
JAZ1-3 and MYC2-1 Synergistically Regulate the Transformation from Completely Mixed Flower Buds to Female Flower Buds in Castanea mollisima

Abstract
:1. Introduction
2. Results
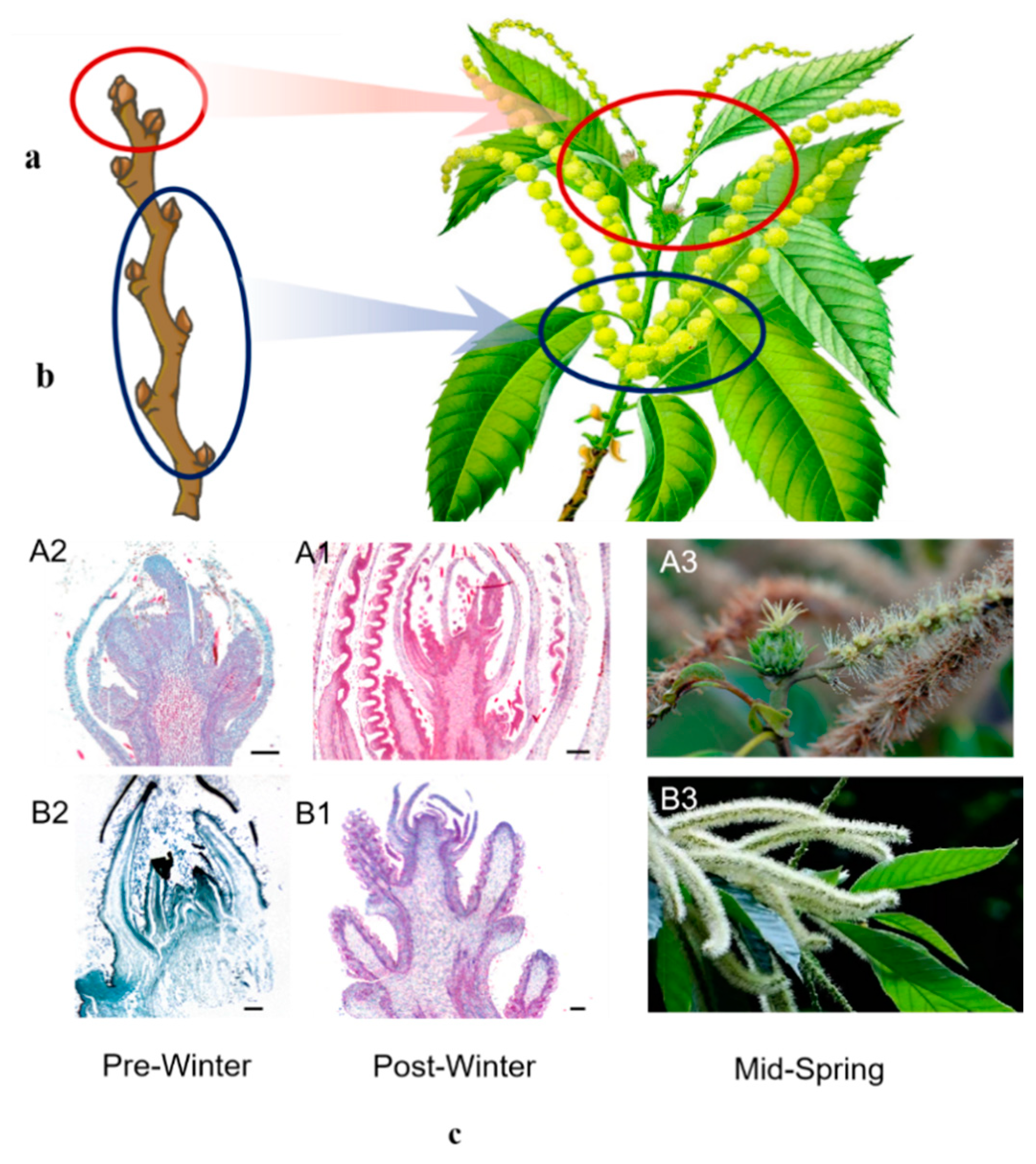
2.1. Characteristics of Flower Buds of Chestnut Pre and Post Winter
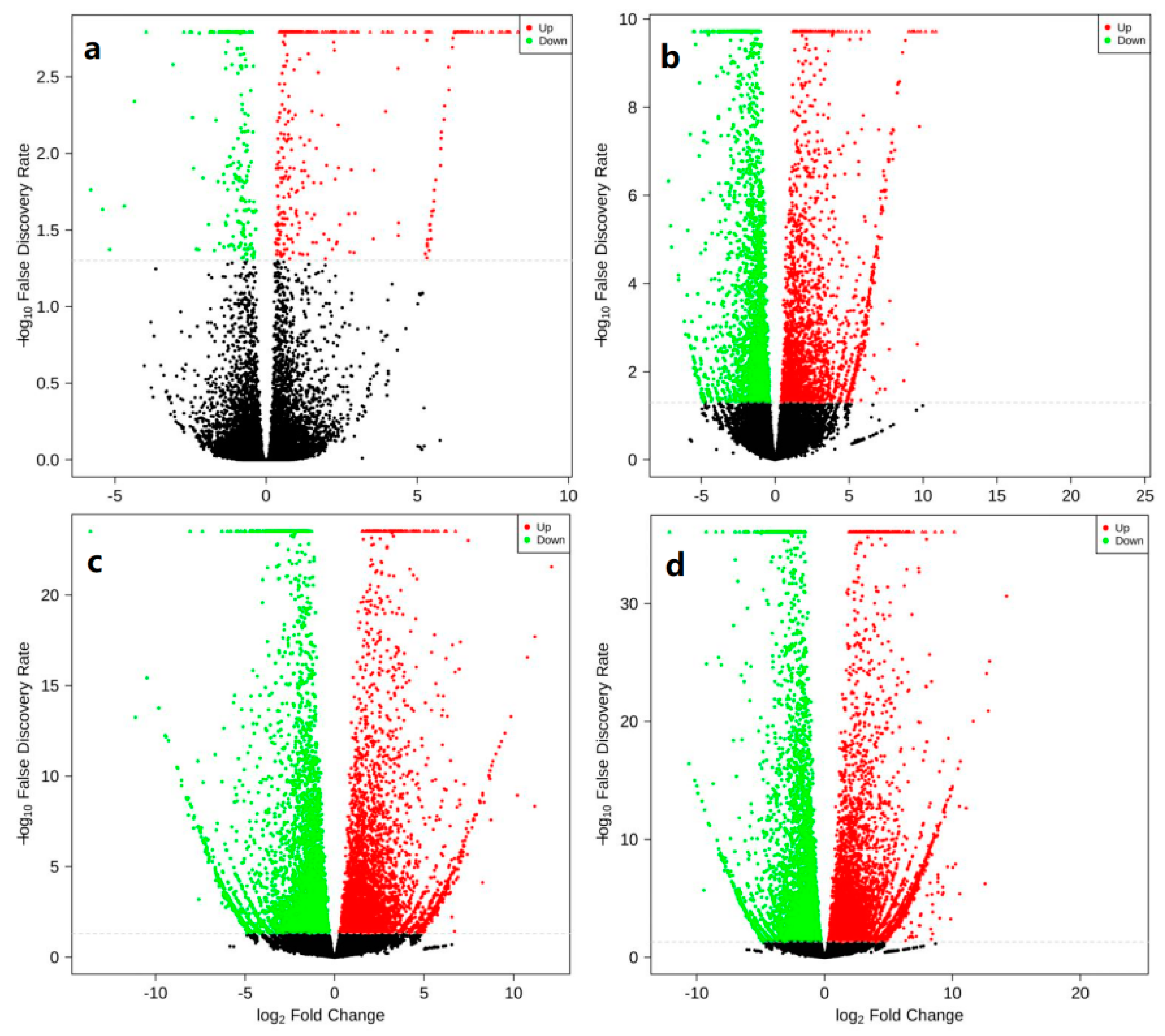
2.2. Basic Data of Transcriptome Sequencing of Two Flower Buds
2.3. KOG Functional Annotation of DEGs
2.4. GO Enrichment Analysis of DEGs in Two Mixed Flower Buds
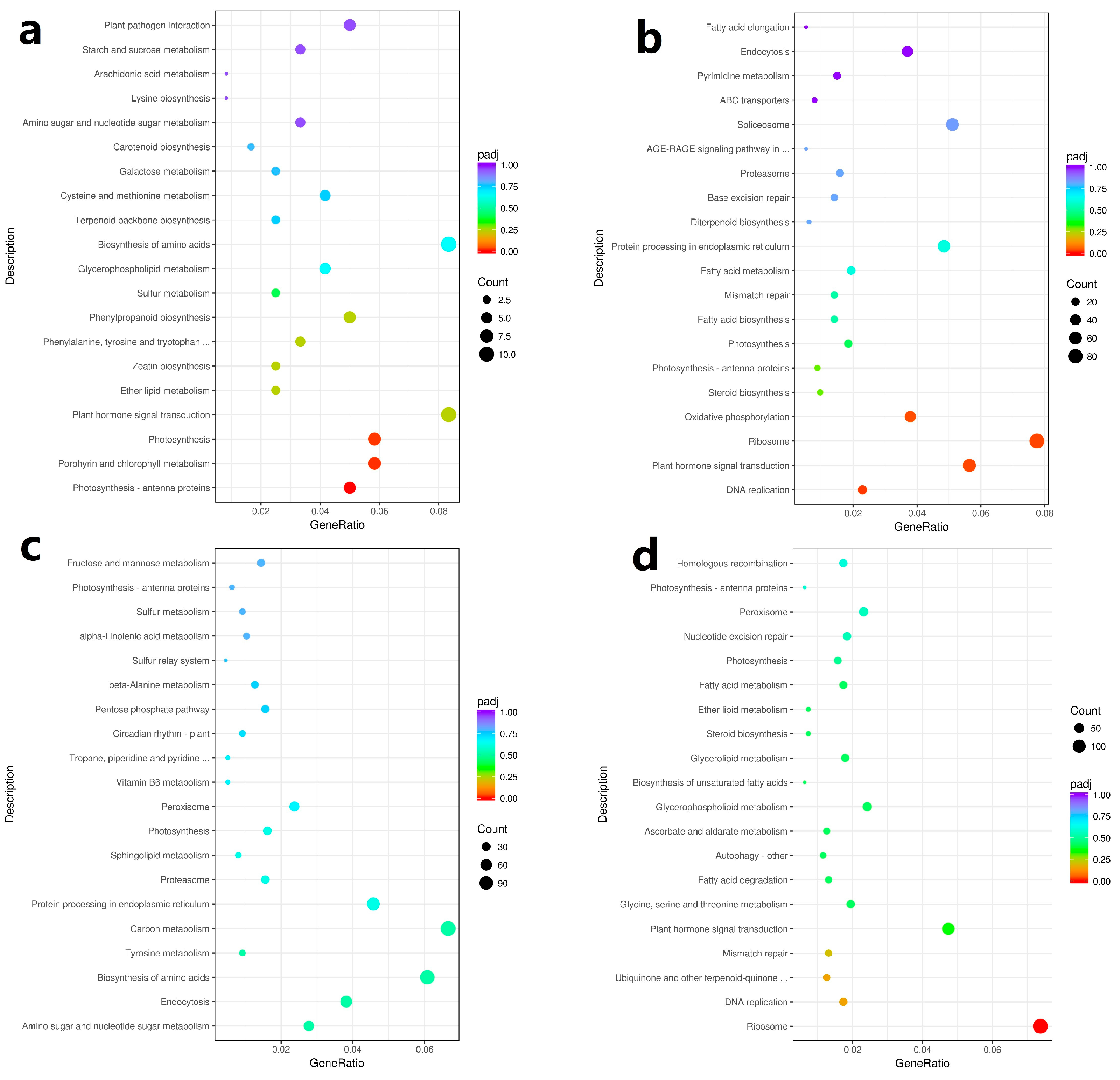
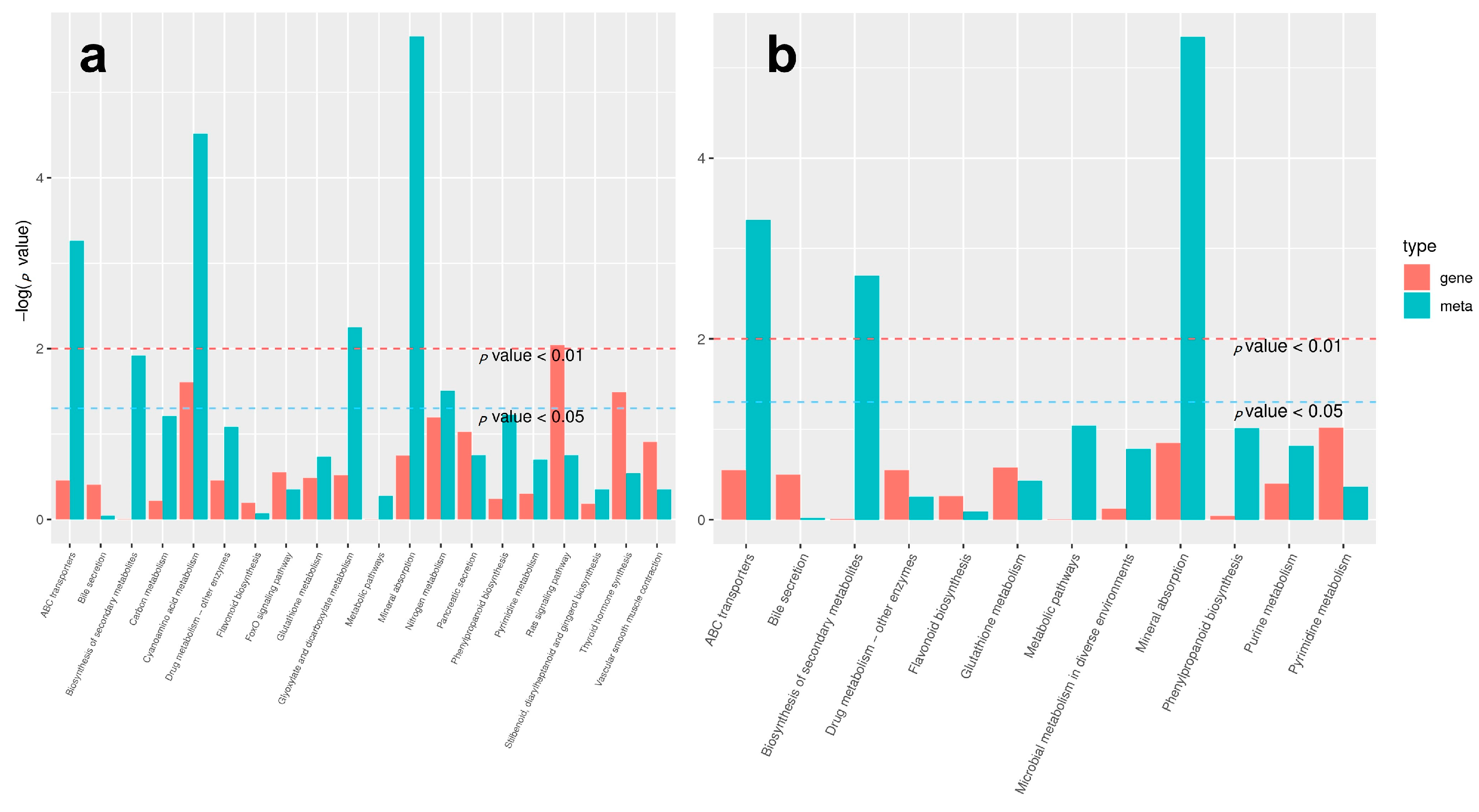
2.5. Analysis of KEGG Pathway of DEGs
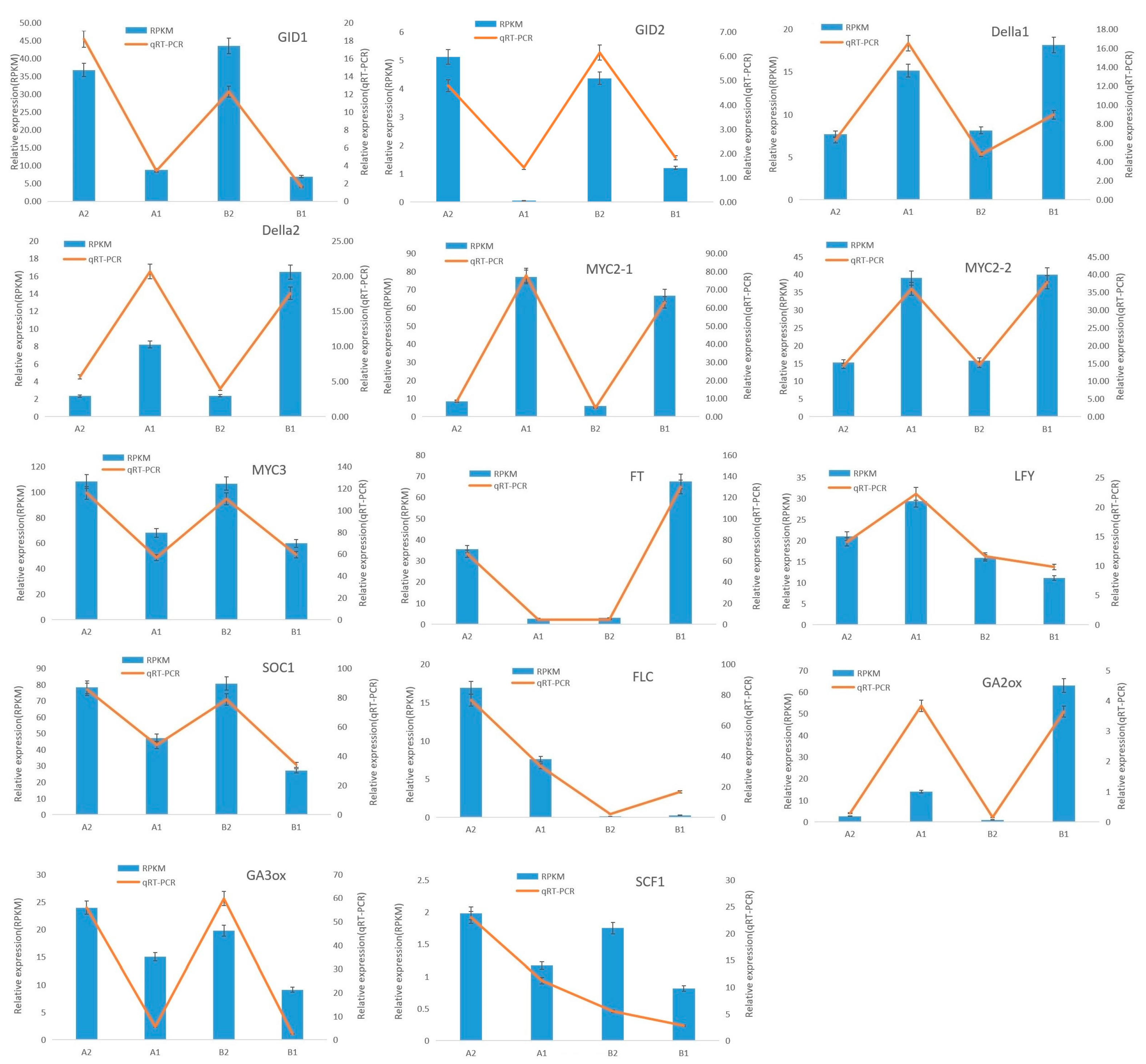
2.6. Confirmation of Expression Using qRT-PCR
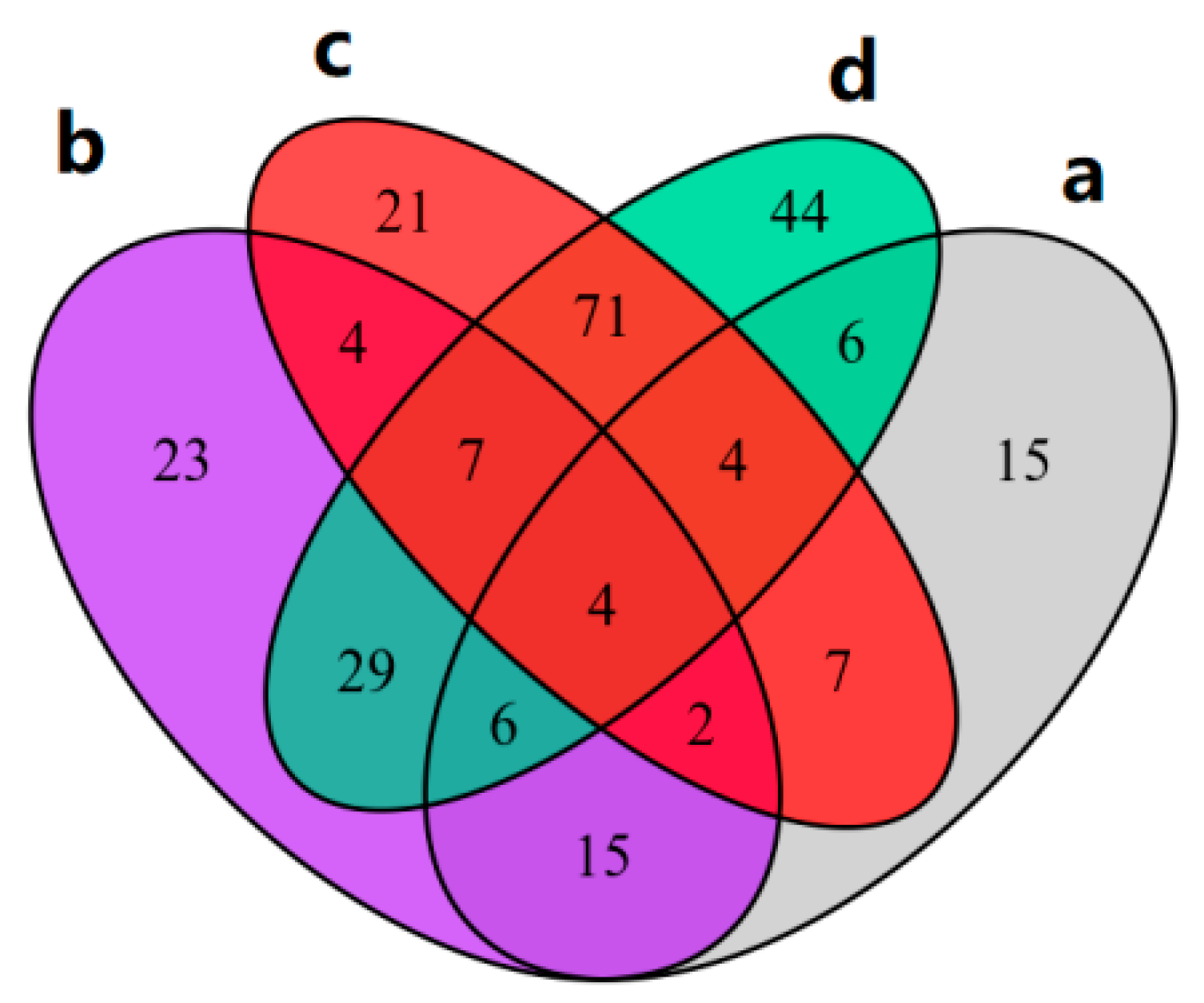
2.7. Differential Metabolite Screening
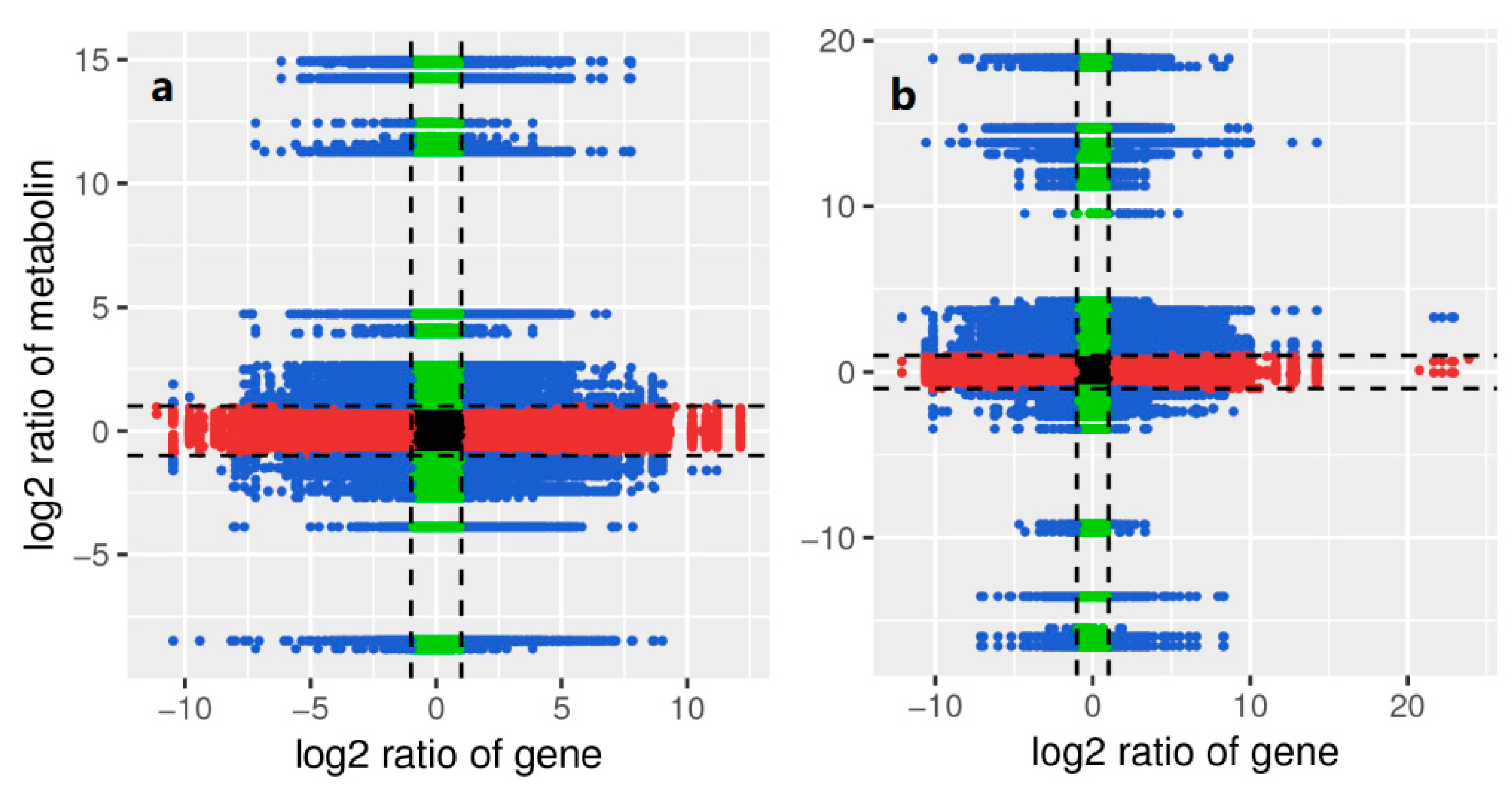
2.8. Combined Analysis of Transcriptome and Metabolome
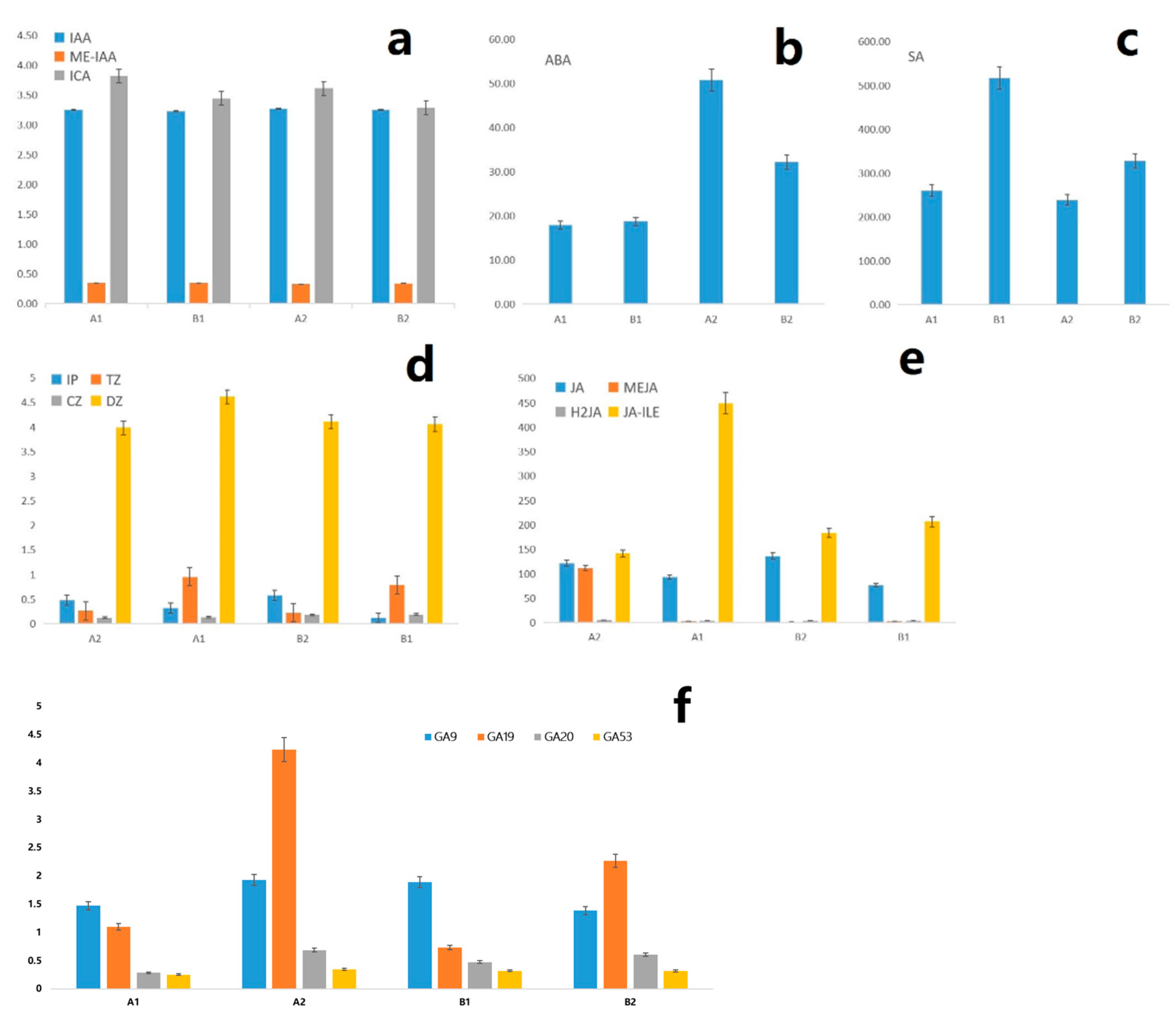
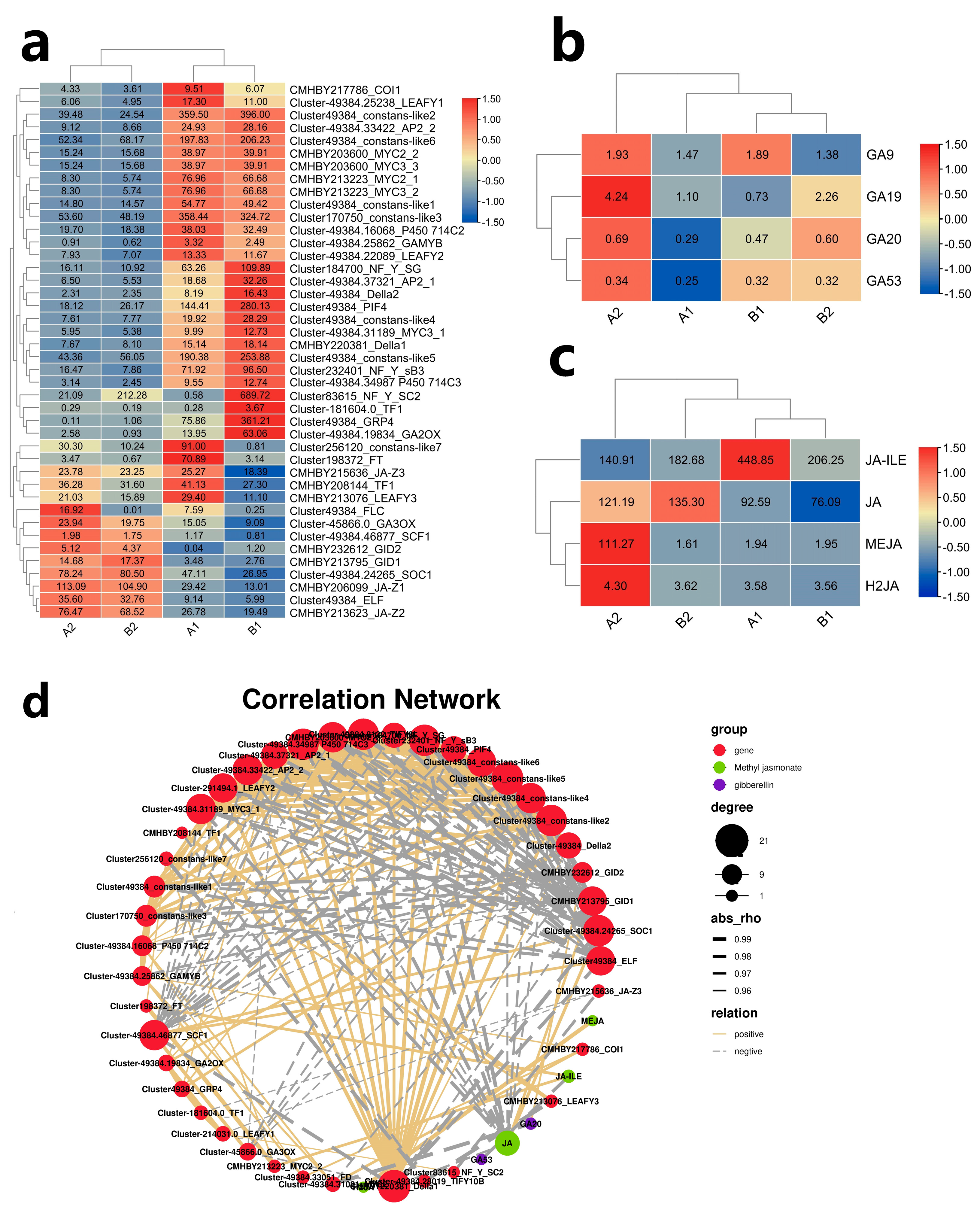
2.9. Analysis of Differential Hormone Content
2.10. Combined Analysis of Flower Bud Differentiation Hormones and Related Genes
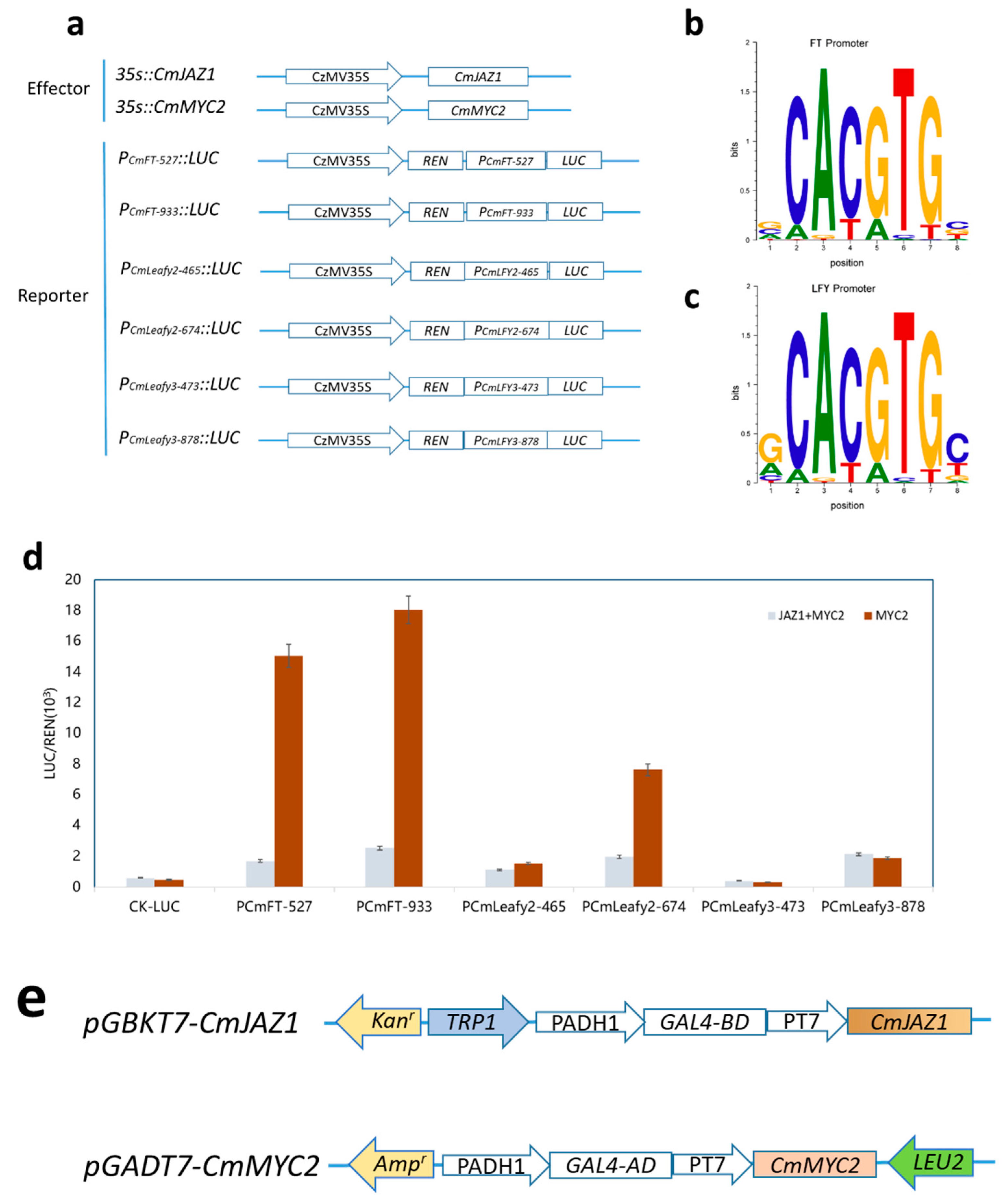
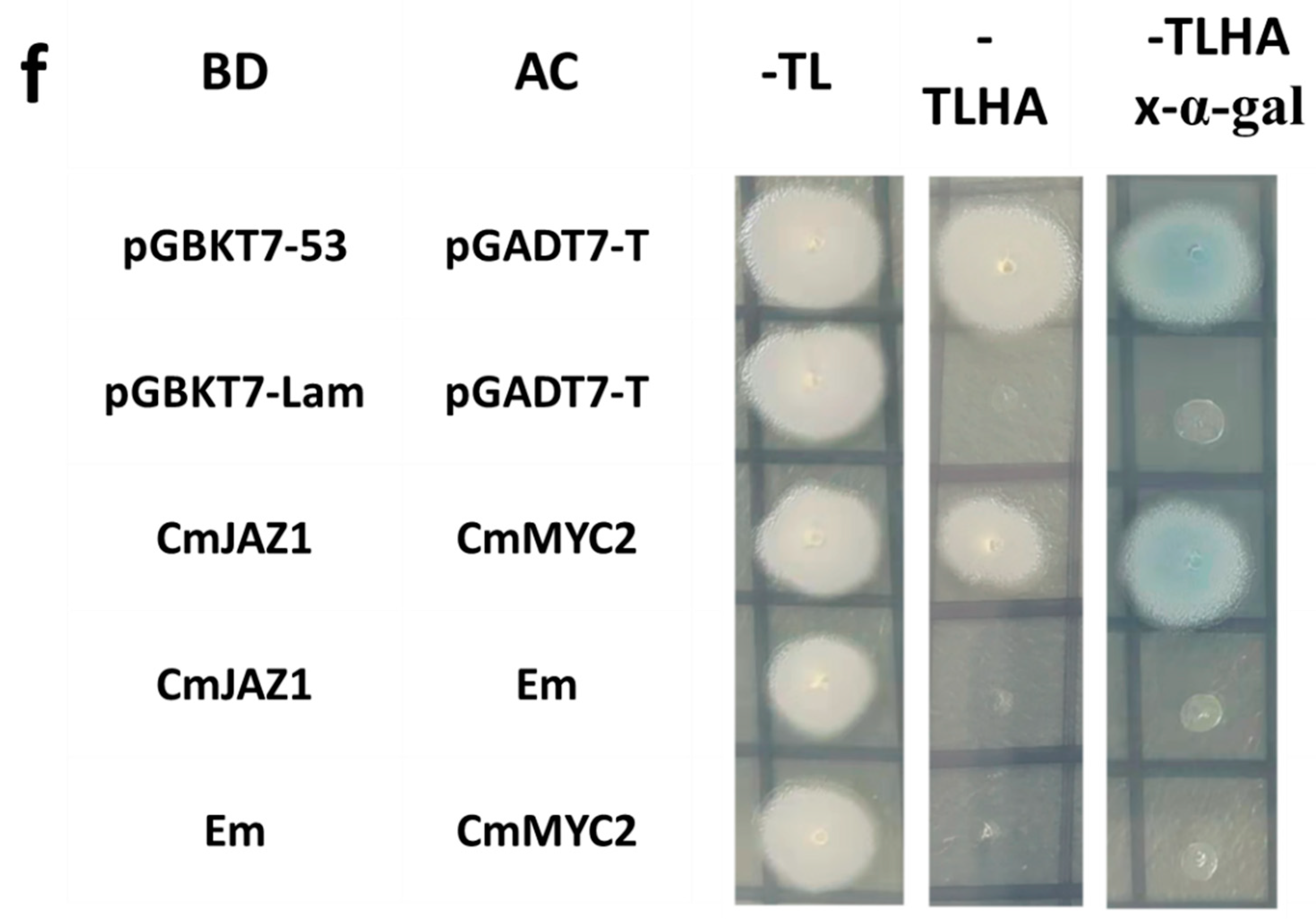
2.11. JAZ1-3 and MYC2-1 Synergistically Regulate the Expression of FT
3. Discussion
3.1. Critical Period of Flower Bud Differentiation in Chestnut
3.2. Metabolites and Flower bud Differentiation
3.3. GA and JA Jointly Regulate the Floral Transition in Chestnut
4. Materials and Methods
4.1. Plant Materials
4.2. Chemicals and Reagents
4.3. RNA Extraction, Clustering, Sequencing, and Quality Control
4.4. De Novo Assembly, Functional Annotation and DEG Analysis
4.5. Real-Time PCR Validation
4.6. Metabolome Sample Preparation and Extraction
4.7. HPLC Conditions
4.8. ESI-Q TRAP-MS/MS
4.9. Yeast Two-Hybrid Assays
4.10. Dual-Luciferase Assay of Transiently Transformed Tobacco Leaves
