
The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity

 , , ,
, , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
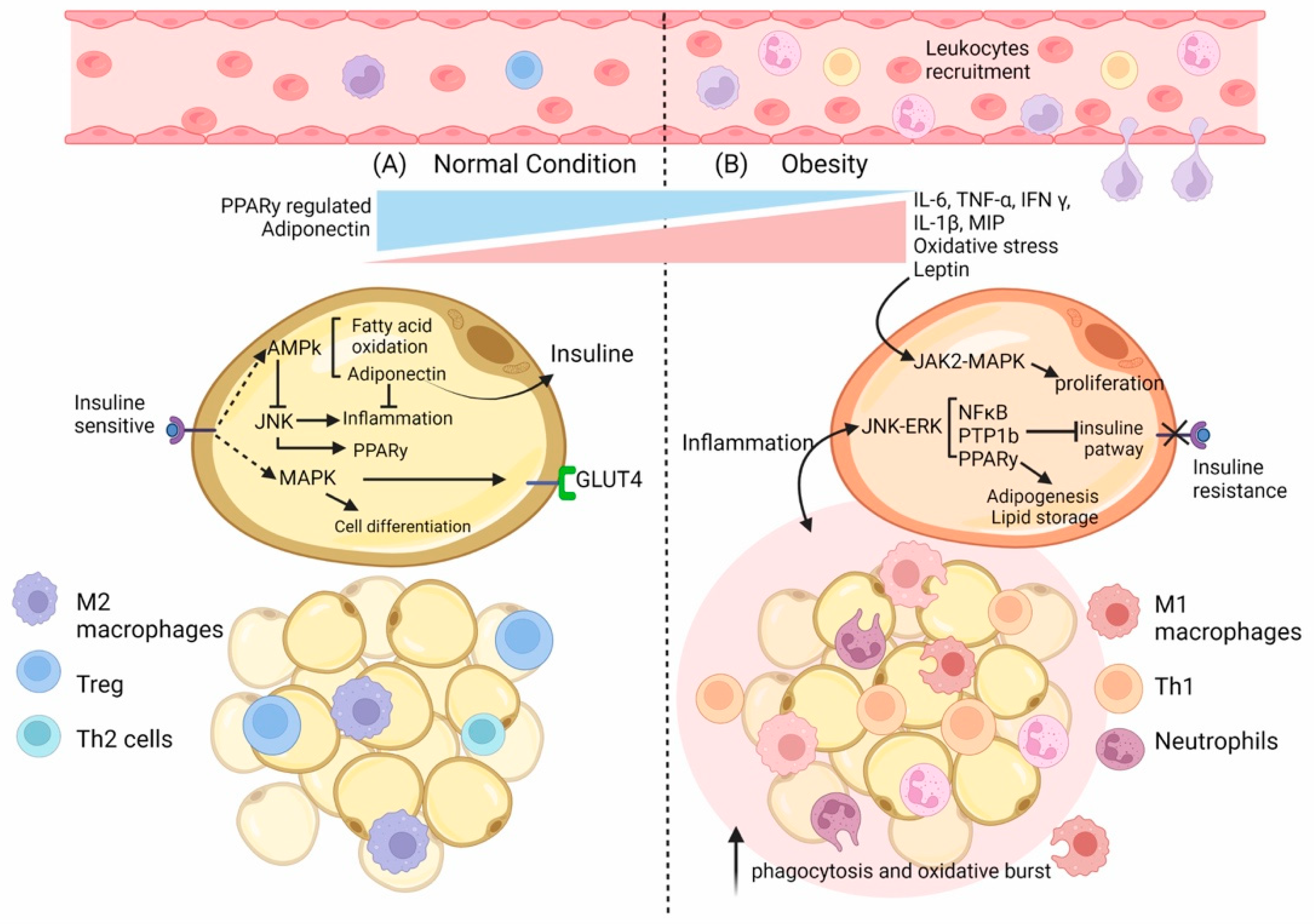
2. The Role of Adipose Tissue in the Immune Response
2.1. Adipose Tissue’s Composition
2.2. Adipokines
2.2.1. Leptin
2.2.2. Adiponectin
2.3. Adipocyte Mass and Function and Their Relationship with the Immune Cells
2.4. IFN Response
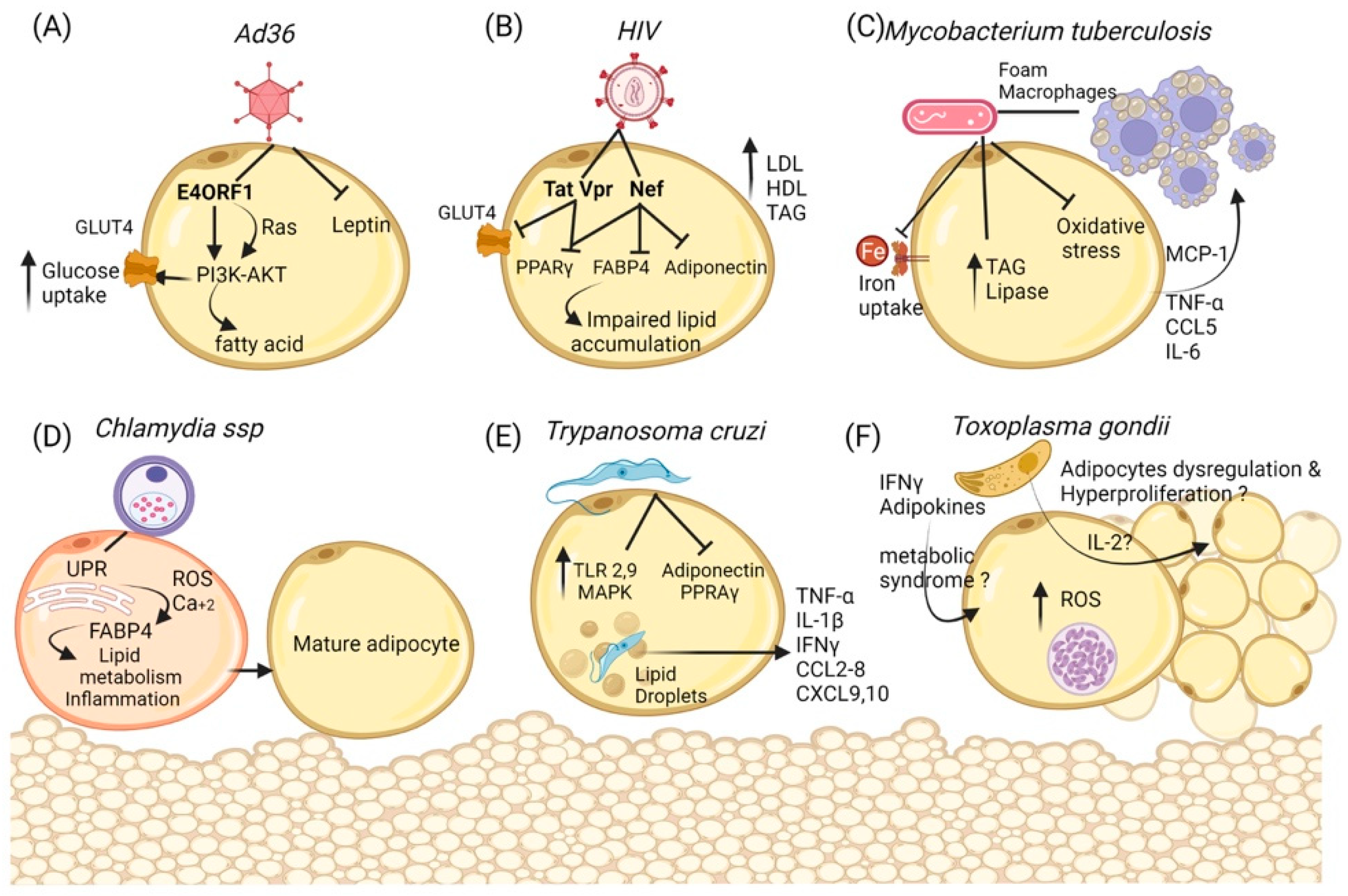
3. Adipocytes as Infection Targets and Their Relationship with Infectobesity
3.1. Adenovirus-36
