
Inhibition of N-Acetyltransferase 10 Suppresses the Progression of Prostate Cancer through Regulation of DNA Replication


Abstract
:1. Introduction
2. Results
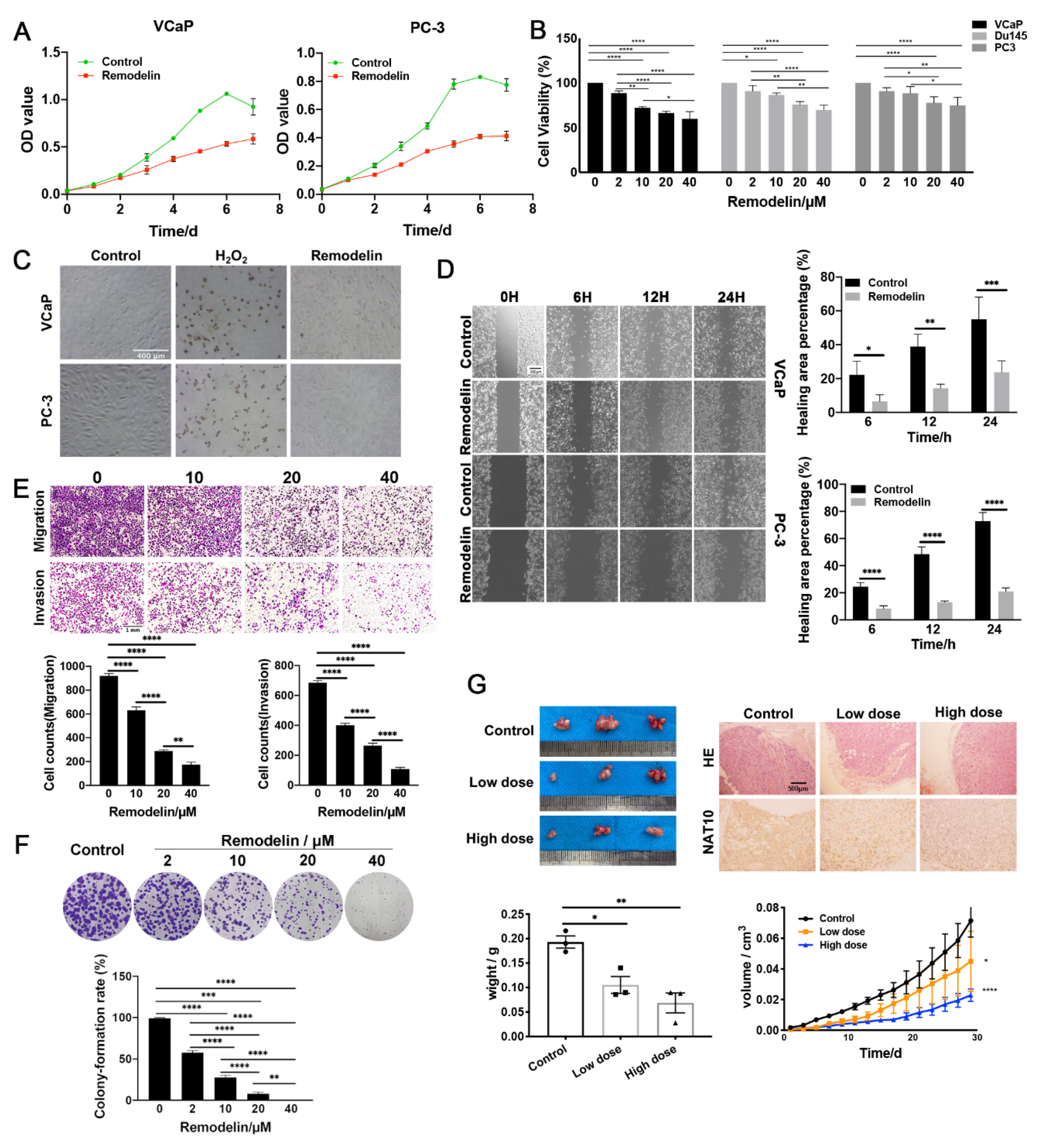
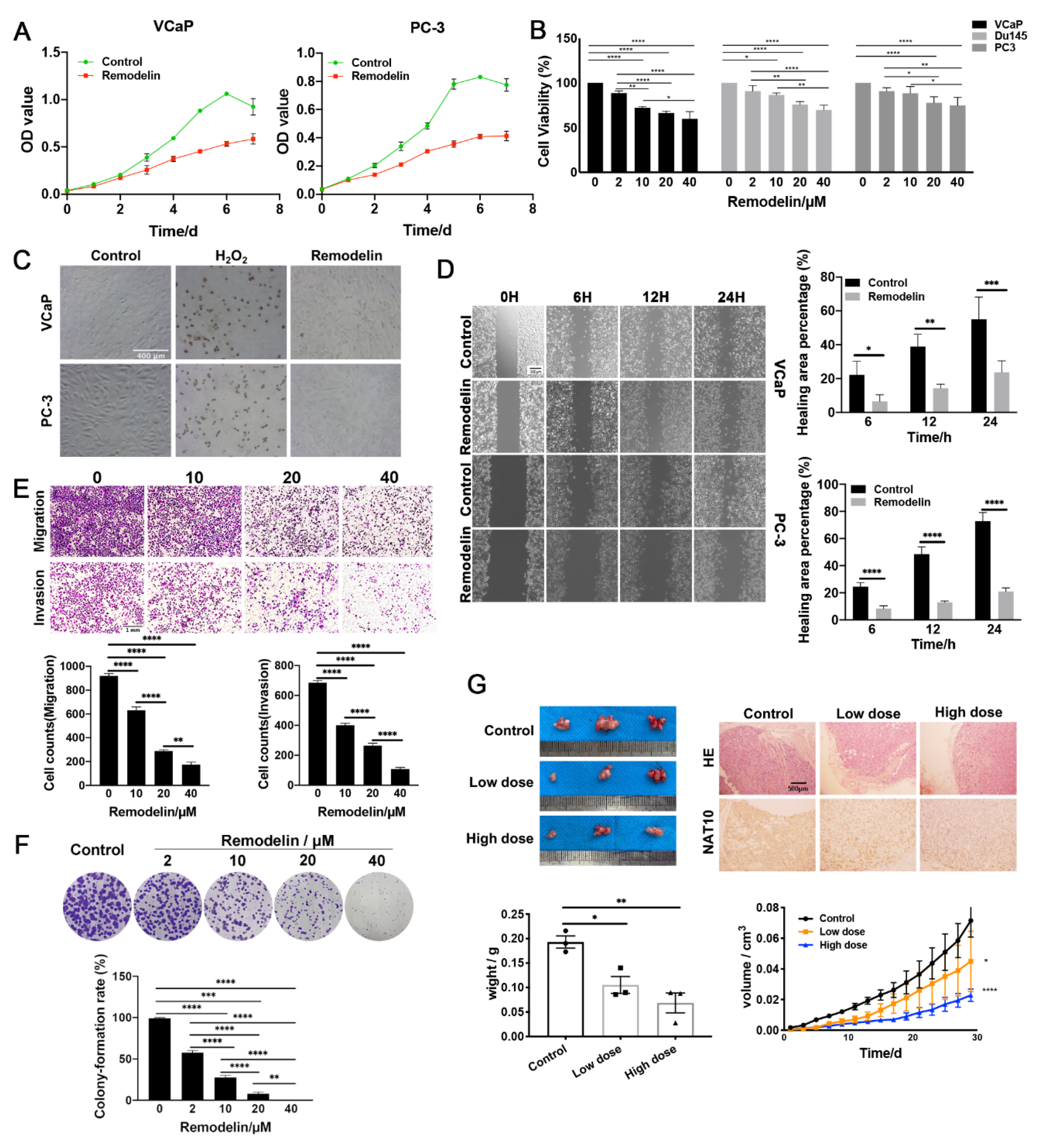
2.1. Inhibition of NAT10 Suppresses the Growth of Both AR-Positive and AR-Negative Prostate Cancer Cells
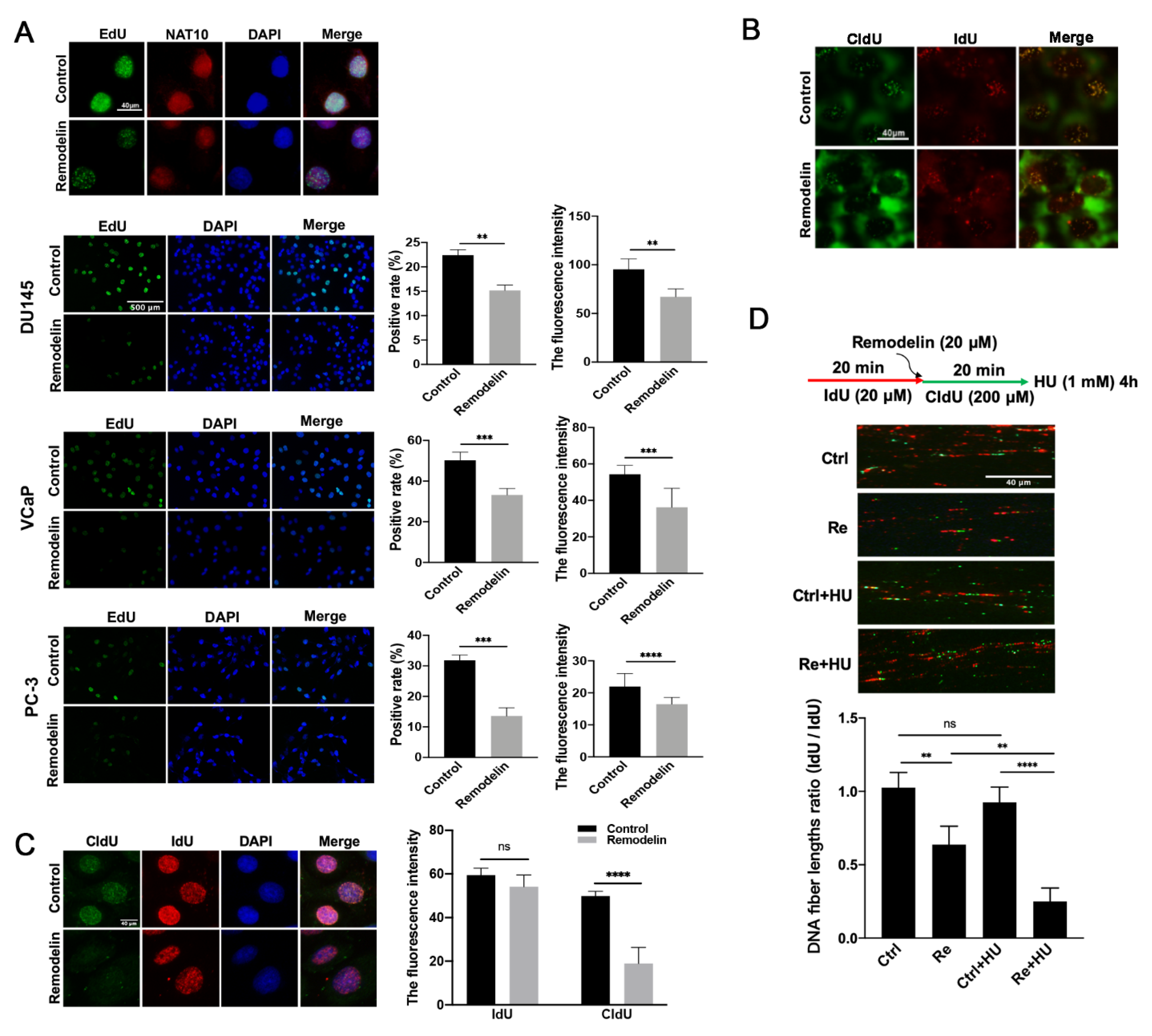
2.2. Inhibition of NAT10 Slows DNA Replication in Prostate Cancer Cells
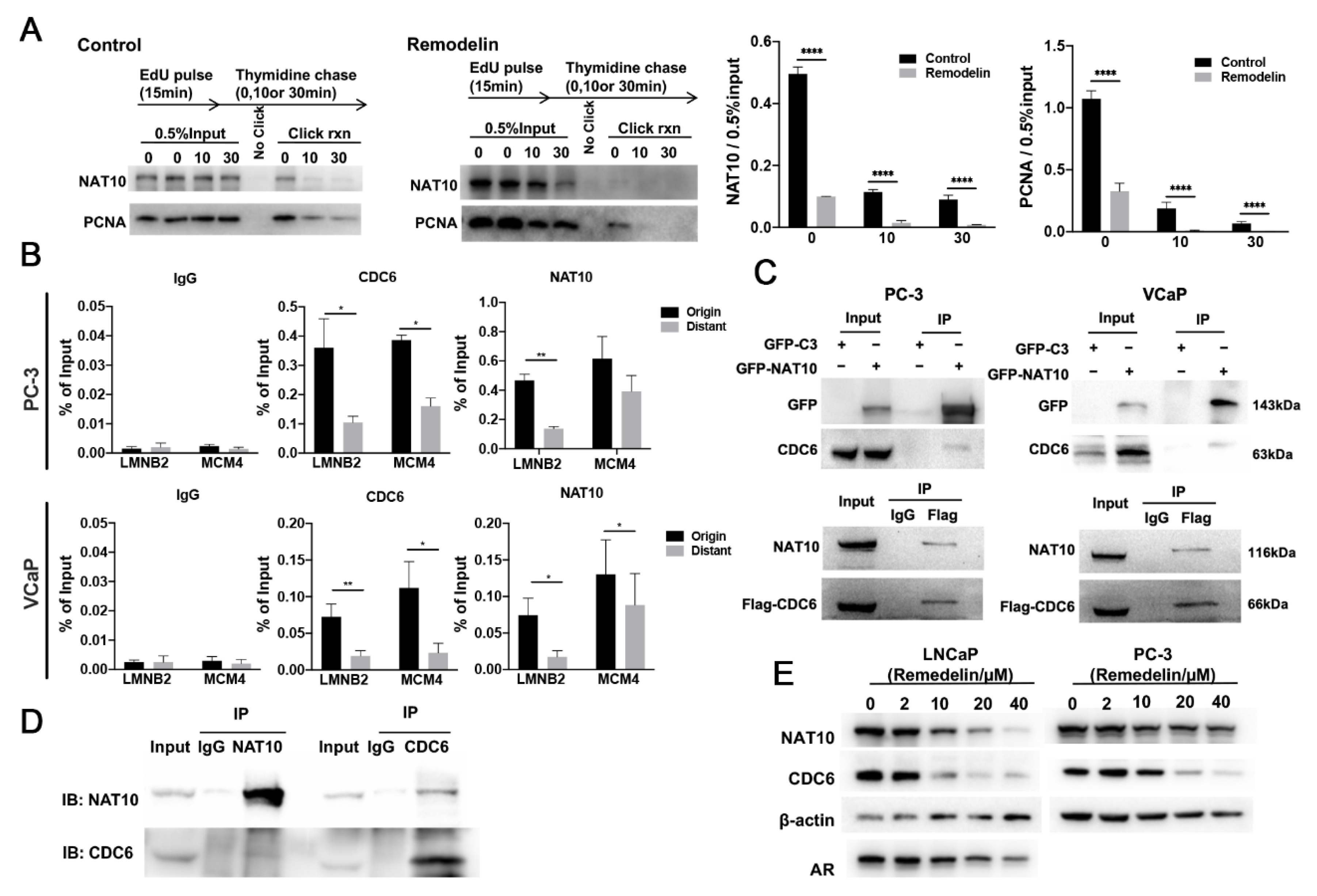
2.3. NAT10 Is Involved in DNA Replication
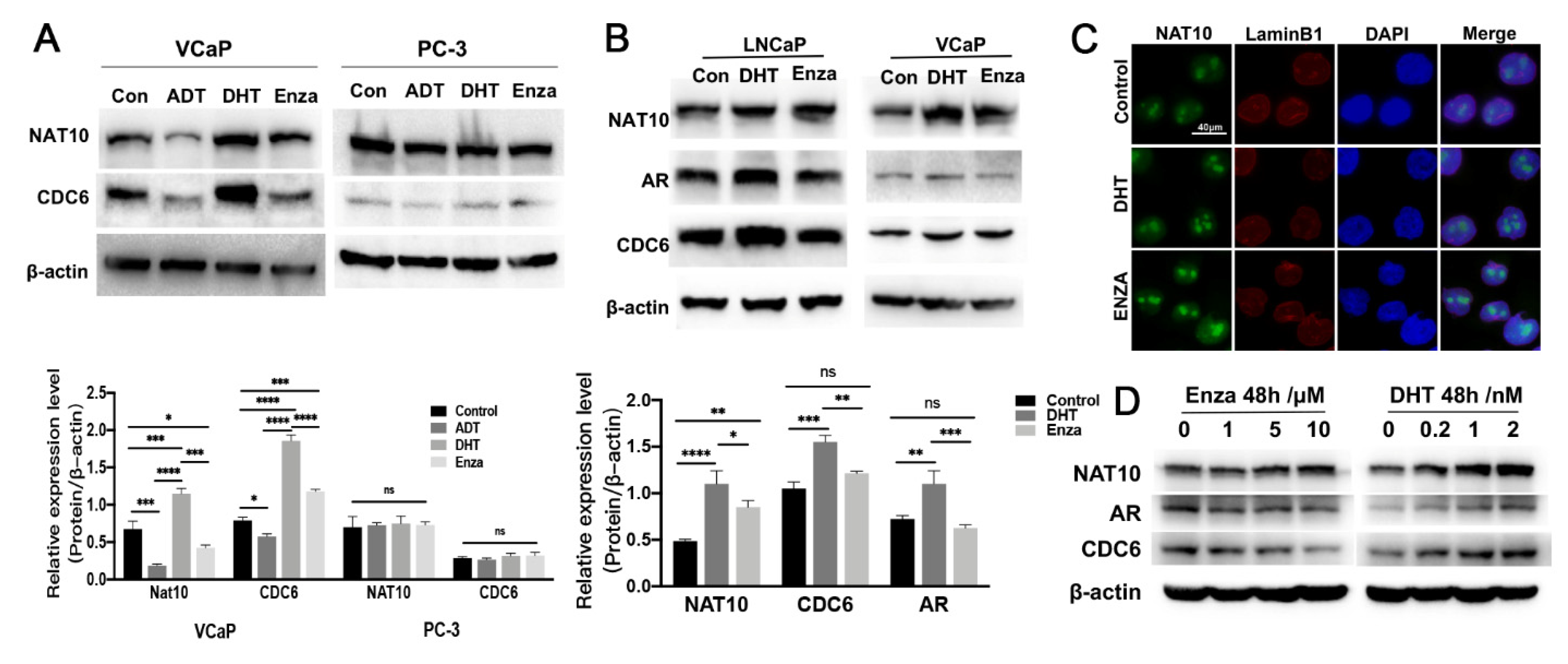
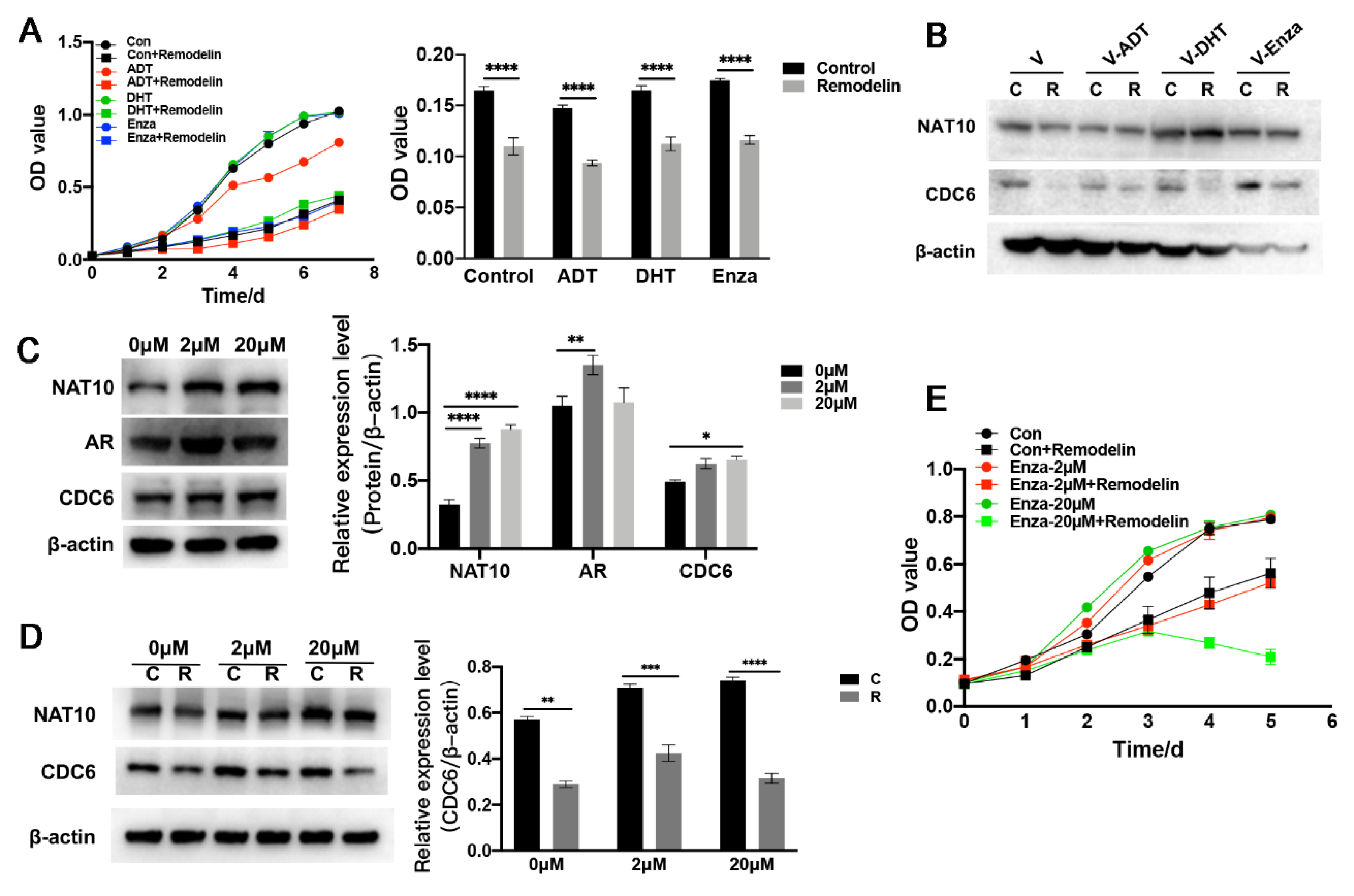
2.4. Expression of NAT10 Could Be Altered by Treatment with AR Modulators
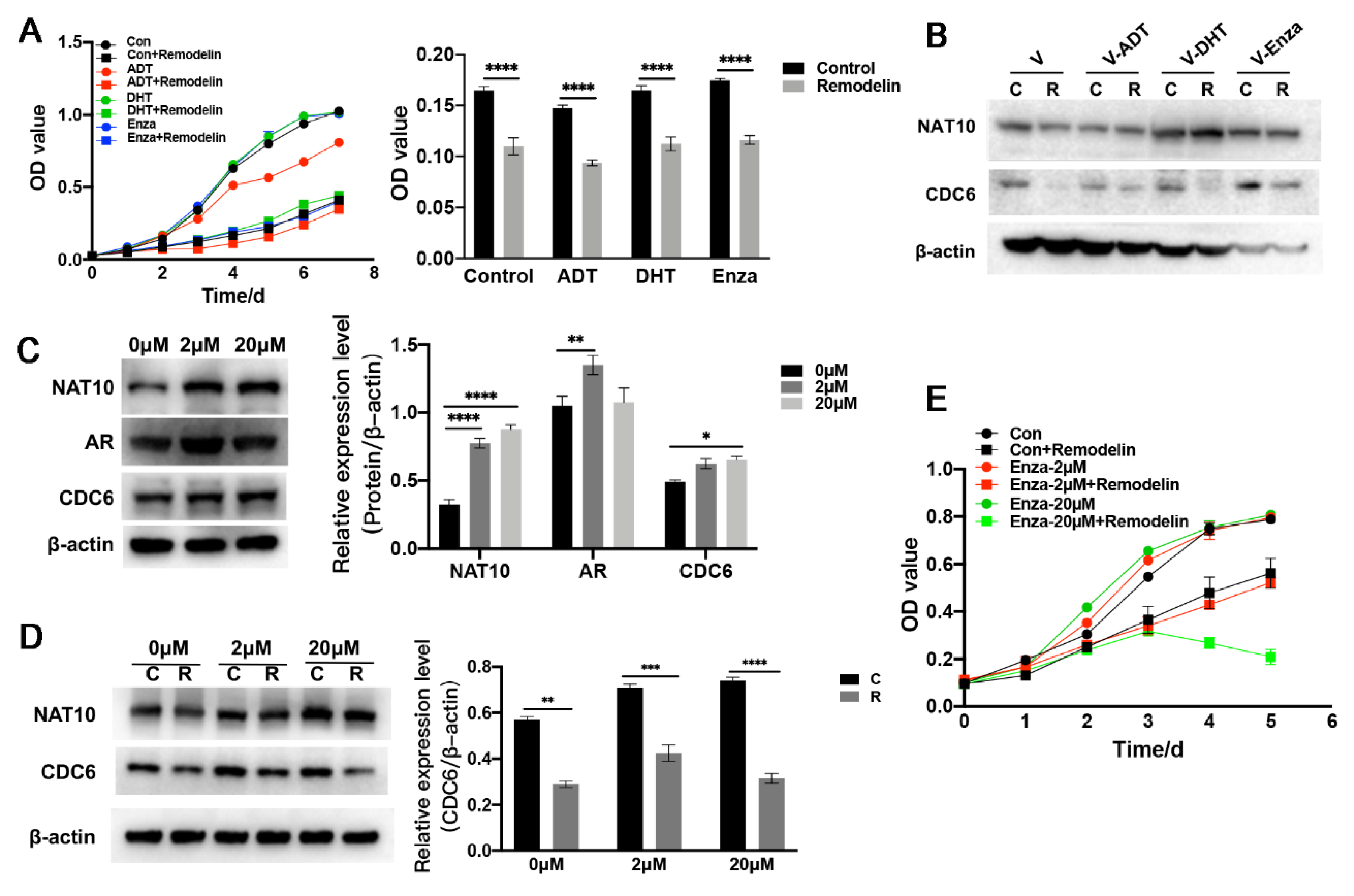
2.5. Inhibition of NAT10 Suppresses Castration-Resistant Prostate Cancer Cells In Vitro
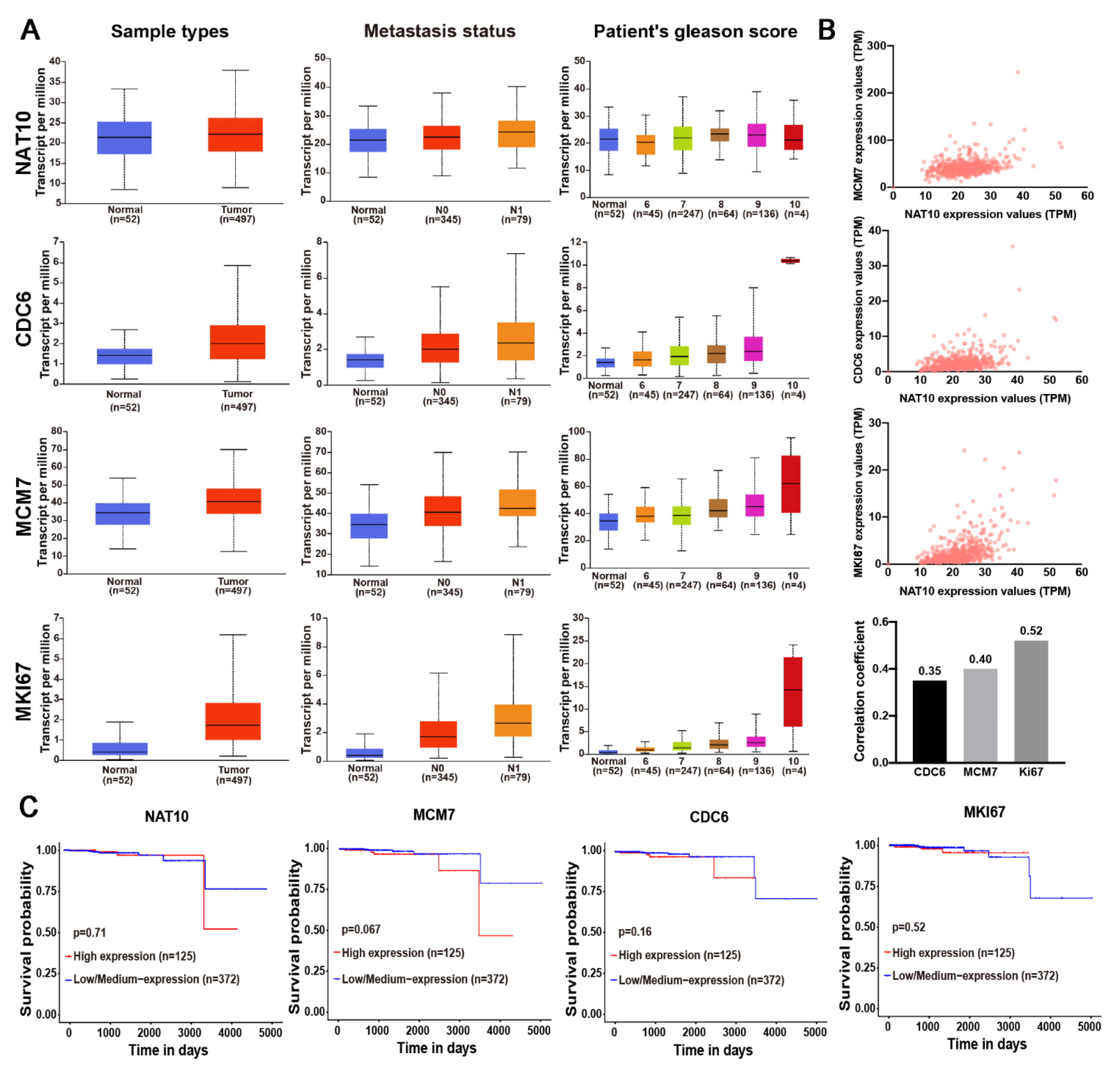
2.6. NAT10 Expression and its Correlation with DNA Replication Factors and Clinicopathological Features in Prostate Cancer
3. Discussion
4. Materials and Methods
4.1. Plasmids and Reagents
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Colony Formation Assay
4.5. Wound Healing Assay
4.6. Cell Migration and Invasion Assays
4.7. Mouse Transplantation Experiment
4.8. Hematoxylin and Eosin (HE) Staining and Immunohistochemistry
4.9. Western Blotting
4.10. Immunofluorescence
4.11. 5-Ethynyl-2′-deoxyuridine (EdU) Analysis