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, X.P.; Li, X.L.; Duan, X.W.; Shen, Y.Y.; Xing, Y.; Cao, Q.Q.; Qin, L. Characterization of sck1, a novel Castanea mollissima mutant with the extreme short catkins and decreased gibberellin. PLoS ONE 2012, 7, e43181. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Li, J.; Liu, Y.; Zhang, Q.; Gao, Y.; Fang, K.; Cao, Q.; Qin, L.; Xing, Y. Roles of the GA-mediated SPL gene family and miR156 in the floral development of Chinese chestnut (Castanea mollissima). Int. J. Mol. Sci. 2019, 20, 1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.Q.; Shen, Y.Y.; Qin, L.; Cao, Q.Q.; Han, Z.H. Short catkin1, a novel mutant of Castanea mollissima, is associated with programmed cell death during chestnut staminate flower differentiation. Sci. Hortic. 2011, 130, 431–435. [Google Scholar] [CrossRef]
- Tanurdzic, M.; Banks, J.A. Sex-determining mechanisms in land plants. Plant Cell 2004, 16, S61–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, S.S.; Niels, A.M. Plant sex chromosomes defy evolutionary models of expanding recombination suppression and genetic degeneration. Nat. Plants 2021, 4, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Marsch-Martínez, N.; de Folter, S. Hormonal control of the development of the gynoecium. Curr. Opin. Plant Biol. 2016, 29, 104–114. [Google Scholar] [CrossRef]
- Deb, J.; Bland, H.M.; Østergaard, L. Developmental cartography: Coordination via hormonal and genetic interactions during gynoecium formation. Curr. Opin. Plant Biol. 2018, 41, 54–60. [Google Scholar] [CrossRef]
- Song, S.; Qi, T.; Huang, H.; Xie, D. Regulation of stamen development by coordinated actions of jasmonate, auxin, and gibberellin in Arabidopsis. Mol. Plant 2013, 6, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Hua, C.; Shen, L.; Yu, H. New insights into gibberellin signaling in regulating flowering in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, H. Current aspects of auxin biosynthesis in plants. Biosci. Biotechnol. Biochem. 2016, 80, 34–42. [Google Scholar] [CrossRef]