3.2. Human Immunodeficiency Virus
3.3. Mycobacterium tuberculosis
3.4. Chlamydia ssp.
3.5. Trypanosoma cruzi
3.6. Toxoplasma gondii
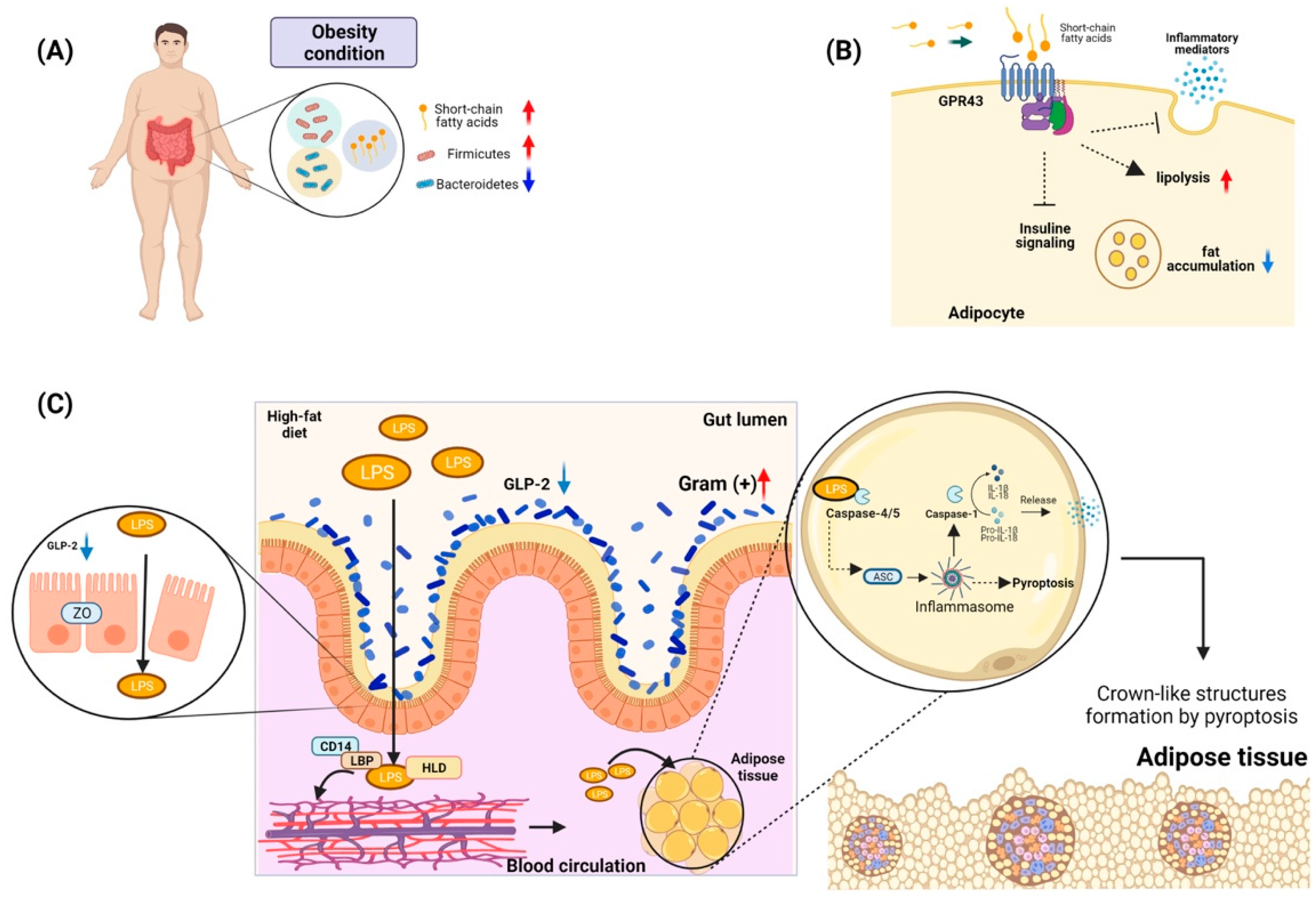
4. Gut Microbiota and Its Relationship with Obesity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tarantino, G.; Citro, V.; Cataldi, M. Findings from Studies Are Congruent with Obesity Having a Viral Origin, but What about Obesity-Related NAFLD? Viruses 2021, 13, 1285. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 28 April 2022).
