
Soluble Epoxide Hydrolase and Diabetes Complications


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
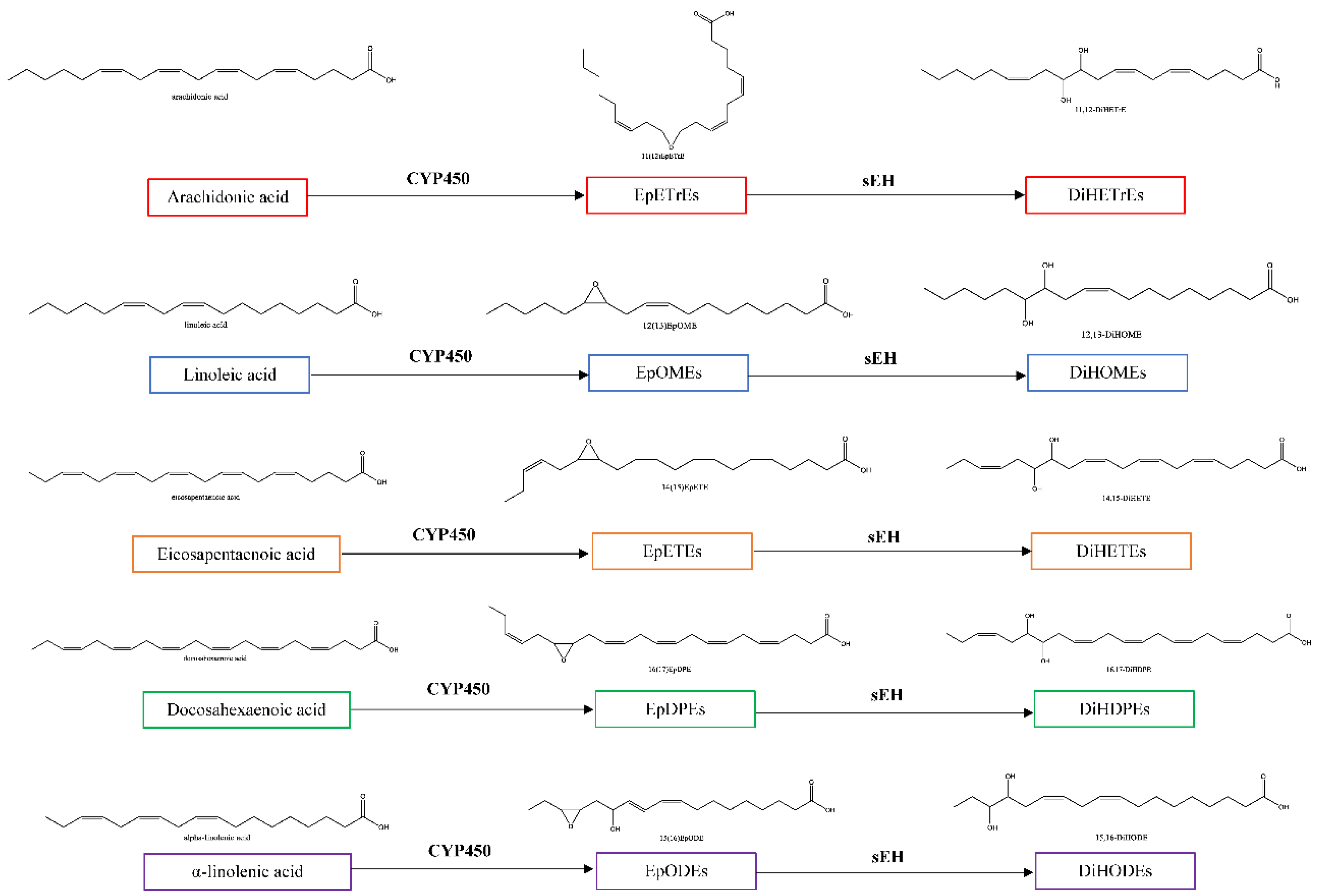
1.1. Oxylipins of the CYP450-sEH Pathway
1.2. sEH Oxylipins in Diabetes
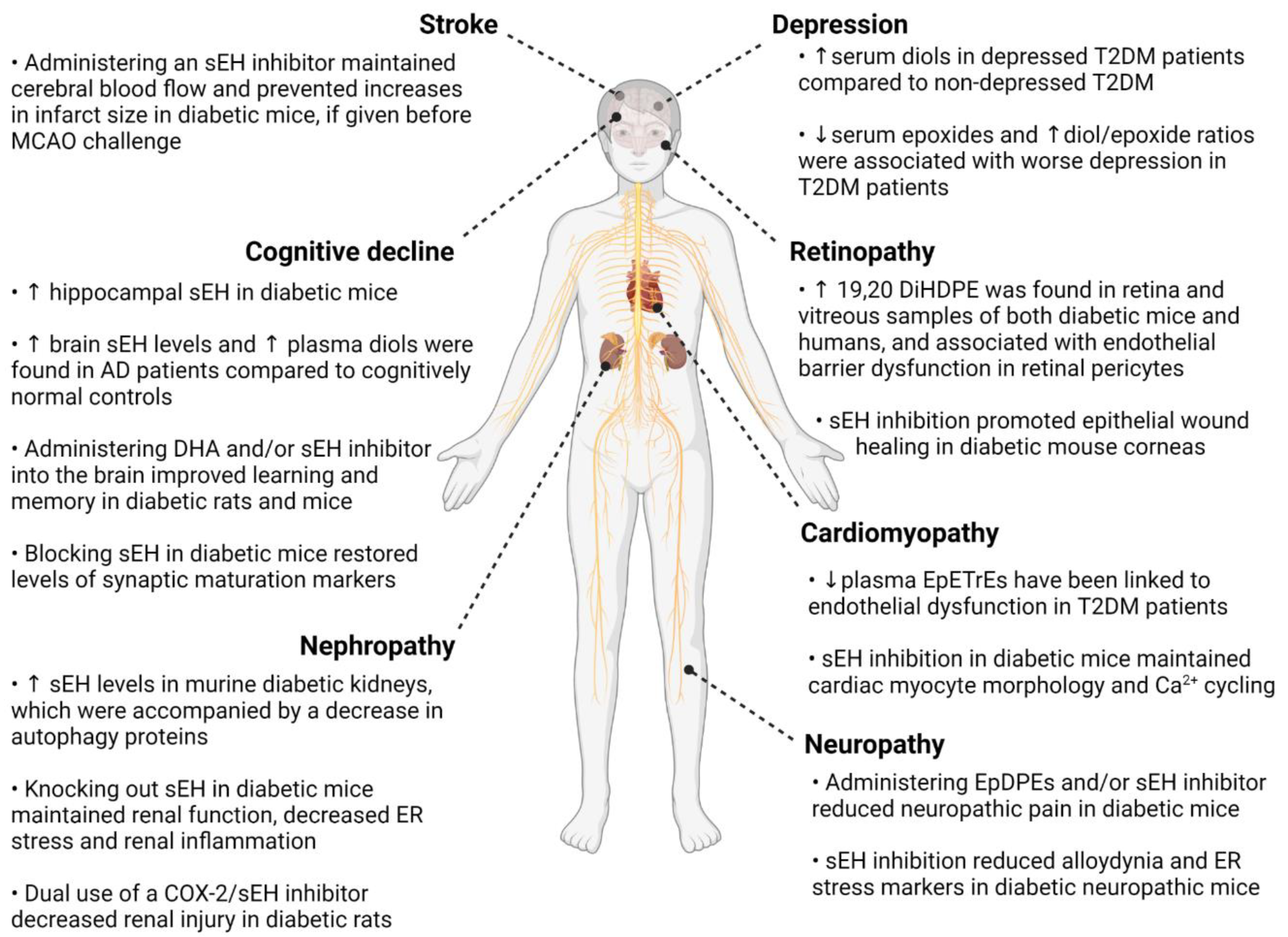
2. Oxylipins and Complications of Diabetes
2.1. Retinopathy
2.2. Neuropathy
2.3. Nephropathy
2.4. Heart Failure with Preserved Ejection Fraction and Cardiomyopathy
2.5. Large Vessel Stroke
2.6. Cognitive Decline and Dementia
2.7. Depression
3. Synthesis and Clinical Translation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas; International Diabetes Federation: Basel, Switzerland, 2019. [Google Scholar]
- Papatheodorou, K.; Banach, M.; Bekiari, E.; Rizzo, M.; Edmonds, M. Complications of Diabetes 2017. J. Diabetes Res. 2018, 2018, 3086167. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Callaghan, B.C.; Feldman, E.L. The emerging role of dyslipidemia in diabetic microvascular complications. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Molecular, Cellular, and Clinical Evidence That Sodium-Glucose Cotransporter 2 Inhibitors Act as Neurohormonal Antagonists When Used for the Treatment of Chronic Heart Failure. J. Am. Heart Assoc. 2020, 9, e016270. [Google Scholar] [CrossRef]
