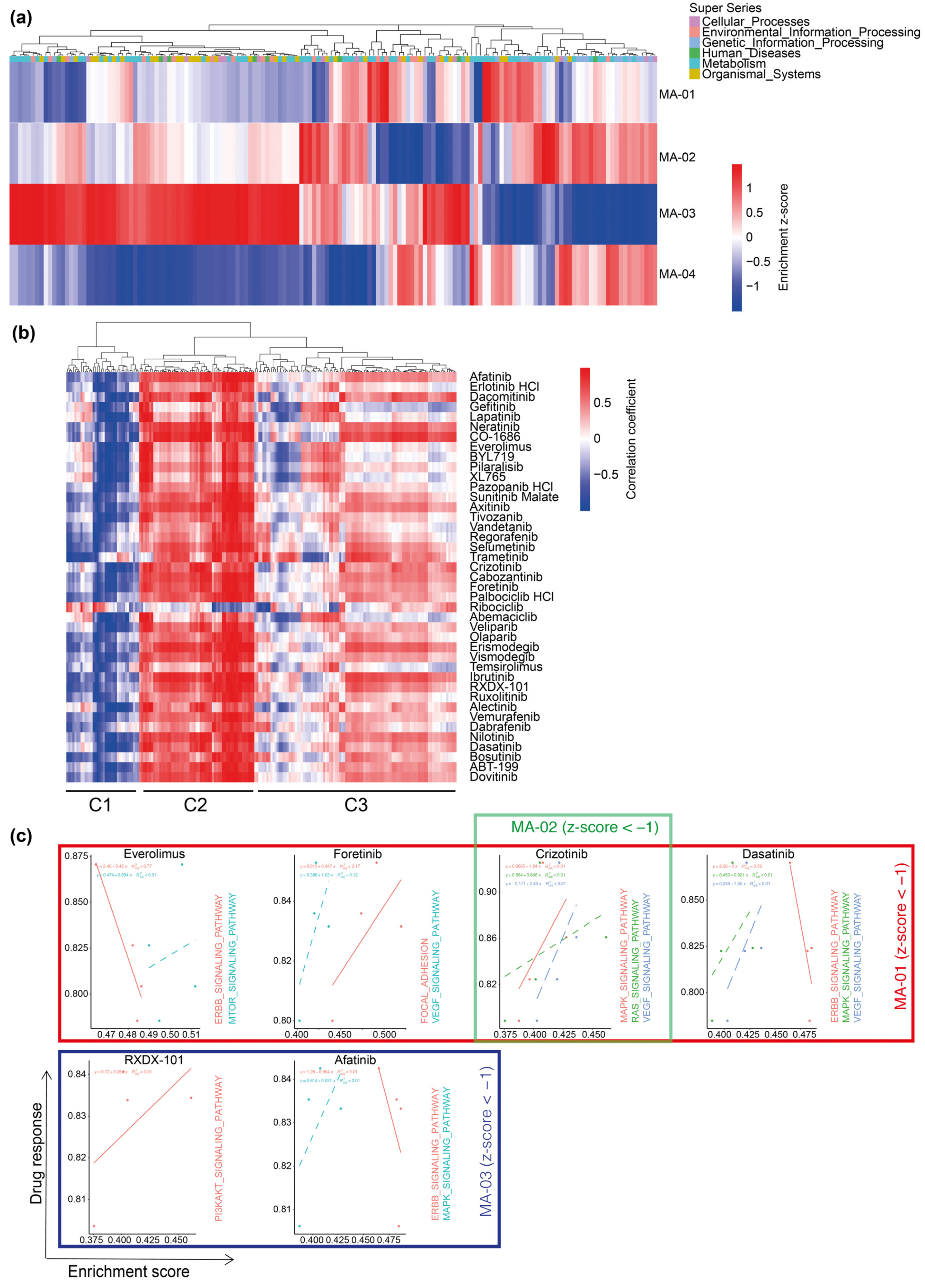
2.4. Transcriptome Analysis of Parental Tumor Tissue
3. Discussion
4. Materials and Methods
4.1. Drug Library Preparation
4.2. Glioblastoma Resection and Tissue Processing
4.3. Establishment of Acute Glioblastoma Spheroids
4.4. Cell Printing and Drug Screening on the 384-Pillar Array
4.5. Immunofluorescence, Histology, and Immunohistochemistry
4.6. Live-Cell Image Acquisition and Analysis
4.7. Analysis of Functional Drug Testing
4.8. RNA-Sequencing Data Processing
4.9. Pathway Enrichment Analysis
4.10. Correlation Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20 (Suppl. S4), iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef]
- Alshami, J.; Guiot, M.C.; Owen, S.; Kavan, P.; Gibson, N.; Solca, F.; Cseh, A.; Reardon, D.A.; Muanza, T. Afatinib, an irreversible ErbB family blocker, with protracted temozolomide in recurrent glioblastoma: A case report. Oncotarget 2015, 6, 34030–34037. [Google Scholar] [CrossRef] [PubMed]
- da Silva, B.; Mathew, R.K.; Polson, E.S.; Williams, J.; Wurdak, H. Spontaneous Glioblastoma Spheroid Infiltration of Early-Stage Cerebral Organoids Models Brain Tumor Invasion. SLAS Discov. 2018, 23, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Rubenstein, B.M.; Kaufman, L.J. The role of extracellular matrix in glioma invasion: A cellular Potts model approach. Biophys. J. 2008, 95, 5661–5680. [Google Scholar] [CrossRef]
- Shroyer, N.F. Tumor Organoids Fill the Niche. Cell Stem. Cell 2016, 18, 686–687. [Google Scholar] [CrossRef][Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef] [PubMed]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture conditions defining glioblastoma cells behavior: What is the impact for novel discoveries? Oncotarget 2017, 8, 69185–69197. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Osswald, M.; Blaes, J.; Wiestler, B.; Sahm, F.; Schmenger, T.; Solecki, G.; Deumelandt, K.; Kurz, F.T.; Xie, R.; et al. Tweety-Homolog 1 Drives Brain Colonization of Gliomas. J. Neurosci. 2017, 37, 6837–6850. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Korber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro. Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef]
- Osswald, M.; Solecki, G.; Wick, W.; Winkler, F. A malignant cellular network in gliomas: Potential clinical implications. Neuro. Oncol. 2016, 18, 479–485. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Atlasy, N.; Dieteren, C.E.J.; Navis, A.C.; Venhuizen, J.H.; Veelken, C.; Schubert, D.; Acker-Palmer, A.; Westerman, B.A.; Wurdinger, T.; et al. p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat. Cell Biol. 2020, 22, 97–107. [Google Scholar] [CrossRef]