4.12. IdU-CldU In Situ Labeling Assay
4.13. DNA Fiber Assay
4.14. iPOND Assay
4.15. TUNEL Assay
4.16. Co-immunoprecipitation
4.17. Chromatin Immunoprecipitation
4.18. Quantitative Real-Time PCR
4.19. TCGA Analysis
4.20. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Mediwala, S.N.; Sun, H.; Szafran, A.T.; Hartig, S.M.; Sonpavde, G.; Hayes, T.G.; Thiagarajan, P.; Mancini, M.A.; Marcelli, M. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate 2013, 73, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.Q.; Huang, H.J. Interplay Among PI3K/AKT, PTEN/FOXO and AR Signaling in Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 319–331. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886.e1824. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Horikawa, S.; Suzuki, T.; Kawauchi, H.; Tanaka, Y.; Suzuki, T.; Suzuki, T. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 2014, 289, 35724–35730. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Liu, H.; Wang, Q.; Tang, Z.; Hou, L.; Zhang, B. Molecular cloning of a novel human gene encoding histone acetyltransferase-like protein involved in transcriptional activation of hTERT. Biochem. Biophys. Res. Commun. 2003, 311, 506–513. [Google Scholar] [CrossRef]
- Liu, H.; Ling, Y.; Gong, Y.; Sun, Y.; Hou, L.; Zhang, B. DNA damage induces N-acetyltransferase NAT10 gene expression through transcriptional activation. Mol. Cell. Biochem. 2007, 300, 249–258. [Google Scholar] [CrossRef]