- Grapov, D.; Adams, S.H.; Pedersen, T.L.; Garvey, W.T.; Newman, J.W. Type 2 Diabetes Associated Changes in the Plasma Non-Esterified Fatty Acids, Oxylipins and Endocannabinoids. PLoS ONE 2012, 7, e48852. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Hammock, B.D. Distribution and properties of a mammalian soluble epoxide hydrase. Biochem. Pharmacol. 1980, 29, 389–395. [Google Scholar] [CrossRef]
- Hashimoto, K. Role of Soluble Epoxide Hydrolase in Metabolism of PUFAs in Psychiatric and Neurological Disorders. Front. Pharmacol. 2019, 9, 36. [Google Scholar] [CrossRef]
- Nelson, J.W.; Young, J.M.; Borkar, R.N.; Woltjer, R.L.; Quinn, J.F.; Silbert, L.C.; Grafe, M.R.; Alkayed, N.J. Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat. 2014, 113–115, 30–37. [Google Scholar] [CrossRef]
- Hu, J.; Dziumbla, S.; Lin, J.; Bibli, S.-I.; Zukunft, S.; De Mos, J.; Awwad, K.; Frömel, T.; Jungmann, A.; Devraj, K.; et al. Inhibition of soluble epoxide hydrolase prevents diabetic retinopathy. Nature 2017, 552, 248–252. [Google Scholar] [CrossRef]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef]
- Moghaddam, M.F.; Granf, D.F.; Cheek, J.M.; Greene, F.; Williamson, K.C.; Hammock, B.D. Bioactivation of Leukotoxins to Their Toxic Dials by Epoxide Hydrolase. 1997. Available online: http://www.nature.com/naturemedicine (accessed on 29 April 2022).