- Aloni, R.; Aloni, E.; Langhans, M.; Ullrich, C.I. Role of auxin in regulating Arabidopsis flower development. Planta 2006, 223, 315–328. [Google Scholar] [CrossRef]
- Wasternack, C.; Hause, B. Jasmonates: Biosynthesis, perception, signal transduction and action in plant stress response, growth and development. Ann. Bot. 2013, 111, 1021–1058. [Google Scholar] [CrossRef]
- Zander, M.; Lewsey, M.G.; Clark, N.M.; Yin, L.; Bartlett, A.; Saldierna Guzmán, J.P.; Hann, E.; Langford, A.E.; Jow, B.; Wise, A.; et al. Integrated multi-omics framework of the plant response to jasmonic acid. Nat. Plants 2020, 6, 290–302. [Google Scholar] [CrossRef]
- Major, I.T.; Yoshida, Y.; Campos, M.L.; Kapali, G.; Xin, X.; Sugimoto, K.; de Oliveira Ferreira, D.; He, S.; Howe, G.A. Regulation of growth-defense balance by the Jasmonate Zim-Domain (JAZ)-MYC transcriptional module. New Phytol. 2017, 215, 1533–1547. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Q.; Zhang, X.; Wu, F.; Feng, H.; Deng, L.; Xu, L.; Zhang, M.; Wang, Q.; Li, C. Transcriptional mechanism of jasmonate receptor COI1-mediated delay of flowering time in Arabidopsis. Plant Cell 2015, 27, 2814–2828. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Sun, L.; Qi, T.; Zhang, B.; Peng, W.; Liu, Y.; Xie, D. The bHLH transcription factor MYC3 interacts with the Jasmonate ZIM-domain proteins to mediate jasmonate response in Arabidopsis. Mol. Plant 2011, 4, 279–288. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Ye, T.; Lu, Y.; Chen, X.; Wu, Y. The inhibitory effect of ABA on floral transition is mediated by ABI5 in Arabidopsis. J. Exp. Bot. 2013, 64, 675–684. [Google Scholar] [CrossRef]
- Shu, K.; Chen, Q.; Wu, Y.; Liu, R.; Zhang, H.; Wang, S.; Tang, S.; Yang, W.; Xie, Q. Abscisic acid-insensitive 4 negatively regulates flowering through directly promoting Arabidopsis flowering locus C transcription. J. Exp. Bot. 2016, 67, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Achard, P.; Baghour, M.; Chapple, A.; Hedden, P.; van der Straeten, D.; Genschik, P.; Moritz, T.; Harberd, N.P. The plant stress hormone ethylene controls floral transition via DELLA-dependent regulation of floral meristem-identity genes. Proc. Natl. Acad. Sci. USA 2007, 104, 6484–6489. [Google Scholar] [CrossRef] [Green Version]