- Blaszczak, A.M.; Jalilvand, A.; Hsueh, W.A. Adipocytes, Innate Immunity and Obesity: A Mini-Review. Front. Immunol. 2021, 12, 650768. [Google Scholar] [CrossRef] [PubMed]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Berge, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Pasarica, M.; Dhurandhar, N.V. Infectobesity: Obesity of infectious origin. Adv. Food Nutr. Res. 2007, 52, 61–102. [Google Scholar] [CrossRef] [PubMed]
- Catalan, V.; Aviles-Olmos, I.; Rodriguez, A.; Becerril, S.; Fernandez-Formoso, J.A.; Kiortsis, D.; Portincasa, P.; Gomez-Ambrosi, J.; Fruhbeck, G. Time to Consider the “Exposome Hypothesis” in the Development of the Obesity Pandemic. Nutrients 2022, 14, 1597. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. (Lausanne) 2016, 7, 30. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef]
- Kim, J.H.; Cho, H.T.; Kim, Y.J. The role of estrogen in adipose tissue metabolism: Insights into glucose homeostasis regulation. Endocr. J. 2014, 61, 1055–1067. [Google Scholar] [CrossRef]
- Cousin, B.; Casteilla, L.; Laharrague, P.; Luche, E.; Lorsignol, A.; Cuminetti, V.; Paupert, J. Immuno-metabolism and adipose tissue: The key role of hematopoietic stem cells. Biochimie 2016, 124, 21–26. [Google Scholar] [CrossRef]
- Chi, J.; Wu, Z.; Choi, C.H.J.; Nguyen, L.; Tegegne, S.; Ackerman, S.E.; Crane, A.; Marchildon, F.; Tessier-Lavigne, M.; Cohen, P. Three-Dimensional Adipose Tissue Imaging Reveals Regional Variation in Beige Fat Biogenesis and PRDM16-Dependent Sympathetic Neurite Density. Cell Metab. 2018, 27, 226–236.e223. [Google Scholar] [CrossRef]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed]
- Rajala, M.W.; Scherer, P.E. Minireview: The adipocyte—At the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003, 144, 3765–3773. [Google Scholar] [CrossRef] [PubMed]
- Benrick, A.; Chanclon, B.; Micallef, P.; Wu, Y.; Hadi, L.; Shelton, J.M.; Stener-Victorin, E.; Wernstedt Asterholm, I. Adiponectin protects against development of metabolic disturbances in a PCOS mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, E7187–E7196. [Google Scholar] [CrossRef] [PubMed]
- Combs, T.P.; Nagajyothi; Mukherjee, S.; de Almeida, C.J.; Jelicks, L.A.; Schubert, W.; Lin, Y.; Jayabalan, D.S.; Zhao, D.; Braunstein, V.L.; et al. The adipocyte as an important target cell for Trypanosoma cruzi infection. J. Biol. Chem. 2005, 280, 24085–24094. [Google Scholar] [CrossRef]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Sakers, A.; De Siqueira, M.K.; Seale, P.; Villanueva, C.J. Adipose-tissue plasticity in health and disease. Cell 2022, 185, 419–446. [Google Scholar] [CrossRef]
- Correa, L.H.; Heyn, G.S.; Magalhaes, K.G. The Impact of the Adipose Organ Plasticity on Inflammation and Cancer Progression. Cells 2019, 8, 662. [Google Scholar] [CrossRef]
- Cinti, S. Pink Adipocytes. Trends Endocrinol. Metab. 2018, 29, 651–666. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Gadgil, A.; Duncan, S.R. Role of T-lymphocytes and pro-inflammatory mediators in the pathogenesis of chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2008, 3, 531–541. [Google Scholar] [CrossRef]
- Ouedraogo, R.; Gong, Y.; Berzins, B.; Wu, X.; Mahadev, K.; Hough, K.; Chan, L.; Goldstein, B.J.; Scalia, R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J. Clin. Investig. 2007, 117, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Surendar, J.; Frohberger, S.J.; Karunakaran, I.; Schmitt, V.; Stamminger, W.; Neumann, A.L.; Wilhelm, C.; Hoerauf, A.; Hubner, M.P. Adiponectin Limits IFN-gamma and IL-17 Producing CD4 T Cells in Obesity by Restraining Cell Intrinsic Glycolysis. Front. Immunol. 2019, 10, 2555. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919, quiz 920. [Google Scholar] [CrossRef] [PubMed]
- Maurya, R.; Bhattacharya, P.; Dey, R.; Nakhasi, H.L. Leptin Functions in Infectious Diseases. Front. Immunol. 2018, 9, 2741. [Google Scholar] [CrossRef]
- Kelesidis, T.; Kelesidis, I.; Chou, S.; Mantzoros, C.S. Narrative review: The role of leptin in human physiology: Emerging clinical applications. Ann. Intern. Med. 2010, 152, 93–100. [Google Scholar] [CrossRef]
- Tsoukas, M.A.; Farr, O.M.; Mantzoros, C.S. Leptin in congenital and HIV-associated lipodystrophy. Metabolism 2015, 64, 47–59. [Google Scholar] [CrossRef]
- Corrales, P.; Vidal-Puig, A.; Medina-Gomez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef]
- Ye, J. Regulation of PPARgamma function by TNF-alpha. Biochem. Biophys. Res. Commun. 2008, 374, 405–408. [Google Scholar] [CrossRef]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef]
- Hornung, F.; Rogal, J.; Loskill, P.; Loffler, B.; Deinhardt-Emmer, S. The Inflammatory Profile of Obesity and the Role on Pulmonary Bacterial and Viral Infections. Int. J. Mol. Sci. 2021, 22, 3456. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Bluher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Scudiero, O.; Monaco, M.L.; Palmieri, A.; Mazzarella, G.; Costagliola, C.; Bianco, A.; Daniele, A. New insight into adiponectin role in obesity and obesity-related diseases. Biomed Res. Int. 2014, 2014, 658913. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Dong, L.Q. Adiponectin signaling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef]
- Dong, Z.M.; Gutierrez-Ramos, J.C.; Coxon, A.; Mayadas, T.N.; Wagner, D.D. A new class of obesity genes encodes leukocyte adhesion receptors. Proc. Natl. Acad. Sci. USA 1997, 94, 7526–7530. [Google Scholar] [CrossRef]
- Pires-Lapa, M.A.; Tamura, E.K.; Salustiano, E.M.; Markus, R.P. Melatonin synthesis in human colostrum mononuclear cells enhances dectin-1-mediated phagocytosis by mononuclear cells. J. Pineal Res. 2013, 55, 240–246. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Zhang, S.S.; Yang, X.J.; Ma, Q.H.; Xu, Y.; Chen, X.; Wang, P.; Pan, C.W. Leukocyte related parameters in older adults with metabolically healthy and unhealthy overweight or obesity. Sci. Rep. 2021, 11, 4652. [Google Scholar] [CrossRef]
- Rabot, S.; Membrez, M.; Bruneau, A.; Gerard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010, 24, 4948–4959. [Google Scholar] [CrossRef]