- Behl, T.; Grover, M.; Shah, K.; Makkar, R.; Kaur, L.; Sharma, S.; Gupta, J. Role of Omega-3-Fatty Acids in the Management of Diabetes and Associated Complications. In Bioactive Food as Dietary Interventions for Diabetes; Academic Press: Cambridge, MA, USA, 2019; pp. 185–192. [Google Scholar] [CrossRef]
- Ramirez, C.E.; Shuey, M.M.; Milne, G.; Gilbert, K.; Hui, N.; Yu, C.; Luther, J.; Brown, N.J. Arg287Gln variant of EPHX2 and epoxyeicosatrienoic acids are associated with insulin sensitivity in humans. Prostaglandins Other Lipid Mediat. 2014, 113–115, 38–44. [Google Scholar] [CrossRef]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, e93751. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Ontko, C.D.; Capozzi, M.E.; Kim, M.J.; McCollum, G.W.; Penn, J.S. Cytochrome P450-epoxygenated fatty acids inhibit Müller glial inflammation. Sci. Rep. 2021, 11, 9677. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lee, P.; Yan, C.; Gao, N.; Wang, J.; Fan, X.; Yu, F.-S. Inhibition of Soluble Epoxide Hydrolase 2 Ameliorates Diabetic Keratopathy and Impaired Wound Healing in Mouse Corneas. Diabetes 2018, 67, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Bibli, S.I.; Wittig, J.; Zukunft, S.; Lin, J.; Hammes, H.-P.; Popp, R.; Fleming, I. Soluble epoxide hydrolase promotes astrocyte survival in retinopathy of prematurity. J. Clin. Investig. 2019, 129, 5204–5218. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Schmelzer, K.; Lee, T.-S.; Fang, X.; Zhu, Y.; Spector, A.A.; Gill, S.; Morisseau, C.; Hammock, B.D.; et al. The antiinflammatory effect of laminar flow: The role of PPARγ, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2005, 102, 16747–16752. [Google Scholar] [CrossRef]
- Pang, W.; Li, N.; Ai, D.; Niu, X.-L.; Guan, Y.-F.; Zhu, Y. Activation of peroxisome proliferator-activated receptor-γ downregulates soluble epoxide hydrolase in cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 2011, 38, 358–364. [Google Scholar] [CrossRef]
- Roche, C.; Guerrot, D.; Harouki, N.; Duflot, T.; Besnier, M.; Rémy-Jouet, I.; Renet, S.; Dumesnil, A.; Lejeune, A.; Morisseau, C.; et al. Impact of soluble epoxide hydrolase inhibition on early kidney damage in hyperglycemic overweight mice. Prostaglandins Other Lipid Mediat. 2015, 120, 148–154. [Google Scholar] [CrossRef]
- Chen, G.; Xu, R.; Wang, Y.; Wang, P.; Zhao, G.; Xu, X.; Gruzdev, A.; Zeldin, D.C.; Wang, D.W. Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. Am. J. Physiol. Metab. 2012, 303, E563–E575. [Google Scholar] [CrossRef]
- Inceoglu, B.; Bettaieb, A.; Haj, F.G.; Gomes, A.V.; Hammock, B.D. Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors. Prostaglandins Other Lipid Mediat. 2017, 133, 68–78. [Google Scholar] [CrossRef]
- Wu, J.; Fan, Z.; Zhao, Y.; Chen, Q.; Xiao, Q. Inhibition of soluble epoxide hydrolase (sEH) protects hippocampal neurons and reduces cognitive decline in type 2 diabetic mice. Eur. J. Neurosci. 2021, 53, 2532–2540. [Google Scholar] [CrossRef]
- Schreiber, A.K. Diabetic neuropathic pain: Physiopathology and treatment. World J. Diabetes 2015, 6, 432–444. [Google Scholar] [CrossRef]
- Wagner, K.; Lee, K.S.S.; Yang, J.; Hammock, B. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur. J. Pain 2016, 21, 456–465. [Google Scholar] [CrossRef]
- Inceoglu, B.; Bettaieb, A.; da Silva, C.A.T.; Lee, K.S.S.; Haj, F.G.; Hammock, B.D. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. USA 2015, 112, 9082–9087. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Alam Riaz, T.; Kim, H.-R.; Chae, H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Lupachyk, S.; Watcho, P.; Obrosov, A.A.; Stavniichuk, R.; Obrosova, I.G. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp. Neurol. 2013, 247, 342–348. [Google Scholar] [CrossRef]
