
No Time to Die: How Kidney Cancer Evades Cell Death

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Programmed Cell Deaths in Renal Cancers
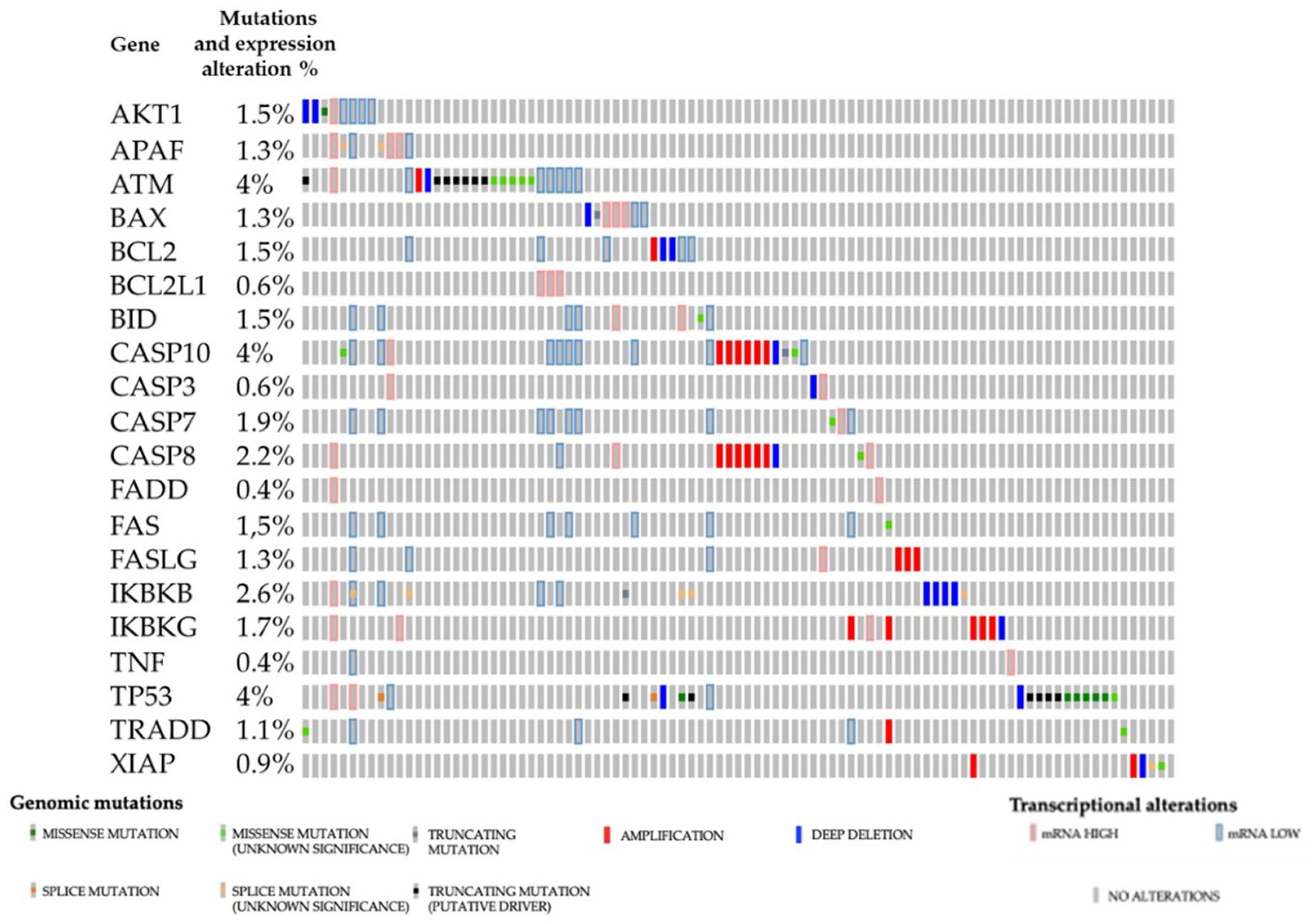
3. Apoptosis Regulation in Renal Cancers
3.1. Main Signaling Pathways
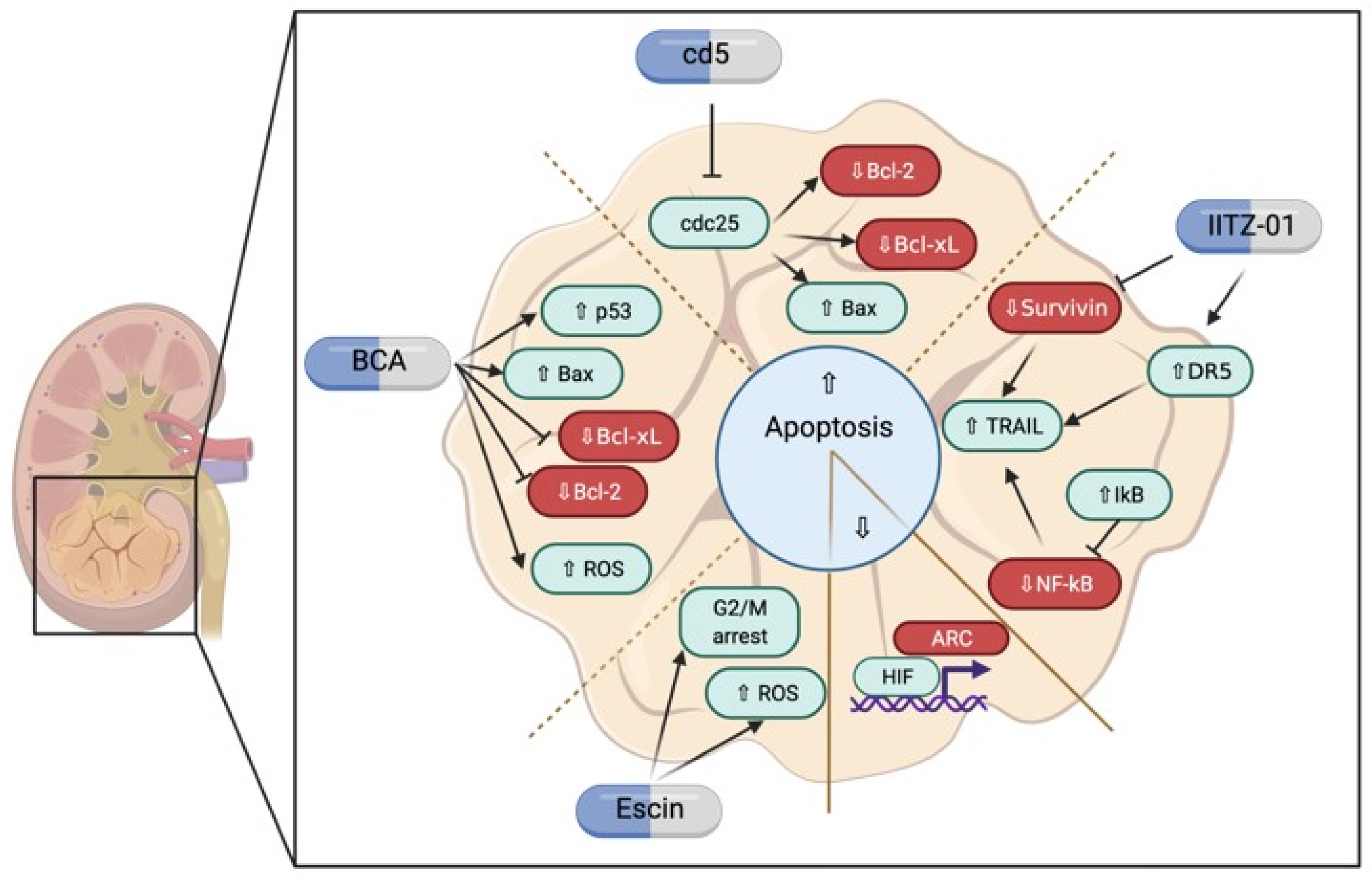
3.2. Possible Therapeutical Approaches
4. Ferroptosis in Renal Cancers
4.1. Main Signaling Pathways

{kind=link}
{kind=link}
| Molecules | Biological Function | Type of Programmed Cell Death | References |
|---|---|---|---|
| glutathione peroxidase 4 (GPX4) | GPX4, an antioxidant defense enzyme, repairs oxidative damage to lipids, and is a leading inhibitor of ferroptosis. | Ferroptosis | [59] |
| RSL3 | Transcription factor that increases the expression of iron metabolism inhibitors, such as ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1). | Ferroptosis | [61] |
| RSL5 | Transcription factor that increases the expression of iron metabolism inhibitors, such as ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1). | Ferroptosis | [61] |
| p53 | p53 has a paradoxical effect on ferroptosis: p53 may induce ferroptosis through both solute-carrier family 7 member 11 (SLC7A11) inhibition, and spermidine/spermine N1-acetyltransferase 1 (SAT1) or glutaminase 2 (GLS2) overexpression; p53 can inhibit ferroptosis by upregulating cyclin-dependent kinase inhibitor 1A (CDKN1A). | Ferroptosis, apoptosis | [62,63] |
| glutathione peroxidase 3 (GPX3) | GPX3, an antioxidant defense enzyme, repairs oxidative damage to lipids. | Ferroptosis | [68] |
| nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) | Multi-subunit enzyme complex that utilizes nicotinamide adenine dinucleotide phosphate to produce superoxide anions and other reactive oxygen species. Excess reactive oxygen species generated by NOX promotes ferroptosis. | Ferroptosis, apoptosis | [71] |
| SLC7A11 | SLC7A11 overexpression is associated with the inhibition of ferroptosis, and the consequent increase in RCCs proliferation, migration, and invasion. | [72] |
4.2. Possible Therapeutical Approaches
| Key Points | References |
| [57] |
| [58,59] |
| [61] |
| [72] |
| [73,74] |
5. Pyroptosis in Kidney Cancer
5.1. Main Signaling Pathways
| Molecules | Biological Function | Type of Programmed Cell Death | Mechanisms | References |
|---|---|---|---|---|
| Apoptosis-associated, speck-like protein | Involved in the caspase-1-dependent inflammatory pyroptosis, and is the major constituent of the pyroptosome. | Pyroptosis | Canonical inflammasome pathway | [88,89] |
| Caspase-1 | Pyroptosis inducer through cleavage of gasdermin-D (GSDMD) into the active mature peptides. | Pyroptosis | Canonical inflammasome pathway | [84,89] |
| Gasdermin D | Precursor of the pore-forming protein, allowing the release of mature interleukin-1 (IL1B and IL18), and triggering pyroptosis. | Pyroptosis | Canonical and non-canonical inflammasome pathway | [88,89] |
| IL-1β | Involved in the transduction of inflammation downstream during pyroptosis processes, and is released through the gasdermin-D pore. | Pyroptosis | Canonical and non-canonical inflammasome pathway | [75,89,90,91,92] |
| IL-18 | Involved in the transduction of inflammation downstream during pyroptosis processes, and is released through the gasdermin-D pore. | Pyroptosis | Canonical and non-canonical inflammasome pathway | [75,89,90,91,92] |
| Caspase 4 | Inflammatory caspase able to promote pyroptosis through NLRP3 and NLRP6 inflammasomes and GSDMD cleavage, in response to non-canonical inflammasome activators. | Pyroptosis | Non-canonical inflammasome pathway | [84,85,86,87,96] |
| Caspase 5 | Responsible for starting pyroptosis through cleavage of GSDMD and the consequent pore formation. | Pyroptosis | Non-canonical inflammasome pathway | [84,85,86,87,96] |
| Gasdermin E | Pore-forming protein able to both convert non-inflammatory apoptosis to pyroptosis, or promote granzyme-mediated pyroptosis. | Pyroptosis | Non-canonical inflammasome pathway | [98,99] |
| Caspase recruitment domain (T cells) | It mediates inflammasome activation, and leads to subsequent pyroptosis of CD4+ T-cells and macrophages | Pyroptosis | Non-canonical inflammasome pathway | [96] |
| NLRP3 | It initiates the formation of the inflammasome complex in response to pathogens and damage-associated signals. | Pyroptosis | Non-canonical inflammasome pathway | [97] |
| Pannexin-1 | It leads to channel opening and extracellular ATP release, which, in turn, activates P2X7 receptors and causes cytotoxicity. | Pyroptosis | Non-canonical inflammasome pathway | [97] |
| LPS | LPS activates phagocytosis-related NADPH oxidase, and leads to the initiation of ROS and NLRP3 inflammasome formation. | Pyroptosis | Non-canonical inflammasome pathway | [96] |
| P2X7 receptor | Through the formation of membrane pores and K+ efflux through the P2X7-dependent pore, the intracellular Ca2+ concentration increases and ATP-dependent lysis of cells occurs. | Pyroptosis | Non-canonical inflammasome pathway | [97] |
| Caspase-3 | Primary protein responsible for GSDME cleavage and activation, playing an essential role in pyroptosis. | Pyroptosis | Inflammasome non-dependent pathway | [75,98,99] |