- Achard, P.; Cheng, H.; de Grauwe, L.; Decat, J.; Schoutteten, H.; Moritz, T.; van der Straeten, D.; Peng, J.; Harberd, N.P. Integration of plant responses to environmentally activated phytohormonal signals. Science 2006, 311, 91–94. [Google Scholar] [CrossRef]
- Kardailsky, I.; Shukla, V.K.; Ahn, J.H.; Dagenais, N.; Christensen, S.K.; Nguyen, J.T.; Chory, J.; Harrison, M.J.; Weigel, D. Activation tagging of the floral inducer FT. Science 1999, 286, 1962–1965. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Suh, S.S.; Park, E.; Cho, E.; Ahn, J.H.; Kim, S.G.; Lee, J.S.; Kwon, Y.M.; Lee, I. The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev. 2000, 14, 2366–2376. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.; Peng, J. DELLA proteins and GA signalling in Arabidopsis. J. Plant Growth Regul. 2003, 22, 134–140. [Google Scholar] [CrossRef]
- Li, Q.F.; Wang, C.; Jiang, L.; Li, S.; Sun, S.; He, J.X. An interaction between BZR1 and DELLAs mediates direct signaling crosstalk between Brassinosteroids and Gibberellins in Arabidopsis. Sci. Signal. 2012, 5, ra72. [Google Scholar] [CrossRef]
- Kumar, S.V.; Lucyshyn, D.; Jaeger, K.E.; Alós, E.; Alvey, E.; Harberd, N.P.; Wigge, P.A. Transcription factor PIF4 controls the thermosensory activation of flowering. Nature 2012, 484, 242. [Google Scholar] [CrossRef]
- Wang, W.R.; Fan, X.C.; Zhang, W.Y.; Liu, C.H.; Fang, J.G.; Wang, C.; University, N.A. Study progress on gibberellin metabolism and signaling transduction pathway in fruits trees. Biotechnol. Bull. 2017, 33, 1–7. [Google Scholar]
- Guo, C. Study on the Flower Bud Differentiation and the Change of Endogenous Hormones in Chestnut. Master’s Thesis, Northwest Agriculture and Frestry University, Yangling, China, 2010. [Google Scholar]