- Wagner, K.; Yang, J.; Inceoglu, B.; Hammock, B.D. Soluble Epoxide Hydrolase Inhibition Is Antinociceptive in a Mouse Model of Diabetic Neuropathy. J. Pain 2014, 15, 907–914. [Google Scholar] [CrossRef]
- Wagner, K.; Inceoglu, B.; Dong, H.; Yang, J.; Hwang, S.H.; Jones, P.; Morisseau, C.; Hammock, B.D. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur. J. Pharmacol. 2012, 700, 93–101. [Google Scholar] [CrossRef]
- Sasso, O.; Wagner, K.; Morisseau, C.; Inceoglu, B.; Hammock, B.D.; Piomelli, D. Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol. Res. 2015, 97, 7–15. [Google Scholar] [CrossRef]
- Churi, S.B.; Abdel-Aleem, O.S.; Tumber, K.K.; Scuderi-Porter, H.; Taylor, B.K. Intrathecal Rosiglitazone Acts at Peroxisome Proliferator–Activated Receptor-γ to Rapidly Inhibit Neuropathic Pain in Rats. J. Pain 2008, 9, 639–649. [Google Scholar] [CrossRef]
- Lecka-Czernik, B.; Moerman, E.J.; Grant, D.F.; Lehmann, J.M.; Manolagas, S.C.; Jilka, R.L. Divergent Effects of Selective Peroxisome Proliferator-Activated Receptor-γ2 Ligands on Adipocyte Versus Osteoblast Differentiation. Endocrinology 2002, 143, 2376–2384. [Google Scholar] [CrossRef]
- Cataldi, S.; Costa, V.; Ciccodicola, A.; Aprile, M. PPARγ and Diabetes: Beyond the Genome and Towards Personalized Medicine. Curr. Diabetes Rep. 2021, 21, 18. [Google Scholar] [CrossRef]
- Kim, T. Peroxisome-proliferator-activated receptors regulate redox signaling in the cardiovascular system. World J. Cardiol. 2013, 5, 164–174. [Google Scholar] [CrossRef]
- Hasegawa-Moriyama, M.; Kurimoto, T.; Nakama, M.; Godai, K.; Kojima, M.; Kuwaki, T.; Kanmura, Y. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates inflammatory pain through the induction of heme oxygenase-1 in macrophages. Pain 2013, 154, 1402–1412. [Google Scholar] [CrossRef]
- Abraham, N.G.; Sodhi, K.; Silvis, A.M.; Vanella, L.; Favero, G.; Rezzani, R.; Lee, C.; Zeldin, D.C.; Schwartzman, M.L. CYP2J2 Targeting to Endothelial Cells Attenuates Adiposity and Vascular Dysfunction in Mice Fed a High-Fat Diet by Reprogramming Adipocyte Phenotype. Hypertension 2014, 64, 1352–1361. [Google Scholar] [CrossRef]
- Iliff, J.J.; Fairbanks, S.L.; Balkowiec, A.; Alkayed, N.J. Epoxyeicosatrienoic acids are endogenous regulators of vasoactive neuropeptide release from trigeminal ganglion neurons. J. Neurochem. 2010, 115, 1530–1542. [Google Scholar] [CrossRef]
- Shuba, Y.M. Beyond Neuronal Heat Sensing: Diversity of TRPV1 Heat-Capsaicin Receptor-Channel Functions. Front. Cell. Neurosci. 2021, 14, 612480. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, C.; Li, H.; Tang, C.; Kan, H.; Yang, Z.; Mao, A.; Ma, X. TRPV4 (Transient Receptor Potential Vanilloid 4) Mediates Endothelium-Dependent Contractions in the Aortas of Hypertensive Mice. Hypertension 2018, 71, 134–142. [Google Scholar] [CrossRef]
- Lim, A. Diabetic nephropathy-complications and treatment. Int. J. Nephrol. Renov. Dis. 2014, 7, 361–381. [Google Scholar] [CrossRef]
- Jiang, X.-S.; Xiang, X.-Y.; Chen, X.-M.; He, J.-L.; Liu, T.; Gan, H.; Du, X.-G. Inhibition of soluble epoxide hydrolase attenuates renal tubular mitochondrial dysfunction and ER stress by restoring autophagic flux in diabetic nephropathy. Cell Death Dis. 2020, 11, 385. [Google Scholar] [CrossRef]
- Bettaieb, A.; Koike, S.; Hsu, M.-F.; Ito, Y.; Chahed, S.; Bachaalany, S.; Gruzdev, A.; Calvo-Rubio, M.; Lee, K.S.S.; Inceoglu, B.; et al. Soluble epoxide hydrolase in podocytes is a significant contributor to renal function under hyperglycemia. Biochim. Biophys. Acta (BBA) - Gen. Subj. 2017, 1861, 2758–2765. [Google Scholar] [CrossRef]
- Abdul, M.; Khan, H.; Hwang, S.H.; Sharma, A.; Corbett, J.A.; Hammock, B.D.; Imig, J.D. A dual cox-2/seh inhibitor improves the metabolic profile and reduces kidney injury in zucker diabetic fatty rat hhs public Access. Prostaglandins Other Lipid Mediat. 2016, 125, 40–47. [Google Scholar] [CrossRef]