| Gasdermin-E | Precursor of the pore-forming protein. | Pyroptosis | Inflammasome non-dependent pathway | [98,99] |
| Granzyme-B | Protease delivered into target cells to catalyze cleavage of GSDME and activate caspase-independent pyroptosis. | Pyroptosis | Inflammasome non-dependent pathway | [100,101] |
| Granzyme A | Protease delivered into target cells to catalyze cleavage of GSDMB and activate caspase-independent pyroptosis. | Pyroptosis | Inflammasome non-dependent pathway | [100,101,102] |
5.2. Possible Therapeutical Approaches
| Key Points | References |
| [77] |
| [79,80,81,82] |
| [111] |
| [109,111,113,114,115,116,117,120,121,122] |
6. Necroptosis in Kidney Cancer
6.1. Main Signaling Pathways
| Molecules | Biological Function | Type of Programmed Cell Death | References |
|---|---|---|---|
| IFN-γ | Responsible for triggering necroptosis. | Necroptosis | [136,140,141,142] |
| LPS | Responsible for triggering necroptosis. | Necroptosis | [136,140,141,142,151] |
| RIPK1 | A key regulator of the assembly of complex IIb (RIPK1-RIPK3-MLKL) during necroptosis. | Necroptosis | [127,131,137,138,139,140,141,142,143,145,146,147,148,149,150,151,152,155] |
| RIPK3 | Serine/threonine-protein kinase that activates necroptosis. | Necroptosis | [127,131,137,138,139,140,141,142,146,147,149,152,155] |
| MLKL | Pseudokinase that plays a key role in TNF-induced necroptosis. | Necroptosis | [127,131,137,138,139,140,141,142,146,147,155] |
| TNFR1 | Its activation allows the recruitment of TRADD, RIP1, and TRAF2. | Necroptosis | [140,141,142,145,146,147] |
| TRADD | It is identified as a target protein for TNF-induced necroptosis in the absence of RIPK1. | Necroptosis | [140,141,142,145,146,147] |
| FADD | It recruits the initiator caspase-8, forming the death-inducing signaling complex (DISC). | Necroptosis | [148] |
| TRAF2 | Through TNF-induced NF-κB activation, it is able to protect cells, inhibiting necroptotic cell death. | Necroptosis | [140,141,142] |
| TRAF5 | Through TNF-induced NF-κB activation, it is able to protect cells, inhibiting necroptotic cell death. | Necroptosis | [140,141,142] |
| LUBAC | It regulates necrosome-associated RIPK1 ubiquitination. | Necroptosis | [140,141,142,143] |
| cIAP1 | It ubiquitinates NF-kB, inducing kinase (NIK) to suppress non-canonical NF-kB signaling and RIPK1 to promote cell survival. | Necroptosis | [140,141,142,143,148] |
| cIAP2 | It ubiquitinates NF-kB, inducing kinase (NIK) to suppress non-canonical NF-kB signaling and RIPK1 to promote cell survival. | Necroptosis | [140,141,142,143,148] |
| IKKα | Together with IKKβ, it constitutes IkB kinase complex. | Necroptosis | [143,144,148] |
| IKKβ | Together with IKKα, it constitutes IkB kinase complex. | Necroptosis | [143,144,148] |
| NEMO | Together with IKKα and IKKβ, it constitutes IkB kinase complex. | Necroptosis | [143,144] |
| TAK1 | Serine/threonine kinase, which phosphorylates RIPK1, regulating its interaction with RIPK3 and promoting necroptosis. It constitutes TAK1 complex. | Necroptosis | [127,143,144,148] |
| TAB1 | Together with TAK1 and TAB2, it constitutes TAK1 complex. | Necroptosis | [143,144] |
| TAB2 | Together with TAK1 and TAB1, it constitutes TAK1 complex. | Necroptosis | [143,144] |
| NF-kB | Its activation, through TAK1 and IKK complexes, allows the cell survival. | Necroptosis | [144] |
| CYLD | Deubiquitinase, which induces TNF-alpha-induced necroptosis. | Necroptosis | [145,146,147] |
| E3-ligase PELI1 | Negatively regulates necroptosis by reducing RIPK3 expression. | Necroptosis | [149] |
6.2. Possible Therapeutical Approaches
| Key Points | References |
| [12,127,130,131,132,133] |
| [131,137,138,139,146,147] |
| [174] |
| [128] |
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F.; et al. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef]
- Inamura, K. Renal Cell Tumors: Understanding Their Molecular Pathological Epidemiology and the 2016 WHO Classification. Int. J. Mol. Sci. 2017, 18, 2195. [Google Scholar] [CrossRef]
- Candi, E.; Terrinoni, A.; Rufini, A.; Chikh, A.; Lena, A.M.; Suzuki, Y.; Sayan, B.S.; Knight, R.A.; Melino, G. p63 is upstream of IKKα in epidermal development. J. Cell Sci. 2006, 119, 4617–4622. [Google Scholar] [CrossRef]
- Tomasini, R.; Tsuchihara, K.; Tsuda, C.; Lau, S.K.; Wilhelm, M.; Rufini, A.; Tsao, M.-S.; Iovanna, J.L.; Jurisicova, A.; Melino, G.; et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc. Natl. Acad. Sci. USA 2009, 106, 797–802. [Google Scholar] [CrossRef]
- Di Rita, A.; Peschiaroli, A.; D’acunzo, P.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef]
- Lai, Y.; Zeng, T.; Liang, X.; Wu, W.; Zhong, F.; Wu, W. Cell death-related molecules and biomarkers for renal cell carcinoma targeted therapy. Cancer Cell Int. 2019, 19, 221. [Google Scholar] [CrossRef]
- Candi, E.; Agostini, M.; Melino, G.; Bernassola, F. How theTP53Family ProteinsTP63andTP73Contribute to Tumorigenesis: Regulators and Effectors. Hum. Mutat. 2014, 35, 702–714. [Google Scholar] [CrossRef]
- Inoue, S.; Hao, Z.; Elia, A.J.; Cescon, D.; Zhou, L.; Silvester, J.; Snow, B.; Harris, I.S.; Sasaki, M.; Li, W.Y.; et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013, 27, 1101–1114. [Google Scholar] [CrossRef]