- Xing, Y.; Liu, Y.; Zhang, Q.; Nie, X.; Qin, L. Hybrid de novo genome assembly of Chinese chestnut (Castanea mollissima). GigaScience 2019, 8, giz112. [Google Scholar] [CrossRef]
- Collani, S.; Neumann, M.; Yant, L.; Schmid, M. FT modulates genome-wide DNA-binding of the bZIP transcription factor FD. Plant Physiol. 2019, 180, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhou, Y.; Chen, Q.; Huang, X.; Tian, C. Molecular basis of flowering time regulation in Arabidopsis. Chin. Bull. Bot. 2014, 49, 469. [Google Scholar]
- Mouradov, A.; Cremer, F.; Coupland, G. Control of flowering time: Interacting pathways as a basis for diversity. Plant Cell 2002, 14, S111–S130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Yu, F. Research progress on flower bud differentiation of trees. Sci. Silvae Sin. 2020, 56, 119–129. [Google Scholar]
- Lester, D.T. Variation in sex expression in Populus tremuloides Michx. Silvae Genet. 1963, 12, 141–151. [Google Scholar]
- Zha, S. Research on Endogenous Hormones and Molecular Regulation Mechanism in Female Inflorescence Differentiation of Castanea mollisima Blume. Master’s Thesis, Wuhan Polytechnic University, Wuhan, China, 2019. [Google Scholar]
- Qiu, W.; He, X.; Xu, Y. Research progress on sex control of chestnut flower buds. J. Fruit Sci. 2015, 32, 142–149. [Google Scholar]
- Zhang, L.; Li, B.; Bai, Z.; Liu, M.; Guohui, Q. Observation of the female flower cluster differentiation process of chestnut. J. Fruit Sci. 1999, 16, 280–283. [Google Scholar]
- Dms, A.; Crz, B.; Vht, A.; Rp, A. Floral characterization and pollen germination protocol for Castanea crenata Siebold & Zucc. S. Afr. J. Bot. 2020, 130, 389–395. [Google Scholar]
- Altamura, M.M.; Tomassi, M. Auxin, photoperiod and putrescine affect flower neoformation in normal and rolB-transformed tobacco thin cell layers. Plant Physiol. Biochem. 1998, 36, 441–448. [Google Scholar] [CrossRef]