- Lee, J.K.; Liu, Z.; Sa, J.K.; Shin, S.; Wang, J.; Bordyuh, M.; Cho, H.J.; Elliott, O.; Chu, T.; Choi, S.W.; et al. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy. Nat. Genet. 2018, 50, 1399–1411. [Google Scholar] [CrossRef]
- Doh, I.; Kwon, Y.J.; Ku, B.; Lee, D.W. Drug Efficacy Comparison of 3D Forming and Preforming Sphere Models with a Micropillar and Microwell Chip Platform. SLAS Discov. 2019, 24, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Doh, I.; Nam, D.H.; Lee, D.W. 3D Cell-Based High-Content Screening (HCS) Using a Micropillar and Microwell Chip Platform. Anal. Chem. 2018, 90, 8354–8361. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Zureick, A.H.; McFadden, K.A.; Mody, R.; Koschmann, C. Successful treatment of a TSC2-mutant glioblastoma with everolimus. BMJ Case Rep. 2019, 12, e227734. [Google Scholar] [CrossRef]
- Chang, S.M.; Wen, P.; Cloughesy, T.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; De Angelis, L.; Raizer, J.; et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest. New Drugs 2005, 23, 357–361. [Google Scholar] [CrossRef]
- Babak, S.; Mason, W.P. mTOR inhibition in glioblastoma: Requiem for a dream? Neuro. Oncol. 2018, 20, 584–585. [Google Scholar] [CrossRef]
- International Cancer Genome Consortium PedBrain Tumor Project. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar] [CrossRef]
- Cruickshanks, N.; Zhang, Y.; Hine, S.; Gibert, M.; Yuan, F.; Oxford, M.; Grello, C.; Pahuski, M.; Dube, C.; Guessous, F.; et al. Discovery and Therapeutic Exploitation of Mechanisms of Resistance to MET Inhibitors in Glioblastoma. Clin. Cancer Res. 2019, 25, 663–673. [Google Scholar] [CrossRef]
- Knubel, K.H.; Pernu, B.M.; Sufit, A.; Nelson, S.; Pierce, A.M.; Keating, A.K. MerTK inhibition is a novel therapeutic approach for glioblastoma multiforme. Oncotarget 2014, 5, 1338–1351. [Google Scholar] [CrossRef]
- de Groot, J.; Milano, V. Improving the prognosis for patients with glioblastoma: The rationale for targeting Src. J. Neurooncol. 2009, 95, 151–163. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.M.; Gladson, C.L. Targeting SRC in glioblastoma tumors and brain metastases: Rationale and preclinical studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; DeAngelis, L.M.; Gaur, R.; et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro Oncol. 2015, 17, 992–998. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Drake, C.G.; Sznol, M.; Choueiri, T.K.; Powderly, J.D.; Smith, D.C.; Brahmer, J.R.; Carvajal, R.D.; Hammers, H.J.; Puzanov, I.; et al. Survival, Durable Response, and Long-Term Safety in Patients with Previously Treated Advanced Renal Cell Carcinoma Receiving Nivolumab. J. Clin. Oncol. 2015, 33, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Mahringer, A.; Fricker, G. BCRP at the blood-brain barrier: Genomic regulation by 17beta-estradiol. Mol. Pharm. 2010, 7, 1835–1847. [Google Scholar] [CrossRef]