- Shen, Q.; Zheng, X.; McNutt, M.A.; Guang, L.; Sun, Y.; Wang, J.; Gong, Y.; Hou, L.; Zhang, B. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp. Cell Res. 2009, 315, 1653–1667. [Google Scholar] [CrossRef] [PubMed]
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical Inhibition of NAT10 Corrects Defects of Laminopathic Cells. Science 2014, 344, 527–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, T.I.; Lee, Y.M.; Lim, B.O.; Lim, J.H. Inhibition of NAT10 Suppresses Melanogenesis and Melanoma Growth by Attenuating Microphthalmia-Associated Transcription Factor (MITF) Expression. Int. J. Mol. Sci. 2017, 18, 1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalhat, M.H.; Mohammed, M.R.S.; Ahmad, A.; Khan, M.I.; Choudhry, H. Remodelin, a N-acetyltransferase 10 (NAT10) inhibitor, alters mitochondrial lipid metabolism in cancer cells. J. Cell. Biochem. 2021, 122, 1936–1945. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yao, M.; Wu, Y.; Ma, N.; Liu, H.; Zhang, B. N-Acetyltransferase 10 Promotes Micronuclei Formation to Activate the Senescence-Associated Secretory Phenotype Machinery in Colorectal Cancer Cells. Transl. Oncol. 2020, 13, 100783. [Google Scholar] [CrossRef] [PubMed]
- Akerblom, L.; Ehrenberg, A.; Graslund, A.; Lankinen, H.; Reichard, P.; Thelander, L. Overproduction of the free radical of ribonucleotide reductase in hydroxyurea-resistant mouse fibroblast 3T6 cells. Proc. Natl. Acad. Sci. USA 1981, 78, 2159–2163. [Google Scholar] [CrossRef] [Green Version]