- Davis, C.D. The Gut Microbiome and Its Role in Obesity. Nutr. Today 2016, 51, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Cho, K.W.; Delproposto, J.L.; Oatmen, K.E.; Geletka, L.M.; Martinez-Santibanez, G.; Singer, K.; Lumeng, C.N. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes 2013, 62, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Tantiwong, P.; Shanmugasundaram, K.; Monroy, A.; Ghosh, S.; Li, M.; DeFronzo, R.A.; Cersosimo, E.; Sriwijitkamol, A.; Mohan, S.; Musi, N. NF-kappaB activity in muscle from obese and type 2 diabetic subjects under basal and exercise-stimulated conditions. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E794–E801. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.A.; Anderson-Baucum, E.K.; Kennedy, A.J.; Webb, C.D.; Yull, F.E.; Hasty, A.H. Activation of NF-kappaB drives the enhanced survival of adipose tissue macrophages in an obesogenic environment. Mol. Metab. 2015, 4, 665–677. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Yuan, M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int. J. Obes. 2003, 27 (Suppl. 3), S49–S52. [Google Scholar] [CrossRef]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NFkappaB signaling in myeloid cells is required for the glioblastoma growth. Sci. Rep. 2017, 7, 13754. [Google Scholar] [CrossRef]
- Popko, K.; Gorska, E.; Stelmaszczyk-Emmel, A.; Plywaczewski, R.; Stoklosa, A.; Gorecka, D.; Pyrzak, B.; Demkow, U. Proinflammatory cytokines Il-6 and TNF-alpha and the development of inflammation in obese subjects. Eur. J. Med. Res. 2010, 15 (Suppl. 2), 120–122. [Google Scholar] [CrossRef]
- Kern, L.; Mittenbuhler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-Induced TNFalpha and IL-6 Signaling: The Missing Link between Obesity and Inflammation-Driven Liver and Colorectal Cancers. Cancers 2018, 11, 24. [Google Scholar] [CrossRef]
- Szretter, K.J.; Gangappa, S.; Lu, X.; Smith, C.; Shieh, W.J.; Zaki, S.R.; Sambhara, S.; Tumpey, T.M.; Katz, J.M. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 2007, 81, 2736–2744. [Google Scholar] [CrossRef]
- Albrecht, L.J.; Tauber, S.C.; Merres, J.; Kress, E.; Stope, M.B.; Jansen, S.; Pufe, T.; Brandenburg, L.O. Lack of Proinflammatory Cytokine Interleukin-6 or Tumor Necrosis Factor Receptor-1 Results in a Failure of the Innate Immune Response after Bacterial Meningitis. Mediat. Inflamm. 2016, 2016, 7678542. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.; Vanier, G.; Al-Numani, D.; Lacouture, S.; Olivier, M.; Gottschalk, M. Proinflammatory cytokine and chemokine modulation by Streptococcus suis in a whole-blood culture system. FEMS Immunol. Med. Microbiol. 2006, 47, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.M.; Weschenfelder, J.; Sander, C.; Minkwitz, J.; Thormann, J.; Chittka, T.; Mergl, R.; Kirkby, K.C.; Fasshauer, M.; Stumvoll, M.; et al. Inflammatory cytokines in general and central obesity and modulating effects of physical activity. PLoS ONE 2015, 10, e0121971. [Google Scholar] [CrossRef] [PubMed]
- Harkins, J.M.; Moustaid-Moussa, N.; Chung, Y.J.; Penner, K.M.; Pestka, J.J.; North, C.M.; Claycombe, K.J. Expression of interleukin-6 is greater in preadipocytes than in adipocytes of 3T3-L1 cells and C57BL/6J and ob/ob mice. J. Nutr. 2004, 134, 2673–2677. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J. Clin. Endocrinol. Metab. 1997, 82, 4196–4200. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I.; Chambless, L.E.; Folsom, A.R.; Carpenter, M.; Heiss, G. Fibrinogen, other putative markers of inflammation, and weight gain in middle-aged adults—The ARIC study. Atherosclerosis Risk in Communities. Obes. Res. 2000, 8, 279–286. [Google Scholar] [CrossRef]
- Tuomisto, K.; Jousilahti, P.; Havulinna, A.S.; Borodulin, K.; Mannisto, S.; Salomaa, V. Role of inflammation markers in the prediction of weight gain and development of obesity in adults—A prospective study. Metab. Open 2019, 3, 100016. [Google Scholar] [CrossRef]
- Yang, R.; Barouch, L.A. Leptin signaling and obesity: Cardiovascular consequences. Circ. Res. 2007, 101, 545–559. [Google Scholar] [CrossRef]
- Richard, A.J.; Stephens, J.M. The role of JAK-STAT signaling in adipose tissue function. Biochim. Biophys. Acta 2014, 1842, 431–439. [Google Scholar] [CrossRef]
- Donohoe, F.; Wilkinson, M.; Baxter, E.; Brennan, D.J. Mitogen-Activated Protein Kinase (MAPK) and Obesity-Related Cancer. Int. J. Mol. Sci. 2020, 21, 1241. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Davis, W.; Stephens, L.R.; Hawkins, P.T.; Saklatvala, J. Synergistic activation of JNK/SAPK by interleukin-1 and platelet-derived growth factor is independent of Rac and Cdc42. Biochem. J. 1999, 338 Pt 2, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lu, S.; Ou, B.; Liu, Q.; Dai, J.; Ji, C.; Zhou, H.; Huang, H.; Ma, Y. The Role of JNk Signaling Pathway in Obesity-Driven Insulin Resistance. Diabetes Metab. Syndr. Obes. 2020, 13, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wilcox, D.M.; Haasch, D.L.; Jung, P.M.; Nguyen, P.T.; Voorbach, M.J.; Doktor, S.; Brodjian, S.; Bush, E.N.; Lin, E.; et al. Liver-specific knockdown of JNK1 up-regulates proliferator-activated receptor gamma coactivator 1 beta and increases plasma triglyceride despite reduced glucose and insulin levels in diet-induced obese mice. J. Biol. Chem. 2007, 282, 22765–22774. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Shen, J.; Yang, T.; Xu, Y.; Luo, Y.; Zhong, X.; Shi, L.; Hu, T.; Guo, T.; Nie, Y.; Luo, F.; et al. delta-Tocotrienol, Isolated from Rice Bran, Exerts an Anti-Inflammatory Effect via MAPKs and PPARs Signaling Pathways in Lipopolysaccharide-Stimulated Macrophages. Int. J. Mol. Sci. 2018, 19, 3022. [Google Scholar] [CrossRef]
- Feng, J.; Zhao, H.; Du, M.; Wu, X. The effect of apelin-13 on pancreatic islet beta cell mass and myocardial fatty acid and glucose metabolism of experimental type 2 diabetic rats. Peptides 2019, 114, 1–7. [Google Scholar] [CrossRef]
- Mullen, M.; Gonzalez-Perez, R.R. Leptin-Induced JAK/STAT Signaling and Cancer Growth. Vaccines 2016, 4, 26. [Google Scholar] [CrossRef]
- Gong, T.W.; Meyer, D.J.; Liao, J.; Hodge, C.L.; Campbell, G.S.; Wang, X.; Billestrup, N.; Carter-Su, C.; Schwartz, J. Regulation of glucose transport and c-fos and egr-1 expression in cells with mutated or endogenous growth hormone receptors. Endocrinology 1998, 139, 1863–1871. [Google Scholar] [CrossRef][Green Version]
- Min, I.M.; Pietramaggiori, G.; Kim, F.S.; Passegue, E.; Stevenson, K.E.; Wagers, A.J. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2008, 2, 380–391. [Google Scholar] [CrossRef]