- Khan, A.H.; Kolb, L.; Skibba, M.; Hartmann, M.; Blöcher, R.; Proschak, E.; Imig, J.D. A novel dual PPAR-γ agonist/sEH inhibitor treats diabetic complications in a rat model of type 2 diabetes. Diabetologia 2018, 61, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Roumeliotis, A.; Stamou, A.; Panagoutsos, S.; Manolopoulos, V.G.; Tsetsos, F.; Georgitsi, M.; Liakopoulos, V. Association of rs11780592 Polymorphism in the Human Soluble Epoxide Hydrolase Gene (EPHX2) with Oxidized LDL and Mortality in Patients with Diabetic Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2021, 2021, 8817502. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Kapadia, P.; Bikkina, P.; Landicho, M.A.; Parekh, S.; Haas, M.J.; Mooradian, A.D. Effect of anti-hyperglycemic drugs on endoplasmic reticulum (ER) stress in human coronary artery endothelial cells. Eur. J. Pharmacol. 2021, 907, 174249. [Google Scholar] [CrossRef]
- Maayah, Z.H.; McGinn, E.; Al Batran, R.; Gopal, K.; Ussher, J.R.; El-Kadi, A.O.S. Role of Cytochrome p450 and Soluble Epoxide Hydrolase Enzymes and Their Associated Metabolites in the Pathogenesis of Diabetic Cardiomyopathy. J. Cardiovasc. Pharmacol. 2019, 74, 235–245. [Google Scholar] [CrossRef]
- Duflot, T.; Moreau-Grangé, L.; Roche, C.; Iacob, M.; Wils, J.; Rémy-Jouet, I.; Cailleux, A.-F.; Leuillier, M.; Renet, S.; Li, D.; et al. Altered bioavailability of epoxyeicosatrienoic acids is associated with conduit artery endothelial dysfunction in type 2 diabetic patients. Cardiovasc. Diabetol. 2019, 18, 35. [Google Scholar] [CrossRef]
- Nayeem, M.A. Role of oxylipins in cardiovascular diseases. Acta Pharmacol. Sin. 2018, 39, 1142–1154. [Google Scholar] [CrossRef]
- Merkel, M.J.; Liu, L.; Cao, Z.; Packwood, W.; Young, J.; Alkayed, N.J.; Van Winkle, D.M. Inhibition of soluble epoxide hydrolase preserves cardiomyocytes: Role of STAT3 signaling. Am. J. Physiol. Circ. Physiol. 2010, 298, H679–H687. [Google Scholar] [CrossRef]
- Guglielmino, K.; Jackson, K.; Harris, T.R.; Vu, V.; Dong, H.; Dutrow, G.; Evans, J.E.; Graham, J.; Cummings, B.P.; Havel, P.J.; et al. Pharmacological inhibition of soluble epoxide hydrolase provides cardioprotection in hyperglycemic rats. Am. J. Physiol. Circ. Physiol. 2012, 303, H853–H862. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, L.; Wang, Z.; Chen, S.; Zhang, W.; Ma, M. Relationship between EPHX2 gene polymorphisms and essential hypertension in Uygur, Kazakh, and Han. Genet. Mol. Res. 2015, 14, 3474–3480. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.-H.; Lew, J.; Borschmann, K.; Thijs, V.; Ekinci, E.I. Prevalence of diabetes and its effects on stroke outcomes: A meta-analysis and literature review. J. Diabetes Investig. 2019, 10, 780–792. [Google Scholar] [CrossRef]
- Chen, R.; Ovbiagele, B.; Feng, W. Diabetes and Stroke: Epidemiology, Pathophysiology, Pharmaceuticals and Outcomes. Am. J. Med. Sci. 2016, 351, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Zuloaga, K.L.; Krasnow, S.M.; Zhu, X.; Zhang, W.; Jouihan, S.A.; Shangraw, R.E.; Alkayed, N.J.; Marks, D.L. Mechanism of Protection by Soluble Epoxide Hydrolase Inhibition in Type 2 Diabetic Stroke. PLoS ONE 2014, 9, e97529. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Zhang, W.; Allen, E.; Bah, T.; Shangraw, R.; Alkayed, N. Soluble Epoxide Hydrolase Blockade after Stroke Onset Protects Normal but Not Diabetic Mice. Int. J. Mol. Sci. 2021, 22, 5419. [Google Scholar] [CrossRef]
- Atone, J.; Wagner, K.; Hashimoto, K.; Hammock, B.D. Cytochrome P450 derived epoxidized fatty acids as a therapeutic tool against neuroinflammatory diseases. Prostaglandins Other Lipid Mediat. 2019, 147, 106385. [Google Scholar] [CrossRef]
- Tu, R.; Armstrong, J.; Lee, K.S.S.; Hammock, B.D.; Sapirstein, A.; Koehler, R.C. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci. Rep. 2018, 8, 5279. [Google Scholar] [CrossRef]