- Reichard, P.; Ehrenberg, A. Ribonucleotide reductase—A radical enzyme. Science 1983, 221, 514–519. [Google Scholar] [CrossRef]
- Dungrawala, H.; Cortez, D. Purification of proteins on newly synthesized DNA using iPOND. Methods Mol. Biol. 2015, 1228, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Warbrick, E. The puzzle of PCNA’s many partners. Bioessays 2000, 22, 997–1006. [Google Scholar] [CrossRef]
- Bai, V.U.; Cifuentes, E.; Menon, M.; Barrack, E.R.; Reddy, G.P. Androgen receptor regulates Cdc6 in synchronized LNCaP cells progressing from G1 to S phase. J. Cell. Physiol. 2005, 204, 381–387. [Google Scholar] [CrossRef]
- Jin, F.; Fondell, J.D. A novel androgen receptor-binding element modulates Cdc6 transcription in prostate cancer cells during cell-cycle progression. Nucleic Acids Res. 2009, 37, 4826–4838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanika, S.; Karantanos, T.; Li, L.; Wang, J.; Park, S.; Yang, G.; Zuo, X.; Song, J.H.; Maity, S.N.; Manyam, G.C.; et al. Targeting DNA Damage Response in Prostate Cancer by Inhibiting Androgen Receptor-CDC6-ATR-Chk1 Signaling. Cell Rep. 2017, 18, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gong, Z.; Sun, L.; Li, X. FOXM1 and androgen receptor co-regulate CDC6 gene transcription and DNA replication