- Xia, X.; Fan, X.; Zhao, M.; Zhu, P. The Relationship between Ferroptosis and Tumors: A Novel Landscape for Therapeutic Approach. Curr. Gene Ther. 2019, 19, 117–124. [Google Scholar] [CrossRef]
- Wang, S.; He, M.-F.; Chen, Y.-H.; Wang, M.-Y.; Yu, X.-M.; Bai, J.; Zhu, H.-Y.; Wang, Y.-Y.; Zhao, H.; Mei, Q.; et al. Rapid reuptake of granzyme B leads to emperitosis: An apoptotic cell-in-cell death of immune killer cells inside tumor cells. Cell Death Dis. 2013, 4, e856. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Amaral, M.P.; Branco, L.M.; Strasser, A.; Dixit, V.M.; Bortoluci, K.R. Paradise revealed III: Why so many ways to die? Apoptosis, necroptosis, pyroptosis, and beyond. Cell Death Differ. 2020, 27, 1740–1742. [Google Scholar] [CrossRef]
- Humpton, T.J.; Hall, H.; Kiourtis, C.; Nixon, C.; Clark, W.; Hedley, A.; Shaw, R.; Bird, T.G.; Blyth, K.; Vousden, K.H. p53-mediated redox control promotes liver regeneration and maintains liver function in response to CCl4. Cell Death Differ. 2021, 29, 514–526. [Google Scholar] [CrossRef]
- Yin, K.; Lee, J.; Liu, Z.; Kim, H.; Martin, D.R.; Wu, D.; Liu, M.; Xue, X. Mitophagy protein PINK1 suppresses colon tumor growth by metabolic reprogramming via p53 activation and reducing acetyl-CoA production. Cell Death Differ. 2021, 28, 2421–2435. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef]
- Ganini, C.; Amelio, I.; Bertolo, R.; Bove, P.; Buonomo, O.C.; Candi, E.; Cipriani, C.; Di Daniele, N.; Juhl, H.; Mauriello, A.; et al. Global mapping of cancers: The Cancer Genome Atlas and beyond. Mol. Oncol. 2021, 15, 2823–2840. [Google Scholar] [CrossRef]
- Amelio, I.; Bertolo, R.; Bove, P.; Candi, E.; Chiocchi, M.; Cipriani, C.; Di Daniele, N.; Ganini, C.; Juhl, H.; Mauriello, A.; et al. Cancer predictive studies. Biol. Direct 2020, 15, 18. [Google Scholar] [CrossRef]
- Rizzotto, D.; Villunger, A. P53 clears aneuploid cells by entosis. Cell Death Differ. 2020, 28, 818–820. [Google Scholar] [CrossRef]
- Shalom-Feuerstein, R.; Lena, A.M.; Zhou, H.; Divonne, S.D.L.F.; Van Bokhoven, H.; Candi, E.; Melino, G.; Aberdam, D. ΔNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ. 2010, 18, 887–896. [Google Scholar] [CrossRef]
- Lena, A.M.; Duca, S.; Novelli, F.; Melino, S.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. Amino-terminal residues of ΔNp63, mutated in ectodermal dysplasia, are required for its transcriptional activity. Biochem. Biophys. Res. Commun. 2015, 467, 434–440. [Google Scholar] [CrossRef]
- Rozenberg, J.M.; Zvereva, S.; Dalina, A.; Blatov, I.; Zubarev, I.; Luppov, D.; Bessmertnyi, A.; Romanishin, A.; Alsoulaiman, L.; Kumeiko, V.; et al. The p53 family member p73 in the regulation of cell stress response. Biol. Direct 2021, 16, 23. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, C.; Huang, Y.; Liu, Z.; Yu, X.; Lv, C.; Su, Z. Decreased expression of apoptosis-inducing factor in renal cell carcinoma is associated with poor prognosis and reduced postoperative survival. Oncol. Lett. 2019, 18, 2805–2812. [Google Scholar] [CrossRef]
- Park, S.-H.; Park, J.-Y.; Weiss, R.H. Antisense Attenuation of p21 Sensitizes Kidney Cancer to Apoptosis in Response to Conventional DNA Damaging Chemotherapy Associated With Enhancement of Phospho-p53. J. Urol. 2008, 180, 352–360. [Google Scholar] [CrossRef][Green Version]
- Mizuno, R.; Oya, M.; Hara, S.; Matsumoto, M.; Horiguchi, A.; Ohigashi, T.; Marumo, K.; Murai, M. Modulation of bcl-2 family proteins in MAPK independent apoptosis induced by a cdc25 phosphatase inhibitor Cpd 5 in renal cancer cells. Oncol. Rep. 2005, 14, 639–644. [Google Scholar] [CrossRef]
- Stöhr, D.; Schmid, J.O.; Beigl, T.B.; Mack, A.; Maichl, D.S.; Cao, K.; Budai, B.; Fullstone, G.; Kontermann, R.E.; Mürdter, T.E.; et al. Stress-induced TRAILR2 expression overcomes TRAIL resistance in cancer cell spheroids. Cell Death Differ. 2020, 27, 3037–3052. [Google Scholar] [CrossRef]
- Vetma, V.; Guttà, C.; Peters, N.; Praetorius, C.; Hutt, M.; Seifert, O.; Meier, F.; Kontermann, R.; Kulms, D.; Rehm, M. Convergence of pathway analysis and pattern recognition predicts sensitization to latest generation TRAIL therapeutics by IAP antagonism. Cell Death Differ. 2020, 27, 2417–2432. [Google Scholar] [CrossRef]
- Seki, N.; Hayakawa, Y.; Brooks, A.D.; Wine, J.; Wiltrout, R.H.; Yagita, H.; Tanner, J.E.; Smyth, M.J.; Sayers, T.J. Tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis is an important endogenous mechanism for resistance to liver metastases in murine renal cancer. Cancer Res. 2003, 63, 207–213. [Google Scholar]
- Shahriyar, S.A.; Seo, S.U.; Min, K.-J.; Kubatka, P.; Min, D.S.; Chang, J.-S.; Kim, D.E.; Woo, S.M.; Kwon, T.K. Upregulation of DR5 and Downregulation of Survivin by IITZ-01, Lysosomotropic Autophagy Inhibitor, Potentiates TRAIL-Mediated Apoptosis in Renal Cancer Cells via Ubiquitin-Proteasome Pathway. Cancers 2020, 12, 2363. [Google Scholar] [CrossRef]
- Oya, M.; Ohtsubo, M.; Takayanagi, A.; Tachibana, M.; Shimizu, N.; Murai, M. Constitutive activation of nuclear factor-κB prevents TRAIL-induced apoptosis in renal cancer cells. Oncogene 2001, 20, 3888–3896. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Y.; Xiao, J.; Shang, J.; Tan, Q.; Ping, F.; Huang, W.; Wu, F.; Zhang, H.; Zhang, X. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020, 27, 2635–2650. [Google Scholar] [CrossRef]