- Xu, J.; Chen, H.; Li, X.; Zhang, Z.; Wang, Y. Effect of exogenous polyamines on female and male flower differentiation and content of endogenous polyamines in leaves of walnut. Acta Hortic. Sin. 2004, 31, 437–440. [Google Scholar]
- Guo, X.; Yu, C.; Luo, L.; Wan, H.; Zhen, N.; Xu, T.; Tan, J.; Pan, H.; Zhang, Q. Transcriptome of the floral transition in Rosa chinensis ‘Old Blush’. BMC Genom. 2017, 18, 199. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Zhang, D.; Li, Y.; Shen, Y.; Zhao, C.; Ma, J.; An, N.; Han, M. Transcription profiles reveal sugar and hormone signaling pathways mediating flower induction in apple (Malus domestica Borkh.). Plant Cell Physiol. 2015, 56, 2052–2068. [Google Scholar] [CrossRef] [Green Version]
- Wahl, V.; Ponnu, J.; Schlereth, A.; Arrivault, S.; Langenecker, T.; Franke, A.; Feil, R.; Lunn, J.E.; Stitt, M.; Schmid, M. Regulation of flowering by trehalose-6-phosphate signaling in Arabidopsis thaliana. Science 2013, 339, 704–707. [Google Scholar] [CrossRef]
- Wang, M.; Xi, D.; Chen, Y.; Zhu, C.; Zhao, Y.; Geng, G. Morphological characterization and transcriptome analysis of pistillate flowering in pecan (Carya illinoinensis). Sci. Hortic. 2019, 257, 108674. [Google Scholar] [CrossRef]
- Hui, W.; Yang, Y.; Wu, G.; Peng, C.; Chen, X.; Zayed, M.Z. Transcriptome profile analysis reveals the regulation mechanism of floral sex differentiation in Jatropha curcas L. Sci. Rep. 2017, 7, 16421. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S. Gibberellin metabolism and its regulation. Annu. Rev. Plant Biol. 2008, 59, 225–251. [Google Scholar] [CrossRef]
- Murase, K.; Hlirano, Y.; Sun, T.P.; Hakoshima, T. Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 2008, 456, 459–463. [Google Scholar] [CrossRef]
- Koornneef, M.; Alonso, B.C.; Vries, H.B.; Hanhart, C.; Peeters, A. Genetic interactions among late-flowering mutants of Arabidopsis. Genetics 1998, 148, 885–892. [Google Scholar] [CrossRef]
- Bao, S.; Hua, C.; Huang, G.; Cheng, P.; Gong, X.; Shen, L.; Yu, H. Molecular basis of natural variation in photoperiodic flowering responses. Dev. Cell 2019, 50, 90–101.e3. [Google Scholar] [CrossRef]