- Chang, L.-H.; Lin, H.-C.; Huang, S.-S.; Chen, I.-C.; Chu, K.-W.; Chih, C.-L.; Liang, Y.-W.; Lee, Y.-C.; Chen, Y.-Y.; Lee, Y.-H.; et al. Blockade of soluble epoxide hydrolase attenuates post-ischemic neuronal hyperexcitation and confers resilience against stroke with TrkB activation. Sci. Rep. 2018, 8, 118. [Google Scholar] [CrossRef]
- Demirdöğen, B.C.; Miçooğulları, Y.; Özçelik, A.T.; Adalı, O. Missense Genetic Polymorphisms of Microsomal (EPHX1) and Soluble Epoxide Hydrolase (EPHX2) and Their Relation to the Risk of Large Artery Atherosclerotic Ischemic Stroke in a Turkish Population. Neuropsychiatr. Dis. Treat. 2021, 16, 3251–3265. [Google Scholar] [CrossRef]
- Yi, X.; Wu, L.; Liao, D.; Wang, C.; Zhang, B. Interactions among CYP2C8, EPHX2, and CYP4A11 Variants and CYP Plasma Metabolite Levels in Ischemic Stroke. J. Atheroscler. Thromb. 2016, 23, 1286–1293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yi, X.; Lin, J.; Li, J.; Zhou, Q.; Han, Z. Epoxyeicosatrienoic Acids are Mediated by EPHX2 Variants and may be a Predictor of Early Neurological Deterioration in Acute Minor Ischemic Stroke. J. Atheroscler. Thromb. 2017, 24, 1258–1266. [Google Scholar] [CrossRef]
- Yi, X.; Zhang, B.; Wang, C.; Liao, D.; Lin, J.; Chi, L. CYP2C8 rs17110453 and EPHX2 rs751141 two-locus interaction increases susceptibility to ischemic stroke. Gene 2015, 565, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Munshi, M.N. Cognitive Dysfunction in Older Adults with Diabetes: What a Clinician Needs to Know. Diabetes Care 2017, 40, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Leys, D. Vascular Cognitive Impairment. Circ. Res. 2017, 120, 573–591. [Google Scholar] [CrossRef]
- Biessels, G.J.; Reijmer, Y.D. Brain Changes Underlying Cognitive Dysfunction in Diabetes: What Can We Learn From MRI? Diabetes 2014, 63, 2244–2252. [Google Scholar] [CrossRef]
- Yu, D.; Hennebelle, M.; Sahlas, D.J.; Ramirez, J.; Gao, F.; Masellis, M.; Cogo-Moreira, H.; Swartz, R.H.; Herrmann, N.; Chan, P.C.; et al. Soluble Epoxide Hydrolase-Derived Linoleic Acid Oxylipins in Serum Are Associated with Periventricular White Matter Hyperintensities and Vascular Cognitive Impairment. Transl. Stroke Res. 2018, 10, 522–533. [Google Scholar] [CrossRef]
- Shinto, L.; Lahna, D.; Murchison, C.F.; Dodge, H.; Hagen, K.; David, J.; Kaye, J.; Quinn, J.F.; Wall, R.; Silbert, L.C. Oxidized Products of Omega-6 and Omega-3 Long Chain Fatty Acids Are Associated with Increased White Matter Hyperintensity and Poorer Executive Function Performance in a Cohort of Cognitively Normal Hypertensive Older Adults. J. Alzheimer’s Dis. 2020, 74, 65–77. [Google Scholar] [CrossRef]
- Borkowski, K.; Taha, A.Y.; Pedersen, T.L.; De Jager, P.L.; Bennett, D.A.; Arnold, M.; Kaddurah-Daouk, R.; Newman, J.W. Serum metabolomic biomarkers of perceptual speed in cognitively normal and mildly impaired subjects with fasting state stratification. Sci. Rep. 2021, 11, 18964. [Google Scholar] [CrossRef]
- Morris, J.K.; Piccolo, B.D.; John, C.S.; Green, Z.D.; Thyfault, J.P.; Adams, S.H. Oxylipin Profiling of Alzheimer’s Disease in Nondiabetic and Type 2 Diabetic Elderly. Metabolites 2019, 9, 177. [Google Scholar] [CrossRef]
- Lee, H.-T.; Lee, K.-I.; Chen, C.-H.; Lee, T.-S. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 267. [Google Scholar] [CrossRef]