- Hwang, J.-H.; Joo, J.C.; Kim, D.J.; Jo, E.; Yoo, H.-S.; Lee, K.-B.; Park, S.J.; Jang, I.-S. Cordycepin promotes apoptosis by modulating the ERK-JNK signaling pathway via DUSP5 in renal cancer cells. Am. J. Cancer Res. 2016, 6, 1758–1771. [Google Scholar]
- Amelio, I.; Melino, G. The p53 family and the hypoxia-inducible factors (HIFs): Determinants of cancer progression. Trends Biochem. Sci. 2015, 40, 425–434. [Google Scholar] [CrossRef]
- Piredda, L.; Farrace, M.G.; Bello, M.L.; Malorni, W.; Melino, G.; Petruzzelli, R.; Piacentini, M. Identification of ‘tissue’ transglutaminase binding proteins in neural cells committed to apoptosis. FASEB J. 1999, 13, 355–364. [Google Scholar] [CrossRef]
- Razorenova, O.V.; Castellini, L.; Colavitti, R.; Edgington, L.E.; Nicolau, M.; Huang, X.; Bedogni, B.; Mills, E.M.; Bogyo, M.; Giaccia, A.J. The Apoptosis Repressor with a CARD Domain (ARC) Gene Is a Direct Hypoxia-Inducible Factor 1 Target Gene and Promotes Survival and Proliferation of VHL-Deficient Renal Cancer Cells. Mol. Cell. Biol. 2014, 34, 739–751. [Google Scholar] [CrossRef]
- Khan, M.N.; Bhattacharyya, T.; Andrikopoulos, P.; Esteban, M.A.; Barod, R.; Connor, T.; Ashcroft, M.; Maxwell, P.H.; Kiriakidis, S. Factor inhibiting HIF (FIH-1) promotes renal cancer cell survival by protecting cells from HIF-1α-mediated apoptosis. Br. J. Cancer 2011, 104, 1151–1159. [Google Scholar] [CrossRef]
- Kim, S.; Lee, T.-J.; Park, J.-W.; Kwon, T.K. Overexpression of cFLIPs inhibits oxaliplatin-mediated apoptosis through enhanced XIAP stability and Akt activation in human renal cancer cells. J. Cell. Biochem. 2008, 105, 971–979. [Google Scholar] [CrossRef]
- Lamastra, F.R.; De Angelis, R.; Antonucci, A.; Salvatori, D.; Prosposito, P.; Casalboni, M.; Congestri, R.; Melino, S.; Nanni, F. Polymer composite random lasers based on diatom frustules as scatterers. RSC Adv. 2014, 4, 61809–61816. [Google Scholar] [CrossRef]
- Yuan, S.-Y.; Cheng, C.-L.; Wang, S.-S.; Ho, H.-C.; Chiu, K.-Y.; Chen, C.-S.; Chen, C.-C.; Shiau, M.-Y.; Ou, Y.-C. Escin induces apoptosis in human renal cancer cells through G2/M arrest and reactive oxygen species-modulated mitochondrial pathways. Oncol. Rep. 2017, 37, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Nepravishta, R.; Sabelli, R.; Iorio, E.; Micheli, L.; Paci, M.; Melino, S. Oxidative species and S-glutathionyl conjugates in the apoptosis induction by allyl thiosulfate. FEBS J. 2011, 279, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, T.; Sacchetta, P.; Pennelli, A.; Cornelio, L.; Romagnoli, R.; Melino, S.; Petruzzelli, R.; Di Ilio, C. Characterization of toad liver glutathione transferase. Biochim. Biophys. Acta 1999, 1431, 189–198. [Google Scholar] [CrossRef]
- Mauretti, A.; Neri, A.; Kossover, O.; Seliktar, D.; Di Nardo, P.; Melino, S. Design of a Novel Composite H2S-Releasing Hydrogel for Cardiac Tissue Repair. Macromol. Biosci. 2016, 16, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Cha, H.S.; Nam, M.J.; Park, K.; Yang, Y.-H.; Lee, J.; Park, S.-H. Broussochalcone A Induces Apoptosis in Human Renal Cancer Cells via ROS Level Elevation and Activation of FOXO3 Signaling Pathway. Oxidative Med. Cell. Longev. 2021, 2021, 2800706. [Google Scholar] [CrossRef]
- Asakuma, J.; Sumitomo, M.; Asano, T.; Asano, T.; Hayakawa, M. Selective Akt inactivation and tumor necrosis actor-related apoptosis-inducing ligand sensitization of renal cancer cells by low concentrations of paclitaxel. Cancer Res. 2003, 63, 1365–1370. [Google Scholar]
- Hillert, L.K.; Ivanisenko, N.V.; Busse, D.; Espe, J.; König, C.; Peltek, S.E.; Kolchanov, N.A.; Ivanisenko, V.; Lavrik, I.N. Dissecting DISC regulation via pharmacological targeting of caspase-8/c-FLIPL heterodimer. Cell Death Differ. 2020, 27, 2117–2130. [Google Scholar] [CrossRef]
- Wu, M.-H.; Chiou, H.-L.; Lin, C.-L.; Lin, C.-Y.; Yang, S.-F.; Hsieh, Y.-H. Induction of endoplasmic reticulum stress and mitochondrial dysfunction dependent apoptosis signaling pathway in human renal cancer cells by norcantharidin. Oncotarget 2017, 9, 4787–4797. [Google Scholar] [CrossRef]
- Selka, A.; Doiron, J.A.; Lyons, P.; Dastous, S.; Chiasson, A.; Cormier, M.; Turcotte, S.; Surette, M.E.; Touaibia, M. Discovery of a novel 2,5-dihydroxycinnamic acid-based 5-lipoxygenase inhibitor that induces apoptosis and may impair autophagic flux in RCC4 renal cancer cells. Eur. J. Med. Chem. 2019, 179, 347–357. [Google Scholar] [CrossRef]
- Miyakuni, K.; Nishida, J.; Koinuma, D.; Nagae, G.; Aburatani, H.; Miyazono, K.; Ehata, S. Genome-wide analysis of DNA methylation identifies the apoptosis-related gene UQCRH as a tumor suppressor in renal cancer. Mol. Oncol. 2022, 16, 732–749. [Google Scholar] [CrossRef]