- Doyle, M.R.; Davis, S.J.; Bastow, R.M.; Mc Watters, H.G.; Kozma-Bognár, L.; Nagy, F.; Millar, A.J.; Amasino, R.M. The ELF4 gene controls circadian rhythms and flowering time in Arabidopsis thaliana. Nature 2002, 419, 74–77. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Sureshkumar, S.; Lempe, J.; Weigel, D. Potent induction of Arabidopsis thaliana flowering by elevated growth temperature. PLoS Genet. 2006, 2, e106. [Google Scholar] [CrossRef]
- De Lucas, M.; Daviere, J.M.; Rodríguez-Falcón, M.; Pontin, M.; Iglesias-Pedraz, J.M.; Lorrain, S.; Fankhauser, C.; Blázquez, M.A.; Titarenko, E.; Prat, S. A molecular framework for light and gibberellin control of cell elongation. Nature 2008, 451, 480–484. [Google Scholar] [CrossRef]
- Koo, A.J.; Howe, G.A. The wound hormone jasmonate. Phytochemistry 2009, 70, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Li, Y.; Xia, K.; Yan, Y.; Hao, Y. DELLAs modulate jasmonate signaling via competitive binding to JAZs. Dev. Cell 2010, 19, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Cortés, A.J.; Felipe, L.H. Harnessing crop wild diversity for climate change adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef]
- Fritsche, S.; Klocko, A.L.; Boron, A.; Brunner, A.M.; Thorlby, G. Strategies for engineering reproductive sterility in plantation forests. Front. Plant Sci. 2018, 9, 1671. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Silva-Junior, O.B.; Resende, R.T.; Cappa, E.P.; Müller, B.S.; Tan, B.; Isik, F.; Ratcliffe, B.; EI-Kassaby, Y.A. Quantitative genetics and genomics converge to accelerate forest tree breeding. Front. Plant Sci. 2018, 9, 1693. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Qiao, L.; Chen, J.; Rong, Y.; Zhao, Y.; Cui, X.; Xu, J.; Hou, X.; Dong, C.H. Arabidopsis REM16 acts as a B3 domain transcription factor to promote flowering time via directly binding to the promoters of SOC1 and FT. Plant J. 2020, 103, 1386–1398. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.J.; Manuela, R.M.; Larry, E.B. Modern strategies to assess and breed forest tree adaptation to changing climate. Front. Plant Sci. 2020, 11, 1606. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Number of Annotated Genes |
