
Identification of Robust and Key Differentially Expressed Genes during C2C12 Cell Myogenesis Based on Multiomics Data

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
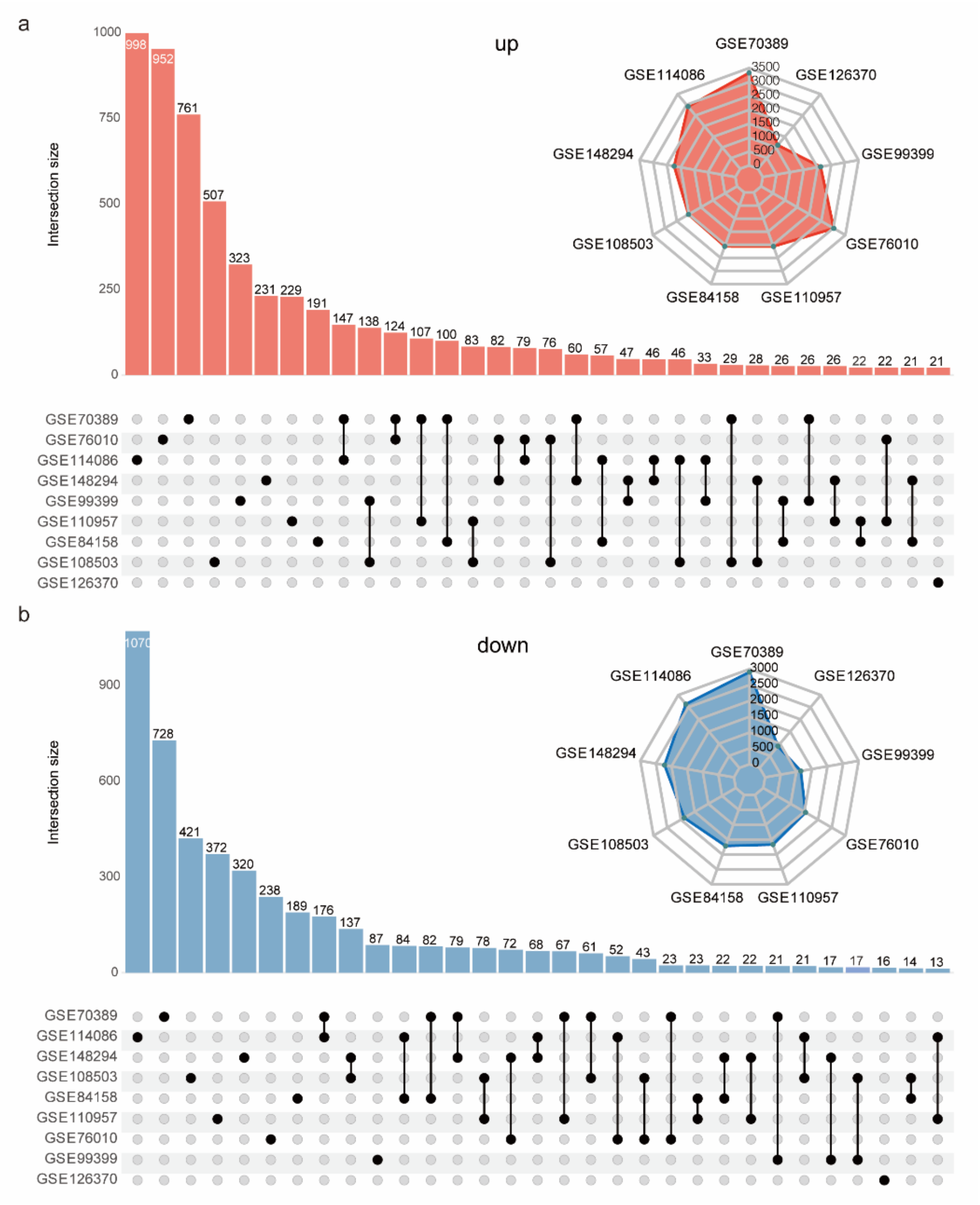