- Dong, L.M.; Brennan, P.; Karami, S.; Hung, R.J.; Menashe, I.; Berndt, S.I.; Yeager, M.; Chanock, S.; Zaridze, D.; Matveev, V.; et al. An Analysis of Growth, Differentiation and Apoptosis Genes with Risk of Renal Cancer. PLoS ONE 2009, 4, e4895. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Hodi, F.S.; Thompson, J.A.; McDermott, D.F.; Hwu, W.-J.; Lawrence, D.P.; Dawson, N.A.; Wong, D.J.L.; Bhatia, S.; James, M.; et al. Pembrolizumab Plus Pegylated Interferon alfa-2b or Ipilimumab for Advanced Melanoma or Renal Cell Carcinoma: Dose-Finding Results from the Phase Ib KEYNOTE-029 Study. Clin. Cancer Res. 2018, 24, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.H.; Kuntz, S.M.; Leibovich, B.C.; Dong, H.; Lohse, C.M.; Webster, W.S.; Sengupta, S.; Frank, I.; Parker, A.S.; Zincke, H.; et al. Tumor B7-H1 Is Associated with Poor Prognosis in Renal Cell Carcinoma Patients with Long-term Follow-up. Cancer Res. 2006, 66, 3381–3385. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021, 28, 2843–2856. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Shou, Y.; Yang, L.; Yang, Y.; Xu, J. Inhibition of keratinocyte ferroptosis suppresses psoriatic inflammation. Cell Death Dis. 2021, 12, 1009. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Pentimalli, F.; Grelli, S.; Di Daniele, N.; Melino, G.; Amelio, I. Cell death pathologies: Targeting death pathways and the immune system for cancer therapy. Genes Immun. 2018, 20, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Panatta, E.; Zampieri, C.; Melino, G.; Amelio, I. Understanding p53 tumour suppressor network. Biol. Direct 2021, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.-C.; Wu, M.-S.; Chou, S.-W.; Jargalsaikhan, G.; Chen, Y.-C. Roles of reactive oxygen species, mitochondrial membrane potential, and p53 in evodiamine-induced apoptosis and G2/M arrest of human anaplastic thyroid carcinoma cells. Chin. Med. 2021, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2018, 133, 162–168. [Google Scholar] [CrossRef]
- Requena, D.O.; Garcia-Buitrago, M. Molecular Insights Into Colorectal Carcinoma. Arch. Med Res. 2020, 51, 839–844. [Google Scholar] [CrossRef]
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 12. [Google Scholar] [CrossRef]
- Tang, S.; Xiao, X. Ferroptosis and kidney diseases. Int. Urol. Nephrol. 2019, 52, 497–503. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Yang, W.-H.; Ding, C.-K.C.; Sun, T.; Rupprecht, G.; Lin, C.-C.; Hsu, D.; Chi, J.-T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501–2508.e4. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Guan, Y.; Xue, L.; Zhang, P.; Li, M.; Gao, M.; Chong, T. The roles of ferroptosis regulatory gene SLC7A11 in renal cell carcinoma: A multi-omics study. Cancer Med. 2021, 10, 9078–9096. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, E.; Guo, T.; Shao, J.; Wang, T.; Zhang, N.; Wang, X.; Zheng, J. A novel ferroptosis-related gene signature associated with cell cycle for prognosis prediction in patients with clear cell renal cell carcinoma. BMC Cancer 2022, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, S.; Ying, Y.; Ma, X.; Shen, H.; Li, J.; Wang, X.; Lin, Y.; Liu, B.; Zheng, X.; et al. Comprehensive Analysis of Ferroptosis Regulators With Regard to PD-L1 and Immune Infiltration in Clear Cell Renal Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 676142. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef]
- Frantz, S. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J. Mol. Cell. Cardiol. 2003, 35, 685–694. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, S.; Zhang, Y.; Li, P.; Wang, K. The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers 2019, 11, 1313. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, W.; Gong, F.; Chen, Y.; Chen, E. The Role and Mechanism of Pyroptosis and Potential Therapeutic Targets in Sepsis: A Review. Front. Immunol. 2021, 12, 711939. [Google Scholar] [CrossRef]
- Aglietti, R.A.; Dueber, E.C. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol. 2017, 38, 261–271. [Google Scholar] [CrossRef]
- Aziz, M.T.A.; Jacob, A.P.; Wang, P. Revisiting caspases in sepsis. Cell Death Dis. 2014, 5, e1526. [Google Scholar] [CrossRef]
- Aziz, M.; Jacob, A.; Yang, W.; Matsuda, A.; Wang, P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J. Leukoc. Biol. 2012, 93, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, Y.; Huang, Z.-X.; Chen, H.; Lan, R.; Wang, Z.; Lai, K.; Chen, H.; Chen, Z.; Zou, Z.; et al. GSDME-mediated pyroptosis promotes inflammation and fibrosis in obstructive nephropathy. Cell Death Differ. 2021, 28, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Yang, Z.; Wang, Z.; Zhang, X.; Zhao, Y.; Yang, H.; Zheng, B.; Tian, W.; Wang, S.; He, Z.; et al. NLRP3/ASC-mediated alveolar macrophage pyroptosis enhances HMGB1 secretion in acute lung injury induced by cardiopulmonary bypass. Lab. Investig. 2018, 98, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Sarkar, A.; Walle, L.V.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.-D.; Dixit, V.M. Inflammasome-Dependent Release of the Alarmin HMGB1 in Endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Muñoz-Planillo, R.; Liu, Q.; Núñez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, E.; Croes, L.; Ibrahim, J.; Pauwels, P.; de Beeck, K.O.; Vandenabeele, P.; Van Camp, G. GSDME and its role in cancer: From behind the scenes to the front of the stage. Int. J. Cancer 2020, 148, 2872–2883. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic Inflammation in Cancer Development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Lieberman, J. Lighting a Fire: Can We Harness Pyroptosis to Ignite Antitumor Immunity? Cancer Immunol. Res. 2021, 9, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef]