- Leifheit-Nestler, M.; Wagner, N.M.; Gogiraju, R.; Didie, M.; Konstantinides, S.; Hasenfuss, G.; Schafer, K. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J. Transl. Med. 2013, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Li, M.M.H. All about the RNA: Interferon-Stimulated Genes that Interfere with Viral RNA Processes. Front. Immunol. 2020, 11, 605024. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Jennings, J.; Gong, Y.; Sang, Y. Viral Infections and Interferons in the Development of Obesity. Biomolecules 2019, 9, 726. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Miller, L.C.; Blecha, F.; Sang, Y. Reduction of infection by inhibiting mTOR pathway is associated with reversed repression of type I interferon by porcine reproductive and respiratory syndrome virus. J. Gen. Virol. 2017, 98, 1316–1328. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Mayuzumi, H.; Kato, S.; Minokoshi, Y.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Matsuzaki, G.; Yoshimura, A. Enhancement of leptin receptor signaling by SOCS3 deficiency induces development of gastric tumors in mice. Oncogene 2014, 33, 74–84. [Google Scholar] [CrossRef]
- Wunderlich, C.M.; Hovelmeyer, N.; Wunderlich, F.T. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAKSTAT 2013, 2, e23878. [Google Scholar] [CrossRef]
- Honce, R.; Schultz-Cherry, S. Impact of Obesity on Influenza A Virus Pathogenesis, Immune Response, and Evolution. Front. Immunol. 2019, 10, 1071. [Google Scholar] [CrossRef]
- Wieser, V.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Moser, P.; Moschen, A.R.; Kaser, S.; Tilg, H. Adipose type I interferon signalling protects against metabolic dysfunction. Gut 2018, 67, 157–165. [Google Scholar] [CrossRef]
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Investig. 2016, 126, 2839–2854. [Google Scholar] [CrossRef]
- O’Rourke, R.W.; White, A.E.; Metcalf, M.D.; Winters, B.R.; Diggs, B.S.; Zhu, X.; Marks, D.L. Systemic inflammation and insulin sensitivity in obese IFN-gamma knockout mice. Metabolism 2012, 61, 1152–1161. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Folco, E.J.; Sukhova, G.; Shimizu, K.; Gotsman, I.; Vernon, A.H.; Libby, P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circ. Res. 2008, 103, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Alsaggar, M.; Mills, M.; Liu, D. Interferon beta overexpression attenuates adipose tissue inflammation and high-fat diet-induced obesity and maintains glucose homeostasis. Gene Ther. 2017, 24, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Guan, K.L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242. [Google Scholar] [CrossRef]
- Nagajyothi, F.; Desruisseaux, M.S.; Thiruvur, N.; Weiss, L.M.; Braunstein, V.L.; Albanese, C.; Teixeira, M.M.; de Almeida, C.J.; Lisanti, M.P.; Scherer, P.E.; et al. Trypanosoma cruzi infection of cultured adipocytes results in an inflammatory phenotype. Obesity 2008, 16, 1992–1997. [Google Scholar] [CrossRef]
- Trovato, G.M.; Martines, G.F.; Garozzo, A.; Tonzuso, A.; Timpanaro, R.; Pirri, C.; Trovato, F.M.; Catalano, D. Ad36 adipogenic adenovirus in human non-alcoholic fatty liver disease. Liver Int. 2010, 30, 184–190. [Google Scholar] [CrossRef]
- Parra-Rojas, I.; Del Moral-Hernandez, O.; Salgado-Bernabe, A.B.; Guzman-Guzman, I.P.; Salgado-Goytia, L.; Munoz-Valle, J.F. Adenovirus-36 seropositivity and its relation with obesity and metabolic profile in children. Int. J. Endocrinol. 2013, 2013, 463194. [Google Scholar] [CrossRef]
- Dhurandhar, N.V.; Israel, B.A.; Kolesar, J.M.; Mayhew, G.F.; Cook, M.E.; Atkinson, R.L. Increased adiposity in animals due to a human virus. Int. J. Obes. 2000, 24, 989–996. [Google Scholar] [CrossRef]
- Dhurandhar, N.V.; Whigham, L.D.; Abbott, D.H.; Schultz-Darken, N.J.; Israel, B.A.; Bradley, S.M.; Kemnitz, J.W.; Allison, D.B.; Atkinson, R.L. Human adenovirus Ad-36 promotes weight gain in male rhesus and marmoset monkeys. J. Nutr. 2002, 132, 3155–3160. [Google Scholar] [CrossRef]
- Kapila, M.; Khosla, P.; Dhurandhar, N.V. Novel short-term effects of adenovirus Ad-36 on hamster lipoproteins. Int. J. Obes. 2004, 28, 1521–1527. [Google Scholar] [CrossRef]
- Na, H.N.; Park, S.; Jeon, H.J.; Kim, H.B.; Nam, J.H. Reduction of adenovirus 36-induced obesity and inflammation by mulberry extract. Microbiol. Immunol. 2014, 58, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Pasarica, M.; Loiler, S.; Dhurandhar, N.V. Acute effect of infection by adipogenic human adenovirus Ad36. Arch. Virol. 2008, 153, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Broderick, M.P.; Hansen, C.J.; Irvine, M.; Metzgar, D.; Campbell, K.; Baker, C.; Russell, K.L. Adenovirus 36 seropositivity is strongly associated with race and gender, but not obesity, among US military personnel. Int. J. Obes. 2010, 34, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Berger, P.K.; Pollock, N.K.; Laing, E.M.; Warden, S.J.; Hill Gallant, K.M.; Hausman, D.B.; Tripp, R.A.; McCabe, L.D.; McCabe, G.P.; Weaver, C.M.; et al. Association of adenovirus 36 infection with adiposity and inflammatory-related markers in children. J. Clin. Endocrinol. Metab. 2014, 99, 3240–3246. [Google Scholar] [CrossRef]
- Atkinson, R.L.; Lee, I.; Shin, H.J.; He, J. Human adenovirus-36 antibody status is associated with obesity in children. Int. J. Pediatr. Obes. 2010, 5, 157–160. [Google Scholar] [CrossRef]
- Dhurandhar, N.V.; Bailey, D.; Thomas, D. Interaction of obesity and infections. Obes. Rev. 2015, 16, 1017–1029. [Google Scholar] [CrossRef]
- Dhurandhar, N.V.; Dhurandhar, E.J.; Ingram, D.K.; Vaughan, K.; Mattison, J.A. Natural infection of human adenovirus 36 in rhesus monkeys is associated with a reduction in fasting glucose 36. J. Diabetes 2014, 6, 614–616. [Google Scholar] [CrossRef]
- Rathod, M.; Vangipuram, S.D.; Krishnan, B.; Heydari, A.R.; Holland, T.C.; Dhurandhar, N.V. Viral mRNA expression but not DNA replication is required for lipogenic effect of human adenovirus Ad-36 in preadipocytes. Int. J. Obes. 2007, 31, 78–86. [Google Scholar] [CrossRef]
- Vangipuram, S.D.; Sheele, J.; Atkinson, R.L.; Holland, T.C.; Dhurandhar, N.V. A human adenovirus enhances preadipocyte differentiation. Obes. Res. 2004, 12, 770–777. [Google Scholar] [CrossRef]
- Vangipuram, S.D.; Yu, M.; Tian, J.; Stanhope, K.L.; Pasarica, M.; Havel, P.J.; Heydari, A.R.; Dhurandhar, N.V. Adipogenic human adenovirus-36 reduces leptin expression and secretion and increases glucose uptake by fat cells. Int. J. Obes. 2007, 31, 87–96. [Google Scholar] [CrossRef]