in prostate cancer cells. Biochim. Biophys. Acta 2014, 1839, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Duncker, B.P.; Chesnokov, I.N.; McConkey, B.J. The origin recognition complex protein family. Genome Biol. 2009, 10, 214. [Google Scholar] [CrossRef] [Green Version]
- Litvinov, I.V.; Vander Griend, D.J.; Antony, L.; Dalrymple, S.; De Marzo, A.M.; Drake, C.G.; Isaacs, J.T. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15085–15090. [Google Scholar] [CrossRef] [Green Version]
- Murthy, S.; Wu, M.; Bai, V.U.; Hou, Z.Z.; Menon, M.; Barrack, E.R.; Kim, S.H.; Reddy, G.P.V. Role of Androgen Receptor in Progression of LNCaP Prostate Cancer Cells from G(1) to S Phase. PLoS ONE 2013, 8, e56692. [Google Scholar] [CrossRef]
- Shi, Y.K.; Yu, Y.P.; Zhu, Z.H.; Han, Y.C.; Ren, B.; Nelson, J.B.; Luo, J.H. MCM7 interacts with androgen receptor. Am. J. Pathol. 2008, 173, 1758–1767. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Sugimoto, N.; Iwahori, S.; Yugawa, T.; Narisawa-Saito, M.; Kiyono, T.; Fujita, M. CDC6 interaction with ATR regulates activation of a replication checkpoint in higher eukaryotic cells. J. Cell. Sci. 2010, 123, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowski, M.C.; Chen, Y.; Feng, Z.; Cullen, J.; Trock, B.J.; Suzman, D.; Antonarakis, E.S.; Paller, C.J.; Rosner, I.; Han, M.; et al. PSA Doubling Time and Absolute PSA Predict Metastasis-free Survival in Men With Biochemically Recurrent Prostate Cancer After Radical Prostatectomy. Clin. Genitourin Cancer 2019, 17, 470–475.e471. [Google Scholar] [CrossRef]