- Duan, X.; Song, Y.; Li, F.; Liao, Y.; Liu, W. Metadherin silencing results in the inhibition of pyroptosis in lipopolysaccharide/adenosine triphosphate-stimulated renal tubular epithelial cells. Tissue Cell 2021, 75, 101722. [Google Scholar] [CrossRef]
- Sun, Z.; Jing, C.; Guo, X.; Zhang, M.; Kong, F.; Wang, Z.; Jiang, S.; Wang, H. Comprehensive Analysis of the Immune Infiltrates of Pyroptosis in Kidney Renal Clear Cell Carcinoma. Front. Oncol. 2021, 11, 716854. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, A.; Feng, Y.; Su, Y.; Wang, X.; Jiang, F.; Ma, J. A Novel Pyroptosis-Related lncRNAs Signature for Predicting the Prognosis of Kidney Renal Clear Cell Carcinoma and Its Associations with Immunity. J. Oncol. 2021, 2021, 9997185. [Google Scholar] [CrossRef]
- Zhang, K.-J.; Wu, Q.; Jiang, S.-M.; Ding, L.; Liu, C.-X.; Xu, M.; Wang, Y.; Zhou, Y.; Li, L. Pyroptosis: A New Frontier in Kidney Diseases. Oxidative Med. Cell. Longev. 2021, 2021, 6686617. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Fu, Q.; Wang, F.; Zhou, X.; Xiang, J.; He, N.; Hu, Z.; Jin, X. Comprehensive analysis of pyroptosis regulators and tumor immune microenvironment in clear cell renal cell carcinoma. Cancer Cell Int. 2021, 21, 667. [Google Scholar] [CrossRef] [PubMed]
- Toth, C.; Funke, S.; Nitsche, V.; Liverts, A.; Zlachevska, V.; Gasis, M.; Wiek, C.; Hanenberg, H.; Mahotka, C.; Schirmacher, P.; et al. The role of apoptosis repressor with a CARD domain (ARC) in the therapeutic resistance of renal cell carcinoma (RCC): The crucial role of ARC in the inhibition of extrinsic and intrinsic apoptotic signalling. Cell Commun. Signal. 2017, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Karki, R.; Kanneganti, T. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Patsos, G.; Germann, A.; Gebert, J.; Dihlmann, S. Restoration of absent in melanoma 2 (AIM2) induces G2/M cell cycle arrest and promotes invasion of colorectal cancer cells. Int. J. Cancer 2009, 126, 1838–1849. [Google Scholar] [CrossRef]
- Qi, M.; Dai, D.; Liu, J.; Li, Z.; Liang, P.; Wang, Y.; Cheng, L.; Zhan, Y.; An, Z.; Song, Y.; et al. AIM2 promotes the development of non-small cell lung cancer by modulating mitochondrial dynamics. Oncogene 2020, 39, 2707–2723. [Google Scholar] [CrossRef]
- Wang, L.; Sun, L.; Byrd, K.M.; Ko, C.-C.; Zhao, Z.; Fang, J. AIM2 Inflammasome’s First Decade of Discovery: Focus on Oral Diseases. Front. Immunol. 2020, 11, 1487. [Google Scholar] [CrossRef]
- Zhou, R.; Sun, J.; He, C.; Huang, C.; Yu, H. CCL19 suppresses gastric cancer cell proliferation, migration, and invasion through the CCL19/CCR7/AIM2 pathway. Hum. Cell 2020, 33, 1120–1132. [Google Scholar] [CrossRef]
- Kurschus, F.C.; Jenne, D.E. Delivery and therapeutic potential of human granzyme B. Immunol. Rev. 2010, 235, 159–171. [Google Scholar] [CrossRef]
- Rathinam, V.A.K.; Zhao, Y.; Shao, F. Innate immunity to intracellular LPS. Nat. Immunol. 2019, 20, 527–533. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, Y.T.; Wang, L.Q.; Li, L.Y.; Bao, Q.; Tian, S.; Chen, M.X.; Chen, H.X.; Cui, J.; Li, C.W. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J. Allergy Clin. Immunol. 2019, 144, 777–787.e9. [Google Scholar] [CrossRef]
- Shi, C.-X.; Wang, Y.; Chen, Q.; Jiao, F.-Z.; Pei, M.-H.; Gong, Z.-J. Extracellular Histone H3 Induces Pyroptosis During Sepsis and May Act through NOD2 and VSIG4/NLRP3 Pathways. Front. Cell. Infect. Microbiol. 2020, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K. Switching from Apoptosis to Pyroptosis: Gasdermin-Elicited Inflammation and Antitumor Immunity. Int. J. Mol. Sci. 2021, 22, 426. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Han, Q.; Cao, Q.; Wang, K.; Zhou, X.; Hou, P.; Liu, C.; Chen, L.; Xu, L. Lefty A is involved in sunitinib resistance of renal cell carcinoma cells via regulation of IL-8. Biol. Chem. 2021, 402, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Numakura, K.; Muto, Y.; Naito, S.; Hatakeyama, S.; Kato, R.; Koguchi, T.; Kojima, T.; Kawasaki, Y.; Kandori, S.; Kawamura, S.; et al. Outcomes of axitinib versus sunitinib as first-line therapy to patients with metastatic renal cell carcinoma in the immune-oncology era. Cancer Med. 2021, 10, 5839–5846. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Tonnus, W.; Meyer, C.; Paliege, A.; Belavgeni, A.; Von Mässenhausen, A.; Bornstein, S.R.; Hugo, C.; Becker, J.U.; Linkermann, A. The pathological features of regulated necrosis. J. Pathol. 2019, 247, 697–707. [Google Scholar] [CrossRef] [PubMed]
- de Almagro, M.C.; Vucic, D. Necroptosis: Pathway diversity and characteristics. Semin. Cell Dev. Biol. 2015, 39, 56–62. [Google Scholar] [CrossRef]
- Höckendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M.; et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell 2016, 30, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Koo, G.-B.; Morgan, M.J.; Lee, D.-G.; Kim, W.-J.; Yoon, J.-H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.-M.; Luz, N.F.; Moriwaki, K. Programmed Necrosis in the Cross Talk of Cell Death and Inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- McQuade, T.; Cho, Y.; Chan, F.K.-M. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 2013, 456, 409–415. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Bellomaria, A.; Barbato, G.; Melino, G.; Paci, M.; Melino, S. Recognition of p63 by the E3 ligase ITCH: Effect of an ectodermal dysplasia mutant. Cell Cycle 2010, 9, 3754–3763. [Google Scholar] [CrossRef]