- Frese, K.K.; Lee, S.S.; Thomas, D.L.; Latorre, I.J.; Weiss, R.S.; Glaunsinger, B.A.; Javier, R.T. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene 2003, 22, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P.; Corvera, S. Signaling mechanisms that regulate glucose transport. J. Biol. Chem. 1999, 274, 1865–1868. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Yamanaka, H.; Fukuoka, T.; Dai, Y.; Obata, K.; Mashimo, T.; Noguchi, K. Expression of HGF and cMet in the peripheral nervous system of adult rats following sciatic nerve injury. Neuroreport 2001, 12, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.M.; Fusinski, K.A.; Rathod, M.A.; Loiler, S.A.; Pasarica, M.; Shaw, M.K.; Kilroy, G.; Sutton, G.M.; McAllister, E.J.; Mashtalir, N.; et al. Human adenovirus Ad-36 induces adipogenesis via its E4 orf-1 gene. Int. J. Obes. 2008, 32, 397–406. [Google Scholar] [CrossRef]
- Dhurandhar, E.J.; Dubuisson, O.; Mashtalir, N.; Krishnapuram, R.; Hegde, V.; Dhurandhar, N.V. E4orf1: A novel ligand that improves glucose disposal in cell culture. PLoS ONE 2011, 6, e23394. [Google Scholar] [CrossRef]
- Damouche, A.; Lazure, T.; Avettand-Fenoel, V.; Huot, N.; Dejucq-Rainsford, N.; Satie, A.P.; Melard, A.; David, L.; Gommet, C.; Ghosn, J.; et al. Adipose Tissue Is a Neglected Viral Reservoir and an Inflammatory Site during Chronic HIV and SIV Infection. PLoS Pathog. 2015, 11, e1005153. [Google Scholar] [CrossRef]
- Bourgeois, C.; Gorwood, J.; Barrail-Tran, A.; Lagathu, C.; Capeau, J.; Desjardins, D.; Le Grand, R.; Damouche, A.; Bereziat, V.; Lambotte, O. Specific Biological Features of Adipose Tissue, and Their Impact on HIV Persistence. Front. Microbiol. 2019, 10, 2837. [Google Scholar] [CrossRef]
- Kosmiski, L.; Kuritzkes, D.; Lichtenstein, K.; Eckel, R. Adipocyte-derived hormone levels in HIV lipodystrophy. Antivir. Ther. 2003, 8, 9–15. [Google Scholar] [CrossRef]
- Dupin, N.; Buffet, M.; Marcelin, A.G.; Lamotte, C.; Gorin, I.; Ait-Arkoub, Z.; Treluyer, J.M.; Bui, P.; Calvez, V.; Peytavin, G. HIV and antiretroviral drug distribution in plasma and fat tissue of HIV-infected patients with lipodystrophy. AIDS 2002, 16, 2419–2424. [Google Scholar] [CrossRef]
- Couturier, J.; Suliburk, J.W.; Brown, J.M.; Luke, D.J.; Agarwal, N.; Yu, X.; Nguyen, C.; Iyer, D.; Kozinetz, C.A.; Overbeek, P.A.; et al. Human adipose tissue as a reservoir for memory CD4+ T cells and HIV. AIDS 2015, 29, 667–674. [Google Scholar] [CrossRef]
- Agarwal, N.; Iyer, D.; Patel, S.G.; Sekhar, R.V.; Phillips, T.M.; Schubert, U.; Oplt, T.; Buras, E.D.; Samson, S.L.; Couturier, J.; et al. HIV-1 Vpr induces adipose dysfunction in vivo through reciprocal effects on PPAR/GR co-regulation. Sci. Transl. Med. 2013, 5, 213ra164. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Lewis, D.E. HIV Persistence in Adipose Tissue Reservoirs. Curr. HIV/AIDS Rep. 2018, 15, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Iyer, D.; Gabbi, C.; Saha, P.; Patel, S.G.; Mo, Q.; Chang, B.; Goswami, B.; Schubert, U.; Kopp, J.B.; et al. HIV-1 viral protein R (Vpr) induces fatty liver in mice via LXRalpha and PPARalpha dysregulation: Implications for HIV-specific pathogenesis of NAFLD. Sci. Rep. 2017, 7, 13362. [Google Scholar] [CrossRef] [PubMed]
- Gorwood, J.; Bourgeois, C.; Mantecon, M.; Atlan, M.; Pourcher, V.; Pourcher, G.; Le Grand, R.; Desjardins, D.; Feve, B.; Lambotte, O.; et al. Impact of HIV/simian immunodeficiency virus infection and viral proteins on adipose tissue fibrosis and adipogenesis. AIDS 2019, 33, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, J.P.; Vinnard, C.; Subbian, S.; Nagajyothi, J.F. Effect of Mycobacterium tuberculosis infection on adipocyte physiology. Microbes Infect. 2018, 20, 81–88. [Google Scholar] [CrossRef]
- Beigier-Bompadre, M.; Montagna, G.N.; Kuhl, A.A.; Lozza, L.; Weiner, J., 3rd; Kupz, A.; Vogelzang, A.; Mollenkopf, H.J.; Lowe, D.; Bandermann, S.; et al. Mycobacterium tuberculosis infection modulates adipose tissue biology. PLoS Pathog. 2017, 13, e1006676. [Google Scholar] [CrossRef]
- Agarwal, P.; Khan, S.R.; Verma, S.C.; Beg, M.; Singh, K.; Mitra, K.; Gaikwad, A.N.; Akhtar, M.S.; Krishnan, M.Y. Mycobacterium tuberculosis persistence in various adipose depots of infected mice and the effect of anti-tubercular therapy. Microbes Infect. 2014, 16, 571–580. [Google Scholar] [CrossRef]
- Neyrolles, O.; Hernandez-Pando, R.; Pietri-Rouxel, F.; Fornes, P.; Tailleux, L.; Barrios Payan, J.A.; Pivert, E.; Bordat, Y.; Aguilar, D.; Prevost, M.C.; et al. Is adipose tissue a place for Mycobacterium tuberculosis persistence? PLoS ONE 2006, 1, e43. [Google Scholar] [CrossRef]
- Rastogi, S.; Agarwal, P.; Krishnan, M.Y. Use of an adipocyte model to study the transcriptional adaptation of Mycobacterium tuberculosis to store and degrade host fat. Int. J. Mycobacteriology 2016, 5, 92–98. [Google Scholar] [CrossRef]
- D’Avila, H.; Melo, R.C.; Parreira, G.G.; Werneck-Barroso, E.; Castro-Faria-Neto, H.C.; Bozza, P.T. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: Intracellular domains for eicosanoid synthesis in vivo. J. Immunol. 2006, 176, 3087–3097. [Google Scholar] [CrossRef]
- Nandy, A.; Mondal, A.K.; Pandey, R.; Arumugam, P.; Dawa, S.; Jaisinghani, N.; Rao, V.; Dash, D.; Gandotra, S. Adipocyte Model of Mycobacterium tuberculosis Infection Reveals Differential Availability of Iron to Bacilli in the Lipid-Rich Caseous Environment. Infect. Immun. 2018, 86, e00041-18. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.; Cheng, C.Y.; Ketheesan, N.; Cullen, A.; Tang, Y.; Lum, J.; West, K.; Poidinger, M.; Guertin, D.A.; Singhal, A.; et al. mTORC2/Akt activation in adipocytes is required for adipose tissue inflammation in tuberculosis. EBioMedicine 2019, 45, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Witkin, S.S.; Minis, E.; Athanasiou, A.; Leizer, J.; Linhares, I.M. Chlamydia trachomatis: The Persistent Pathogen. Clin. Vaccine Immunol. 2017, 24, e00203-17. [Google Scholar] [CrossRef] [PubMed]
- Jaworowska, A.; Bazylak, G. Chlamydophila pneumoniae antibodies may be independently associated with increased BMI and percentage of body fat among women. Int. J. Obes. 2011, 35, 1225–1232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rantala, A.; Lajunen, T.; Juvonen, R.; Bloigu, A.; Paldanius, M.; Silvennoinen-Kassinen, S.; Peitso, A.; Vainio, O.; Leinonen, M.; Saikku, P. Chlamydia pneumoniae infection is associated with elevated body mass index in young men. Epidemiol. Infect. 2010, 138, 1267–1273. [Google Scholar] [CrossRef]