- Wu, Y.; Cao, Y.; Liu, H.; Yao, M.; Ma, N.; Zhang, B. Remodelin, an inhibitor of NAT10, could suppress hypoxia-induced or constitutional expression of HIFs in cells. Mol. Cell. Biochem. 2020, 472, 19–31. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, W.; Wang, H.L.; Liu, H.J.; Jia, X.Y.; Zheng, X.Z.; Zou, Y.X.; Li, X.; Hou, L.; McNutt, M.A.; et al. GSK-3 beta-Regulated N-Acetyltransferase 10 Is Involved in Colorectal Cancer Invasion. Clin. Cancer Res. 2014, 20, 4717–4729. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhang, Q.; Luo, H.; Shao, L.; Liu, R.; Kong, Y.; Zhao, X.; Geng, Y.; Li, C.; Wang, X. Effect of Carbon Ion Radiation Induces Bystander Effect on Metastasis of A549 Cells and Metabonomic Correlation Analysis. Front. Oncol. 2020, 10, 601620. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Western blotting: An introduction. Methods Mol. Biol. 2015, 1312, 17–30. [Google Scholar] [CrossRef]
- Odell, I.D.; Cook, D. Immunofluorescence techniques. J. Investig. Dermatol. 2013, 133, e4. [Google Scholar] [CrossRef] [Green Version]