2.1. Published RNA-Seq Datasets during C2C12 Cell Myogenesis Are Poorly Consistent
2.2. Considerable Robust DEGs Were Identified by Integrating Nine RNA-Seq Datasets
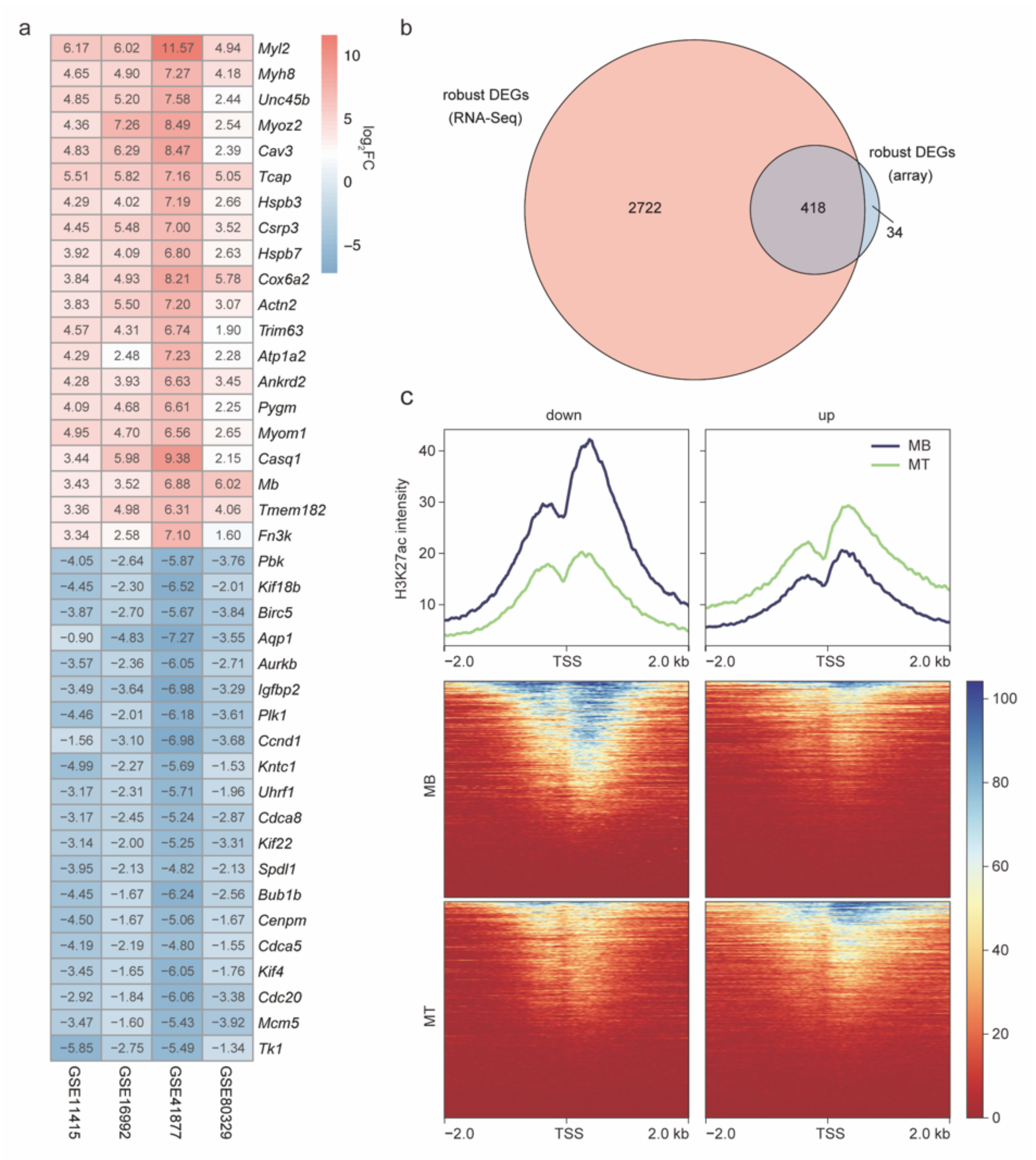