- Petyaev, I.M.; Zigangirova, N.A.; Kapotina, L.N.; Fedina, E.D.; Kyle, N.H. Chlamydia trachomatis promotes 3T3 cell differentiation into adipocytes. Adv. Clin. Exp. Med. 2014, 23, 511–516. [Google Scholar] [CrossRef]
- Goodall, K.J.; Nguyen, A.; McKenzie, C.; Eckle, S.B.G.; Sullivan, L.C.; Andrews, D.M. The murine CD94/NKG2 ligand, Qa-1(b), is a high-affinity, functional ligand for the CD8alphaalpha homodimer. J. Biol. Chem. 2020, 295, 3239–3246. [Google Scholar] [CrossRef]
- Walenna, N.F.; Kurihara, Y.; Chou, B.; Ishii, K.; Soejima, T.; Itoh, R.; Shimizu, A.; Ichinohe, T.; Hiromatsu, K. Chlamydia pneumoniae exploits adipocyte lipid chaperone FABP4 to facilitate fat mobilization and intracellular growth in murine adipocytes. Biochem. Biophys. Res. Commun. 2018, 495, 353–359. [Google Scholar] [CrossRef]
- Engeli, S.; Utz, W.; Haufe, S.; Lamounier-Zepter, V.; Pofahl, M.; Traber, J.; Janke, J.; Luft, F.C.; Boschmann, M.; Schulz-Menger, J.; et al. Fatty acid binding protein 4 predicts left ventricular mass and longitudinal function in overweight and obese women. Heart 2013, 99, 944–948. [Google Scholar] [CrossRef]
- Steverding, D. The history of Chagas disease. Parasites Vectors 2014, 7, 317. [Google Scholar] [CrossRef]
- Urdaneta-Morales, S. Chagas’ disease: An emergent urban zoonosis. The caracas valley (Venezuela) as an epidemiological model. Front. Public Health 2014, 2, 265. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.C.P.; Beaton, A.; Acquatella, H.; Bern, C.; Bolger, A.F.; Echeverria, L.E.; Dutra, W.O.; Gascon, J.; Morillo, C.A.; Oliveira-Filho, J.; et al. Chagas Cardiomyopathy: An Update of Current Clinical Knowledge and Management: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e169–e209. [Google Scholar] [CrossRef] [PubMed]
- Nagajyothi, F.; Desruisseaux, M.S.; Jelicks, L.A.; Machado, F.S.; Chua, S.; Scherer, P.E.; Tanowitz, H.B. Perspectives on adipose tissue, chagas disease and implications for the metabolic syndrome. Interdiscip. Perspect. Infect. Dis. 2009, 2009, 824324. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.B.; Villar, S.R.; Toneatto, J.; Pacini, M.F.; Marquez, J.; D’Attilio, L.; Bottasso, O.A.; Piwien-Pilipuk, G.; Perez, A.R. Immune response triggered by Trypanosoma cruzi infection strikes adipose tissue homeostasis altering lipid storage, enzyme profile and adipokine expression. Med. Microbiol. Immunol. 2019, 208, 651–666. [Google Scholar] [CrossRef]
- Santamaria, M.H.; Rios, L.D.; Corral, R.S. Trypanosoma cruzi down-regulates adiponectin expression in mouse adipocytes via the NFAT signaling pathway. Microbes Infect. 2021, 23, 104757. [Google Scholar] [CrossRef]
- Nagajyothi, F.; Desruisseaux, M.S.; Machado, F.S.; Upadhya, R.; Zhao, D.; Schwartz, G.J.; Teixeira, M.M.; Albanese, C.; Lisanti, M.P.; Chua, S.C., Jr.; et al. Response of adipose tissue to early infection with Trypanosoma cruzi (Brazil strain). J. Infect. Dis. 2012, 205, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Lizardo, K.; Ayyappan, J.P.; Oswal, N.; Weiss, L.M.; Scherer, P.E.; Nagajyothi, J.F. Fat tissue regulates the pathogenesis and severity of cardiomyopathy in murine chagas disease. PLoS Negl. Trop. Dis. 2021, 15, e0008964. [Google Scholar] [CrossRef]
- Tian, Y.M.; Dai, F.Y.; Huang, S.Y.; Deng, Z.H.; Duan, G.; Zhou, D.H.; Yang, J.F.; Weng, Y.B.; Zhu, X.Q.; Zou, F.C. First report of Toxoplasma gondii seroprevalence in peafowls in Yunnan Province, Southwestern China. Parasites Vectors 2012, 5, 205. [Google Scholar] [CrossRef]
- Feustel, S.M.; Meissner, M.; Liesenfeld, O. Toxoplasma gondii and the blood-brain barrier. Virulence 2012, 3, 182–192. [Google Scholar] [CrossRef]
- Persson, C.M.; Lambert, H.; Vutova, P.P.; Dellacasa-Lindberg, I.; Nederby, J.; Yagita, H.; Ljunggren, H.G.; Grandien, A.; Barragan, A.; Chambers, B.J. Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect. Immun. 2009, 77, 970–976. [Google Scholar] [CrossRef]
- Courret, N.; Darche, S.; Sonigo, P.; Milon, G.; Buzoni-Gatel, D.; Tardieux, I. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 2006, 107, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.R.; Zeldovich, V.B.; Poukchanski, A.; Boothroyd, J.C.; Bakardjiev, A.I. Tissue barriers of the human placenta to infection with Toxoplasma gondii. Infect. Immun. 2012, 80, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Remington, J.S.; Cavanaugh, E.N. Isolation of the encysted form of Toxoplasma gondii from human skeletal muscle and brain. N. Engl. J. Med. 1965, 273, 1308–1310. [Google Scholar] [CrossRef] [PubMed]
- Di Cristina, M.; Marocco, D.; Galizi, R.; Proietti, C.; Spaccapelo, R.; Crisanti, A. Temporal and spatial distribution of Toxoplasma gondii differentiation into Bradyzoites and tissue cyst formation in vivo. Infect. Immun. 2008, 76, 3491–3501. [Google Scholar] [CrossRef]
- Shirbazou, S.; Delpisheh, A.; Mokhetari, R.; Tavakoli, G. Serologic Detection of Anti Toxoplasma gondii Infection in Diabetic Patients. Iran. Red Crescent Med. J. 2013, 15, 701–703. [Google Scholar] [CrossRef]
- Salem, D.A.; Salem, N.A.; Hendawy, S.R. Association between Toxoplasma gondii infection and metabolic syndrome in obese adolescents: A possible immune-metabolic link. Parasitol. Int. 2021, 83, 102343. [Google Scholar] [CrossRef]
- Hermes-Uliana, C.; Pereira-Severi, L.S.; Luerdes, R.B.; Franco, C.L.; da Silva, A.V.; Araujo, E.J.; Sant’Ana Dde, M. Chronic infection with Toxoplasma gondii causes myenteric neuroplasticity of the jejunum in rats. Auton. Neurosci. 2011, 160, 3–8. [Google Scholar] [CrossRef]
- Reeves, G.M.; Mazaheri, S.; Snitker, S.; Langenberg, P.; Giegling, I.; Hartmann, A.M.; Konte, B.; Friedl, M.; Okusaga, O.; Groer, M.W.; et al. A Positive Association between T. gondii Seropositivity and Obesity. Front. Public Health 2013, 1, 73. [Google Scholar] [CrossRef]
- Picard, F.; Arsenijevic, D.; Richard, D.; Deshaies, Y. Responses of adipose and muscle lipoprotein lipase to chronic infection and subsequent acute lipopolysaccharide challenge. Clin. Vaccine Immunol. 2002, 9, 771–776. [Google Scholar] [CrossRef]