- Quinet, A.; Carvajal-Maldonado, D.; Lemacon, D.; Vindigni, A. DNA Fiber Analysis: Mind the Gap! Methods Enzymol. 2017, 591, 55–82. [Google Scholar] [CrossRef]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Yasukouchi, S.; Osano, S.; Watanabe, S.; Aizawa, M.; Yugawa, T.; Kiyono, T.; Kurumizaka, H.; et al. Cdt1-binding protein GRWD1 is a novel histone-binding protein that facilitates MCM loading through its influence on chromatin architecture. Nucleic Acids Res. 2015, 43, 5898–5911. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Yugawa, T.; Iizuka, M.; Kiyono, T.; Fujita, M. Chromatin Remodeler Sucrose Nonfermenting 2 Homolog (SNF2H) Is Recruited onto DNA Replication Origins through Interaction with Cdc10 Protein-dependent Transcript 1 (Cdt1) and Promotes Pre-replication Complex Formation. J. Biol. Chem. 2011, 286, 39200–39210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | NAT10 | CDC6 | MCM7 | Ki67 |
|---|---|---|---|---|
| Normal-vs-Tumor | 9.36 × 10−2 | 2.90 × 10−6 (***) | 3.34 × 10−8 (***) | 2.07 × 10−9 (***) |
| Normal vs. Gleason score | ||||
| Normal-vs-Gleason score 6 | 3.55 × 10−1 | 5.42 × 10−1 | 1.43 × 10−2 (*) | 1.49 × 10−1 |
| Normal-vs-Gleason score 7 | 2.44 × 10−1 | 1.09 × 10−2 (*) | 4.22 × 10−3 (**) | 1.17 × 10−4 (***) |
| Normal-vs-Gleason score 8 | 2.51 × 10−2 (**) | 2.76 × 10−3 (**) | 5.01 × 10−7 (***) | 2.29 × 10−5 (***) |
| Normal-vs-Gleason score 9 | 9.64 × 10−3 (***) | 2.26 × 10−7 (***) | 6.16 × 10−11 (***) | 9.07 × 10−13 (***) |
| Normal-vs-Gleason score 10 | 5.01 × 10−1 | 8.51 × 10−2 | 1.89 × 10−1 | 1.09 × 10−1 |
| Node metastasis | ||||