- Hsu, H.; Huang, J.; Shu, H.-B.; Baichwal, V.; Goeddel, D.V. TNF-Dependent Recruitment of the Protein Kinase RIP to the TNF Receptor-1 Signaling Complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Ea, C.-K.; Deng, L.; Xia, Z.-P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFα Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP Is Required for Tumor Necrosis Factor α-induced NF-κB Activation. J. Biol. Chem. 2006, 281, 13636–13643. [Google Scholar] [CrossRef] [PubMed]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944–E5953. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-α Induces Two Distinct Caspase-8 Activation Pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- Shutinoski, B.; Alturki, N.; Rijal, D.; Bertin, J.; Gough, P.J.; Schlossmacher, M.G.; Sad, S. K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ. 2016, 23, 1628–1637. [Google Scholar] [CrossRef]
- Kearney, C.J.; Cullen, S.P.; Clancy, D.; Martin, S.J. RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J. 2014, 281, 4921–4934. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.-M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Chico, J.F.; Falk-Paulsen, M.; Luzius, A.; Saggau, C.; Ruder, B.; Bolik, J.; Schmidt-Arras, D.; Linkermann, A.; Becker, C.; Rosenstiel, P.; et al. The enhanced susceptibility of ADAM-17 hypomorphic mice to DSS-induced colitis is not ameliorated by loss of RIPK3, revealing an unexpected function of ADAM-17 in necroptosis. Oncotarget 2018, 9, 12941–12958. [Google Scholar] [CrossRef] [PubMed]
- Onizawa, M.; Oshima, S.; Schulze-Topphoff, U.; Oses-Prieto, J.A.; Lu, T.; Tavares, R.; Prodhomme, T.; Duong, B.; Whang, M.I.; Advincula, R.; et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 2015, 16, 618–627. [Google Scholar] [CrossRef]
- Huang, D.; Zheng, X.; Wang, Z.-A.; Chen, X.; He, W.-T.; Zhang, Y.; Xu, J.-G.; Zhao, H.; Shi, W.; Wang, X.; et al. The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol. Cell. Biol. 2017, 37, e00497-16. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Liu, L.; Huang, S.; Ren, Y.; Wang, H.; Yao, Z.; Li, L.; Chen, S.; Wang, X.; Zhang, Z. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. 2017, 53, 3637–3640. [Google Scholar] [CrossRef]
- Gallo, M.; Paludi, D.; Cicero, D.; Chiovitti, K.; Millo, E.; Salis, A.; Damonte, G.; Corsaro, A.; Thellung, S.; Schettini, G.; et al. Identification of a Conserved N-Capping Box Important for the Structural Autonomy of the Prion α3-Helix: The Disease Associated D202N Mutation Destabilizes the Helical Conformation. Int. J. Immunopathol. Pharmacol. 2005, 18, 95–112. [Google Scholar] [CrossRef]
- Vitali, A.; Botta, B.; Monache, G.D.; Zappitelli, S.; Ricciardi, P.; Melino, S.; Petruzzelli, R.; Giardina, B. Purification and partial characterization of a peroxidase from plant cell cultures of Cassia didymobotrya and biotransformation studies. Biochem. J. 1998, 331, 513–519. [Google Scholar] [CrossRef]
- Dai, W.; Cheng, J.; Leng, X.; Hu, X.; Ao, Y. The potential role of necroptosis in clinical diseases (Review). Int. J. Mol. Med. 2021, 47, 89. [Google Scholar] [CrossRef]
- Liu, W.; Chen, B.; Wang, Y.; Meng, C.; Huang, H.; Huang, X.-R.; Qin, J.; Mulay, S.R.; Anders, H.-J.; Qiu, A.; et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, E1475–E1484. [Google Scholar] [CrossRef]
- Zhu, Y.; Cui, H.; Gan, H.; Xia, Y.; Wang, L.; Wang, Y.; Sun, Y. Necroptosis mediated by receptor interaction protein kinase 1 and 3 aggravates chronic kidney injury of subtotal nephrectomised rats. Biochem. Biophys. Res. Commun. 2015, 461, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Mei, M.; Pu, Y.; Zhang, H.; Liu, H.; Tang, M.; Pan, Q.; He, Y.; Wu, X.; Zhao, H. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1α/mir-26a/TRPC6/PARP1 Signaling. Mol. Ther. Nucleic Acids 2019, 17, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-N.; Liu, M.-M.; Wang, F.; Wei, B.; Yang, Q.; Cai, Y.-T.; Chen, X.; Liu, X.-Q.; Jiang, L.; Li, C.; et al. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin. Sci. 2019, 133, 1609–1627. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fang, Y.; Wu, J.; Chen, H.; Zou, Z.; Zhang, X.; Shao, J.; Xu, Y. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. 2018, 9, 878. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Desai, J.; Kumar, S.V.; Eberhard, J.N.; Thomasova, D.; Romoli, S.; Grigorescu, M.; Kulkarni, O.P.; Popper, B.; Vielhauer, V.; et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 2016, 7, 10274. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Patino, E.; Ma, K.C.; Laursen, K.; Finkelsztein, E.; Akchurin, O.; Muthukumar, T.; Ryter, S.W.; Gudas, L.; Choi, A.M.K.; et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight 2018, 3, e98411. [Google Scholar] [CrossRef]
- Bozec, D.; Iuga, A.C.; Roda, G.; Dahan, S.; Yeretssian, G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 2016, 7, 46384–46400. [Google Scholar] [CrossRef]