- Wong, S.K.; Nguyen, M.M.; Major-Orfao, C.; Lanctôt, K.L.; Herrmann, N.; Oh, P.; Shah, B.R.; Gilbert, J.; Assal, A.; Halperin, I.; et al. 36-LB: Linoleic Acid Derived Oxylipins, Memory, and Microvascular Retinal Complications in Patients with Type 2 Diabetes Mellitus. Diabetes 2020, 69. [Google Scholar] [CrossRef]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Arfeen, M.; Ahmed, S.; Jamwal, R.; Hammock, B.D.; Lahkar, M.; Goswami, S.K. Docosahexaenoic Acid Increases the Potency of Soluble Epoxide Hydrolase Inhibitor in Alleviating Streptozotocin-Induced Alzheimer’s Disease-Like Complications of Diabetes. Front. Pharmacol. 2019, 10, 288. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, Y.; Fan, Z.; Chen, Q.; Chen, J.; Sun, Y.; Jiang, X.; Xiao, Q. Soluble epoxide hydrolase inhibitor protects against blood-brain barrier dysfunction in a mouse model of type 2 diabetes via the AMPK/HO-1 pathway. Biochem. Biophys. Res. Commun. 2020, 524, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Darwish, L.; Beroncal, E.; Sison, M.V.; Swardfager, W. Depression in people with type 2 diabetes: Current perspectives. Diabetes, Metab. Syndr. Obesity Targets Ther. 2018, 11, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Al Sayah, F.; Wozniak, L.; Rees, S.; Soprovich, A.; Chik, C.L.; Chue, P.; Florence, P.; Jacquier, J.; Lysak, P.; et al. Controlled trial of a collaborative primary care team model for patients with diabetes and depression: Rationale and design for a comprehensive evaluation. BMC Health Serv. Res. 2012, 12, 258. [Google Scholar] [CrossRef]
- Hennebelle, M.; Otoki, Y.; Yang, J.; Hammock, B.D.; Levitt, A.J.; Taha, A.Y.; Swardfager, W. Altered soluble epoxide hydrolase-derived oxylipins in patients with seasonal major depression: An exploratory study. Psychiatry Res. 2017, 252, 94–101. [Google Scholar] [CrossRef]
- Borsini, A.; Nicolaou, A.; Camacho-Muñoz, D.; Kendall, A.C.; Di Benedetto, M.G.; Giacobbe, J.; Su, K.-P.; Pariante, C.M. Omega-3 polyunsaturated fatty acids protect against inflammation through production of LOX and CYP450 lipid mediators: Relevance for major depression and for human hippocampal neurogenesis. Mol. Psychiatry 2021, 26, 6773–6788. [Google Scholar] [CrossRef]
- Anita, N.Z.; Forkan, N.; Kamal, R.; Nguyen, M.M.; Yu, D.; Major-Orfao, C.; Wong, S.K.; Lanctôt, K.L.; Herrmann, N.; Oh, P.I.; et al. Serum soluble epoxide hydrolase related oxylipins and major depression in patients with type 2 diabetes. Psychoneuroendocrinology 2021, 126, 105149. [Google Scholar] [CrossRef]
- Nguyen, M.M.; Perlman, G.; Kim, N.; Wu, C.-Y.; Daher, V.; Zhou, A.; Mathers, E.H.; Anita, N.Z.; Lanctôt, K.L.; Herrmann, N.; et al. Depression in type 2 diabetes: A systematic review and meta-analysis of blood inflammatory markers. Psychoneuroendocrinology 2021, 134, 105448. [Google Scholar] [CrossRef]
- Ren, Q.; Ma, M.; Ishima, T.; Morisseau, C.; Yang, J.; Wagner, K.M.; Zhang, J.-C.; Yang, C.; Yao, W.; Dong, C.; et al. Gene deficiency and pharmacological inhibition of soluble epoxide hydrolase confers resilience to repeated social defeat stress. Proc. Natl. Acad. Sci. USA 2016, 113, E1944–E1952. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Cai, H.; Song, J.; Chang, Q. The effects of sEH inhibitor on depression-like behavior and neurogenesis in male mice. J. Neurosci. Res. 2017, 95, 2483–2492. [Google Scholar] [CrossRef]
- Qin, X.-H.; Wu, Z.; Dong, J.-H.; Zeng, Y.-N.; Xiong, W.-C.; Liu, C.; Wang, M.-Y.; Zhu, M.-Z.; Chen, W.-J.; Zhang, Y.; et al. Liver Soluble Epoxide Hydrolase Regulates Behavioral and Cellular Effects of Chronic Stress. Cell Rep. 2019, 29, 3223–3234.e6. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Fu, Z.; Mui, D.; Zhu, H.; Zhang, Y. Exenatide inhibits NF-κB and attenuates ER stress in diabetic cardiomyocyte models. Aging 2020, 12, 8640–8651. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol. Ther. 2018, 192, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Nagata, N.; AbouBechara, D.; Chahed, S.; Morisseau, C.; Hammock, B.D.; Haj, F.G. Soluble Epoxide Hydrolase Deficiency or Inhibition Attenuates Diet-induced Endoplasmic Reticulum Stress in Liver and Adipose Tissue. J. Biol. Chem. 2013, 288, 14189–14199. [Google Scholar] [CrossRef] [PubMed]