- Kannan, G.; Moldovan, K.; Xiao, J.C.; Yolken, R.H.; Jones-Brando, L.; Pletnikov, M.V. Toxoplasma gondii strain-dependent effects on mouse behaviour. Folia Parasitol 2010, 57, 151–155. [Google Scholar] [CrossRef]
- Thjodleifsson, B.; Olafsson, I.; Gislason, D.; Gislason, T.; Jogi, R.; Janson, C. Infections and obesity: A multinational epidemiological study. Scand. J. Infect. Dis. 2008, 40, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Melchor, S.J.; Ewald, S.E. Disease Tolerance in Toxoplasma Infection. Front. Cell. Infect. Microbiol. 2019, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Kochumon, S.; Al Madhoun, A.; Al-Rashed, F.; Thomas, R.; Sindhu, S.; Al-Ozairi, E.; Al-Mulla, F.; Ahmad, R. Elevated adipose tissue associated IL-2 expression in obesity correlates with metabolic inflammation and insulin resistance. Sci. Rep. 2020, 10, 16364. [Google Scholar] [CrossRef] [PubMed]
- Kloting, N.; Bluher, M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 277–287. [Google Scholar] [CrossRef]
- Gustafson, B.; Gogg, S.; Hedjazifar, S.; Jenndahl, L.; Hammarstedt, A.; Smith, U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E999–E1003. [Google Scholar] [CrossRef]
- Zhuang, H.; Yao, C.; Zhao, X.; Chen, X.; Yang, Y.; Huang, S.; Pan, L.; Du, A.; Yang, Y. DNA double-strand breaks in the Toxoplasma gondii-infected cells by the action of reactive oxygen species. Parasites Vectors 2020, 13, 490. [Google Scholar] [CrossRef]
- Wang, Z.J.; Yu, S.M.; Gao, J.M.; Zhang, P.; Hide, G.; Yamamoto, M.; Lai, D.H.; Lun, Z.R. High resistance to Toxoplasma gondii infection in inducible nitric oxide synthase knockout rats. iScience 2021, 24, 103280. [Google Scholar] [CrossRef]
- Zhu, S.; Li, Q.R.; Du, Y.; Yang, X.; Fan, J.M.; Dong, Z.M. Toxicity of derivatives from semicarbazide-sensitive amine oxidase-mediated deamination of methylamine against Toxoplasma gondii after infection of differentiated 3T3-L1 cells. Toxicol. Vit. 2010, 24, 809–814. [Google Scholar] [CrossRef]
- Han, C.Y. Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue. Diabetes Metab. J. 2016, 40, 272–279. [Google Scholar] [CrossRef]
- Ruff, W.E.; Greiling, T.M.; Kriegel, M.A. Host-microbiota interactions in immune-mediated diseases. Nat. Rev. Microbiol. 2020, 18, 521–538. [Google Scholar] [CrossRef]
- Runge, S.; Rosshart, S.P. The Mammalian Metaorganism: A Holistic View on How Microbes of All Kingdoms and Niches Shape Local and Systemic Immunity. Front. Immunol. 2021, 12, 702378. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Kapitan, M.; Niemiec, M.J.; Steimle, A.; Frick, J.S.; Jacobsen, I.D. Fungi as Part of the Microbiota and Interactions with Intestinal Bacteria. Curr. Top Microbiol. Immunol. 2019, 422, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Matijasic, M.; Mestrovic, T.; Paljetak, H.C.; Peric, M.; Baresic, A.; Verbanac, D. Gut Microbiota beyond Bacteria-Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Ryan, P.M.; Cryan, J.F.; Dinan, T.G.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Gut microbiota, obesity and diabetes. Postgrad. Med. J. 2016, 92, 286–300. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Angelakis, E.; Armougom, F.; Million, M.; Raoult, D. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 2012, 7, 91–109. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [PubMed]
- Bjursell, M.; Admyre, T.; Goransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly, Y.M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef]
- Santos, N.C.; Silva, A.C.; Castanho, M.A.; Martins-Silva, J.; Saldanha, C. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. Chembiochem 2003, 4, 96–100. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef]
- Kallio, K.A.; Buhlin, K.; Jauhiainen, M.; Keva, R.; Tuomainen, A.M.; Klinge, B.; Gustafsson, A.; Pussinen, P.J. Lipopolysaccharide associates with pro-atherogenic lipoproteins in periodontitis patients. Innate Immun. 2008, 14, 247–253. [Google Scholar] [CrossRef]
- Levels, J.H.; Abraham, P.R.; van den Ende, A.; van Deventer, S.J. Distribution and kinetics of lipoprotein-bound endotoxin. Infect. Immun. 2001, 69, 2821–2828. [Google Scholar] [CrossRef]
- Wurfel, M.M.; Kunitake, S.T.; Lichenstein, H.; Kane, J.P.; Wright, S.D. Lipopolysaccharide (LPS)-binding protein is carried on lipoproteins and acts as a cofactor in the neutralization of LPS. J. Exp. Med. 1994, 180, 1025–1035. [Google Scholar] [CrossRef]
- Hersoug, L.G.; Moller, P.; Loft, S. Role of microbiota-derived lipopolysaccharide in adipose tissue inflammation, adipocyte size and pyroptosis during obesity. Nutr. Res. Rev. 2018, 31, 153–163. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Souders, C.P.; Zhao, H.; Ackerman, A.L. Considerations for Bedside Urologic Procedures in Patients with Severe Acute Respiratory Syndrome Coronavirus-2. Urology 2020, 142, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Shao, F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 2015, 32, 78–83. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Ortega, O.; Moreno-Corona, N.C.; Cruz-Holguin, V.J.; Garcia-Gonzalez, L.D.; Helguera-Repetto, A.C.; Romero-Valdovinos, M.; Arevalo-Romero, H.; Cedillo-Barron, L.; León-Juárez, M. The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity. Int. J. Mol. Sci. 2022, 23, 6154. https://doi.org/10.3390/ijms23116154
López-Ortega O, Moreno-Corona NC, Cruz-Holguin VJ, Garcia-Gonzalez LD, Helguera-Repetto AC, Romero-Valdovinos M, Arevalo-Romero H, Cedillo-Barron L, León-Juárez M. The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity. International Journal of Molecular Sciences. 2022; 23(11):6154. https://doi.org/10.3390/ijms23116154
Chicago/Turabian StyleLópez-Ortega, Orestes, Nidia Carolina Moreno-Corona, Victor Javier Cruz-Holguin, Luis Didier Garcia-Gonzalez, Addy Cecilia Helguera-Repetto, Mirza Romero-Valdovinos, Haruki Arevalo-Romero, Leticia Cedillo-Barron, and Moisés León-Juárez. 2022. "The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity" International Journal of Molecular Sciences 23, no. 11: 6154. https://doi.org/10.3390/ijms23116154
APA StyleLópez-Ortega, O., Moreno-Corona, N. C., Cruz-Holguin, V. J., Garcia-Gonzalez, L. D., Helguera-Repetto, A. C., Romero-Valdovinos, M., Arevalo-Romero, H., Cedillo-Barron, L., & León-Juárez, M. (2022). The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity. International Journal of Molecular Sciences, 23(11), 6154. https://doi.org/10.3390/ijms23116154