- Schiff, D.; Sarkaria, J. Dasatinib in recurrent glioblastoma: Failure as a teacher. Neuro Oncol. 2015, 17, 910–911. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—A phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E., 2nd; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neurooncol. 2010, 96, 219–230. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Jones, D.T.; Meyer, J.; Kratz, A.; Reuss, D.; Capper, D.; Koelsche, C.; Korshunov, A.; Wiestler, B.; et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016, 131, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.N.; Kang, S.Y.; Hong, S.; Lee, M.Y. High-throughput metabolism-induced toxicity assays demonstrated on a 384-pillar plate. Arch Toxicol. 2018, 92, 2501–2516. [Google Scholar] [CrossRef] [PubMed]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Weight of Tumor Tissue (g) a | Pattern of Tumor Progression b | Diagnosis | Ki67 | Time Until Lab Report (Days) |
|---|---|---|---|---|---|
| MA01 | 9.15 | Distant | GB WHO grade 4 MGMT methylated, IDH wt | 40% | 14 |
| MA02 | 3.42 | Distant | GB WHO grade 4 MGMT methylated, IDH wt | 0% | 13 |
| MA03 | 8.97 | Local | GB WHO grade 4 MGMT methylated, IDH wt | 2% | 15 |
| MA04 | 16.16 | Distant | IDH mut astrocytoma grade 4 | 70% | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratliff, M.; Kim, H.; Qi, H.; Kim, M.; Ku, B.; Azorin, D.D.; Hausmann, D.; Khajuria, R.K.; Patel, A.; Maier, E.; et al. Patient-Derived Tumor Organoids for Guidance of Personalized Drug Therapies in Recurrent Glioblastoma. Int. J. Mol. Sci. 2022, 23, 6572. https://doi.org/10.3390/ijms23126572
Ratliff M, Kim H, Qi H, Kim M, Ku B, Azorin DD, Hausmann D, Khajuria RK, Patel A, Maier E, et al. Patient-Derived Tumor Organoids for Guidance of Personalized Drug Therapies in Recurrent Glioblastoma. International Journal of Molecular Sciences. 2022; 23(12):6572. https://doi.org/10.3390/ijms23126572
Chicago/Turabian StyleRatliff, Miriam, Hichul Kim, Hao Qi, Minsung Kim, Bosung Ku, Daniel Dominguez Azorin, David Hausmann, Rajiv K. Khajuria, Areeba Patel, Elena Maier, and et al. 2022. "Patient-Derived Tumor Organoids for Guidance of Personalized Drug Therapies in Recurrent Glioblastoma" International Journal of Molecular Sciences 23, no. 12: 6572. https://doi.org/10.3390/ijms23126572
APA StyleRatliff, M., Kim, H., Qi, H., Kim, M., Ku, B., Azorin, D. D., Hausmann, D., Khajuria, R. K., Patel, A., Maier, E., Cousin, L., Ogier, A., Sahm, F., Etminan, N., Bunse, L., Winkler, F., El-Khoury, V., Platten, M., & Kwon, Y.-J. (2022). Patient-Derived Tumor Organoids for Guidance of Personalized Drug Therapies in Recurrent Glioblastoma. International Journal of Molecular Sciences, 23(12), 6572. https://doi.org/10.3390/ijms23126572