| Normal-vs-N0 | 7.01 × 10−2 | 5.76 × 10−5 (***) | 3.16 × 10−7 (***) | 2.80 × 10−8 (***) |
| Normal-vs-N1 | 3.78 × 10−3 (**) | 1.45 × 10−4 (***) | 7.03 × 10−7 (***) | 3.93 × 10−9 (***) |
| N0-vs-N1 | 4.52 × 10−2 (*) | 2.15 × 10−2 (*) | 6.16 × 10−3 (**) | 2.44 × 10−3 (**) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, N.; Liu, H.; Wu, Y.; Yao, M.; Zhang, B. Inhibition of N-Acetyltransferase 10 Suppresses the Progression of Prostate Cancer through Regulation of DNA Replication. Int. J. Mol. Sci. 2022, 23, 6573. https://doi.org/10.3390/ijms23126573
Ma N, Liu H, Wu Y, Yao M, Zhang B. Inhibition of N-Acetyltransferase 10 Suppresses the Progression of Prostate Cancer through Regulation of DNA Replication. International Journal of Molecular Sciences. 2022; 23(12):6573. https://doi.org/10.3390/ijms23126573
Chicago/Turabian StyleMa, Ningning, Haijing Liu, Yaqian Wu, Mengfei Yao, and Bo Zhang. 2022. "Inhibition of N-Acetyltransferase 10 Suppresses the Progression of Prostate Cancer through Regulation of DNA Replication" International Journal of Molecular Sciences 23, no. 12: 6573. https://doi.org/10.3390/ijms23126573
APA StyleMa, N., Liu, H., Wu, Y., Yao, M., & Zhang, B. (2022). Inhibition of N-Acetyltransferase 10 Suppresses the Progression of Prostate Cancer through Regulation of DNA Replication. International Journal of Molecular Sciences, 23(12), 6573. https://doi.org/10.3390/ijms23126573