- Schmidt, S.V.; Seibert, S.; Walch-Rückheim, B.; Vicinus, B.; Kamionka, E.-M.; Pahne-Zeppenfeld, J.; Solomayer, E.-F.; Kim, Y.-J.; Bohle, R.M.; Smola, S. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 2015, 6, 8635–8647. [Google Scholar] [CrossRef]
- Al-Lamki, R.S.; Lu, W.; Manalo, P.; Wang, J.; Warren, A.Y.; Tolkovsky, A.M.; Pober, J.S.; Bradley, J. Tubular epithelial cells in renal clear cell carcinoma express high RIPK1/3 and show increased susceptibility to TNF receptor 1-induced necroptosis. Cell Death Dis. 2016, 7, e2287. [Google Scholar] [CrossRef]
- Thapa, R.J.; Chen, P.; Cheung, M.; Nogusa, S.; Pei, J.; Peri, S.; Testa, J.R.; Balachandran, S. NF-κB Inhibition by Bortezomib Permits IFN-γ–Activated RIP1 Kinase–Dependent Necrosis in Renal Cell Carcinoma. Mol. Cancer Ther. 2013, 12, 1568–1578. [Google Scholar] [CrossRef]
- Wang, K.-J.; Meng, X.-Y.; Chen, J.-F.; Wang, K.-Y.; Zhou, C.; Yu, R.; Ma, Q. Emodin Induced Necroptosis and Inhibited Glycolysis in the Renal Cancer Cells by Enhancing ROS. Oxidative Med. Cell. Longev. 2021, 2021, 8840590. [Google Scholar] [CrossRef]
- Mao, Q.; Zhuang, Q.; Shen, J.; Chen, Z.; Xue, D.; Ding, T.; He, X. MiRNA-124 regulates the sensitivity of renal cancer cells to cisplatin-induced necroptosis by targeting the CAPN4-CNOT3 axis. Transl. Androl. Urol. 2021, 10, 3669–3683. [Google Scholar] [CrossRef]
- Zhao, C.; Zhou, Y.; Ran, Q.; Yao, Y.; Zhang, H.; Ju, J.; Yang, T.; Zhang, W.; Yu, X.; He, S. MicroRNA-381-3p Functions as a Dual Suppressor of Apoptosis and Necroptosis and Promotes Proliferation of Renal Cancer Cells. Front. Cell Dev. Biol. 2020, 8, 290. [Google Scholar] [CrossRef]
- Degterev, A.; Linkermann, A. Generation of small molecules to interfere with regulated necrosis. Cell. Mol. Life Sci. 2016, 73, 2251–2267. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Hu, L.; Shi, S.-N.; Chen, X.; Zhuang, C.; Zhang, W.; Jitkaew, S.; Pang, X.; Yu, J.; Tan, Y.-X.; et al. The Bcr-Abl inhibitor GNF-7 inhibits necroptosis and ameliorates acute kidney injury by targeting RIPK1 and RIPK3 kinases. Biochem. Pharmacol. 2020, 177, 113947. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-H.; Cruz, S.A.; Qin, Z.; Stewart, A. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen. Res. 2018, 13, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wang, Q.; Phan, N.; Ren, J.; Yang, H.; Feldman, C.C.; Feltenberger, J.B.; Ye, Z.; Wildman, S.; Tang, W.; et al. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis. 2019, 10, 226. [Google Scholar] [CrossRef]
- Romera-Giner, S.; Martínez, Z.A.; García-García, F.; Hidalgo, M.R. Common pathways and functional profiles reveal underlying patterns in Breast, Kidney and Lung cancers. Biol. Direct 2021, 16, 9. [Google Scholar] [CrossRef]
- Laurenzi, V.; Melino, G. Evolution of Functions within the p53/p63/p73 Family. Ann. N. Y. Acad. Sci. 2006, 926, 90–100. [Google Scholar] [CrossRef]
- Melino, S.; Nepravishta, R.; Bellomaria, A.; Di Marco, S.; Paci, M. Nucleic Acid Binding of the RTN1-C C-Terminal Region: Toward the Functional Role of a Reticulon Protein. Biochemistry 2008, 48, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Salomoni, P.; Oberst, A.; DiComo, C.; Pagano, M.; Melino, G.; Pandolfi, P.P. Ubiquitin-dependent Degradation of p73 Is Inhibited by PML. J. Exp. Med. 2004, 199, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, E.V.; Petrichuk, S.V.; Kopeina, G.S.; Zhivotovsky, B. A link between mitotic defects and mitotic catastrophe: Detection and cell fate. Biol. Direct 2021, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Feng, C.; Chen, W.; Hou, P.; Liu, Z.; Zuo, M.; Han, Y.; Xu, C.; Melino, G.; Verkhratsky, A.; et al. Redressing the interactions between stem cells and immune system in tissue regeneration. Biol. Direct 2021, 16, 18. [Google Scholar] [CrossRef]
- Mammarella, E.; Zampieri, C.; Panatta, E.; Melino, G.; Amelio, I. NUAK2 and RCan2 participate in the p53 mutant pro-tumorigenic network. Biol. Direct 2021, 16, 11. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganini, C.; Montanaro, M.; Scimeca, M.; Palmieri, G.; Anemona, L.; Concetti, L.; Melino, G.; Bove, P.; Amelio, I.; Candi, E.; et al. No Time to Die: How Kidney Cancer Evades Cell Death. Int. J. Mol. Sci. 2022, 23, 6198. https://doi.org/10.3390/ijms23116198
Ganini C, Montanaro M, Scimeca M, Palmieri G, Anemona L, Concetti L, Melino G, Bove P, Amelio I, Candi E, et al. No Time to Die: How Kidney Cancer Evades Cell Death. International Journal of Molecular Sciences. 2022; 23(11):6198. https://doi.org/10.3390/ijms23116198
Chicago/Turabian StyleGanini, Carlo, Manuela Montanaro, Manuel Scimeca, Giampiero Palmieri, Lucia Anemona, Livia Concetti, Gerry Melino, Pierluigi Bove, Ivano Amelio, Eleonora Candi, and et al. 2022. "No Time to Die: How Kidney Cancer Evades Cell Death" International Journal of Molecular Sciences 23, no. 11: 6198. https://doi.org/10.3390/ijms23116198
APA StyleGanini, C., Montanaro, M., Scimeca, M., Palmieri, G., Anemona, L., Concetti, L., Melino, G., Bove, P., Amelio, I., Candi, E., & Mauriello, A. (2022). No Time to Die: How Kidney Cancer Evades Cell Death. International Journal of Molecular Sciences, 23(11), 6198. https://doi.org/10.3390/ijms23116198