- Ghaemi, F.; Firouzabadi, F.D.; Moosaie, F.; Shadnoush, M.; Poopak, A.; Kermanchi, J.; Abhari, S.M.F.; Forouzanfar, R.; Mansournia, M.A.; Khosravi, A.; et al. Effects of a Mediterranean diet on the development of diabetic complications: A longitudinal study from the nationwide diabetes report of the National Program for Prevention and Control of Diabetes (NPPCD 2016-2020). Maturitas 2021, 153, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.L.; Rudyk, O.; Prysyazhna, O.; Kamynina, A.; Yang, J.; Morisseau, C.; Hammock, B.D.; Freeman, B.A.; Eaton, P. Protection from hypertension in mice by the Mediterranean diet is mediated by nitro fatty acid inhibition of soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2014, 111, 8167–8172. [Google Scholar] [CrossRef]
- Burns-Whitmore, B.; Froyen, E.; Heskey, C.; Parker, T.; Pablo, G.S. Alpha-Linolenic and Linoleic Fatty Acids in the Vegan Diet: Do They Require Dietary Reference Intake/Adequate Intake Special Consideration? Nutrients 2019, 11, 2365. [Google Scholar] [CrossRef]
- Spector, A.A.; Kim, H.-Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. et Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 356–365. [Google Scholar] [CrossRef]
- Capozzi, M.; Mccollum, G.W.; Penn, J.S. The Role of Cytochrome P450 Epoxygenases in Retinal Angiogenesis. Investig. Opthalmology Vis. Sci. 2014, 55, 4253–4260. [Google Scholar] [CrossRef]
- Morisseau, C. Role of epoxide hydrolases in lipid metabolism. Biochime 2013, 95, 91–95. [Google Scholar] [CrossRef]
- Gautheron, J.; Jéru, I. The Multifaceted Role of Epoxide Hydrolases in Human Health and Disease. Int. J. Mol. Sci. 2020, 22, 13. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.M.; Ray, J.; Wei, D.; Koethe, J.R.; Hannah, L.; DeMatteo, A.; Manning, R.; Terker, A.S.; Peng, D.; Nian, H.; et al. GSK2256294 Decreases sEH (Soluble Epoxide Hydrolase) Activity in Plasma, Muscle, and Adipose and Reduces F2-Isoprostanes but Does Not Alter Insulin Sensitivity in Humans. Hypertension 2021, 78, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Anita, N.Z.; Zebarth, J.; Chan, B.; Wu, C.-Y.; Syed, T.; Shahrul, D.; Nguyen, M.M.; Pakosh, M.; Herrmann, N.; Lanctôt, K.L.; et al. Inflammatory markers in type 2 diabetes with vs. without cognitive impairment; a systematic review and meta-analysis. Brain. Behav. Immun. 2022, 100, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Shakya, A.K.; Perez-Pinzon, M.A.; Dave, K.R. Cerebral ischemic damage in diabetes: An inflammatory perspective. J. Neuroinflamm. 2017, 14, 21. [Google Scholar] [CrossRef]
- Duran-Salgado, M.B. Diabetic nephropathy and inflammation. World J. Diabetes 2014, 5, 393–398. [Google Scholar] [CrossRef]
- Baka, P.; Escolano-Lozano, F.; Birklein, F. Systemic inflammatory biomarkers in painful diabetic neuropathy. J. Diabetes Its Complicat. 2021, 35, 108017. [Google Scholar] [CrossRef]
- Kaur, N.; Guan, Y.; Raja, R.; Ruiz-Velasco, A.; Liu, W. Mechanisms and Therapeutic Prospects of Diabetic Cardiomyopathy Through the Inflammatory Response. Front. Physiol. 2021, 12, 694864. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anita, N.Z.; Swardfager, W. Soluble Epoxide Hydrolase and Diabetes Complications. Int. J. Mol. Sci. 2022, 23, 6232. https://doi.org/10.3390/ijms23116232
Anita NZ, Swardfager W. Soluble Epoxide Hydrolase and Diabetes Complications. International Journal of Molecular Sciences. 2022; 23(11):6232. https://doi.org/10.3390/ijms23116232
Chicago/Turabian StyleAnita, Natasha Z., and Walter Swardfager. 2022. "Soluble Epoxide Hydrolase and Diabetes Complications" International Journal of Molecular Sciences 23, no. 11: 6232. https://doi.org/10.3390/ijms23116232
APA StyleAnita, N. Z., & Swardfager, W. (2022). Soluble Epoxide Hydrolase and Diabetes Complications. International Journal of Molecular Sciences, 23(11), 6232. https://doi.org/10.3390/ijms23116232