|---|---|
| NR | 2582 |
| Swissprot | 1707 |
| TrEMBL | 2461 |
| KOG | 2002 |
| KEGG | 226 |
| GO | 1677 |
| Group | Significant Difference in the Number of Metabolites | Down Regulation of the Number of Metabolites | Up Regulation of the Number of Metabolites |
|---|---|---|---|
| A2 vs. B2 | 59 | 20 | 39 |
| A1 vs. B1 | 90 | 16 | 74 |
| A2 vs. A1 | 120 | 30 | 90 |
| B2 vs. B1 | 171 | 34 | 137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.; Zha, S.; Luo, Y.; Li, L.; Wang, S.; Wu, S.; Cheng, S.; Li, L. JAZ1-3 and MYC2-1 Synergistically Regulate the Transformation from Completely Mixed Flower Buds to Female Flower Buds in Castanea mollisima. Int. J. Mol. Sci. 2022, 23, 6452. https://doi.org/10.3390/ijms23126452
Cheng H, Zha S, Luo Y, Li L, Wang S, Wu S, Cheng S, Li L. JAZ1-3 and MYC2-1 Synergistically Regulate the Transformation from Completely Mixed Flower Buds to Female Flower Buds in Castanea mollisima. International Journal of Molecular Sciences. 2022; 23(12):6452. https://doi.org/10.3390/ijms23126452
Chicago/Turabian StyleCheng, Hua, Sanxing Zha, Yanyan Luo, Li Li, Shiyan Wang, Shuai Wu, Shuiyuan Cheng, and Linling Li. 2022. "JAZ1-3 and MYC2-1 Synergistically Regulate the Transformation from Completely Mixed Flower Buds to Female Flower Buds in Castanea mollisima" International Journal of Molecular Sciences 23, no. 12: 6452. https://doi.org/10.3390/ijms23126452
APA StyleCheng, H., Zha, S., Luo, Y., Li, L., Wang, S., Wu, S., Cheng, S., & Li, L. (2022). JAZ1-3 and MYC2-1 Synergistically Regulate the Transformation from Completely Mixed Flower Buds to Female Flower Buds in Castanea mollisima. International Journal of Molecular Sciences, 23(12), 6452. https://doi.org/10.3390/ijms23126452