2.3. The Robust DEGs Were Validated by Array Expression Profiles and H3K27ac Signals
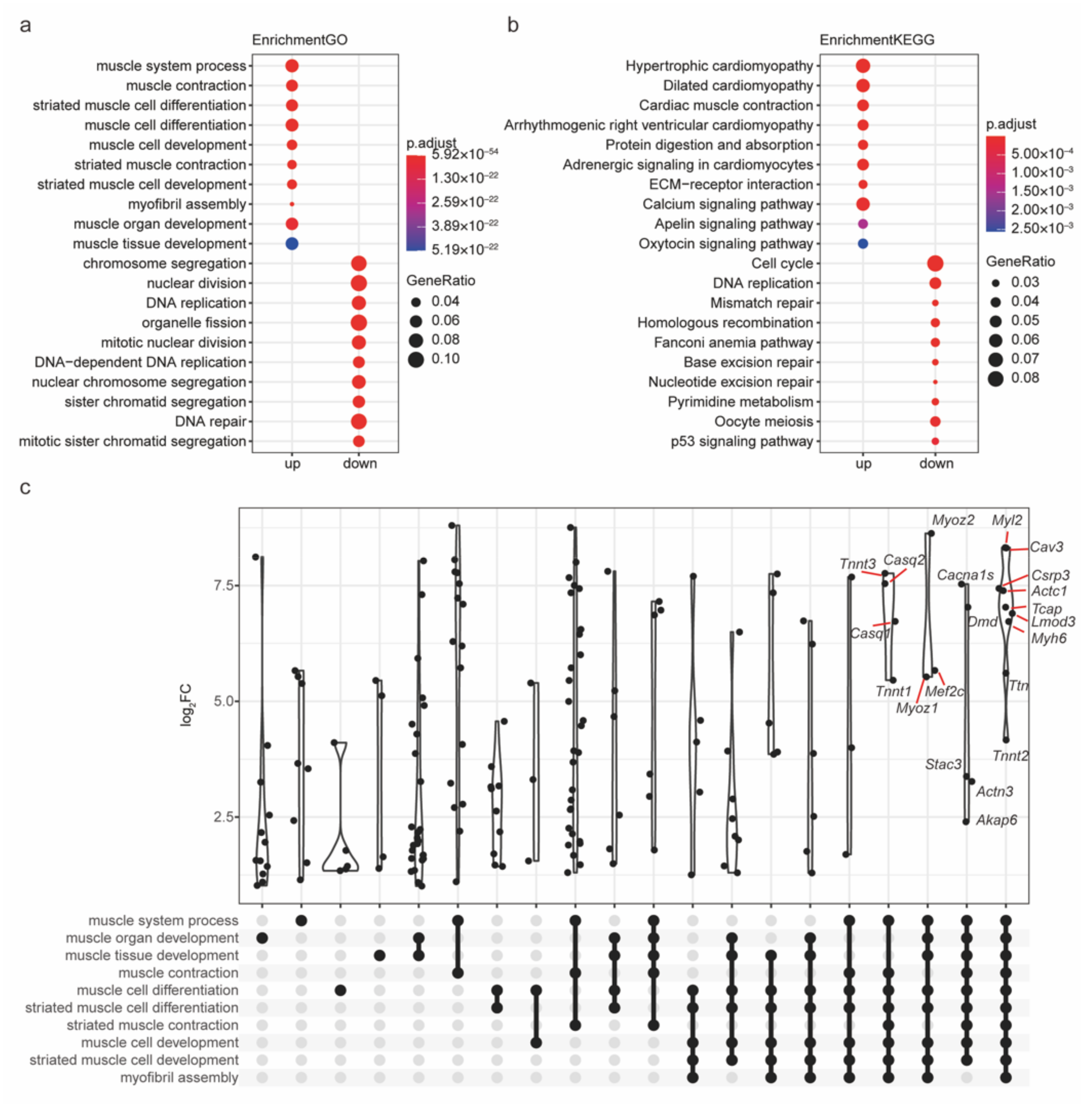
2.4. The Functions of Robust DEGs Were Investigated by GO/KEGG Analysis
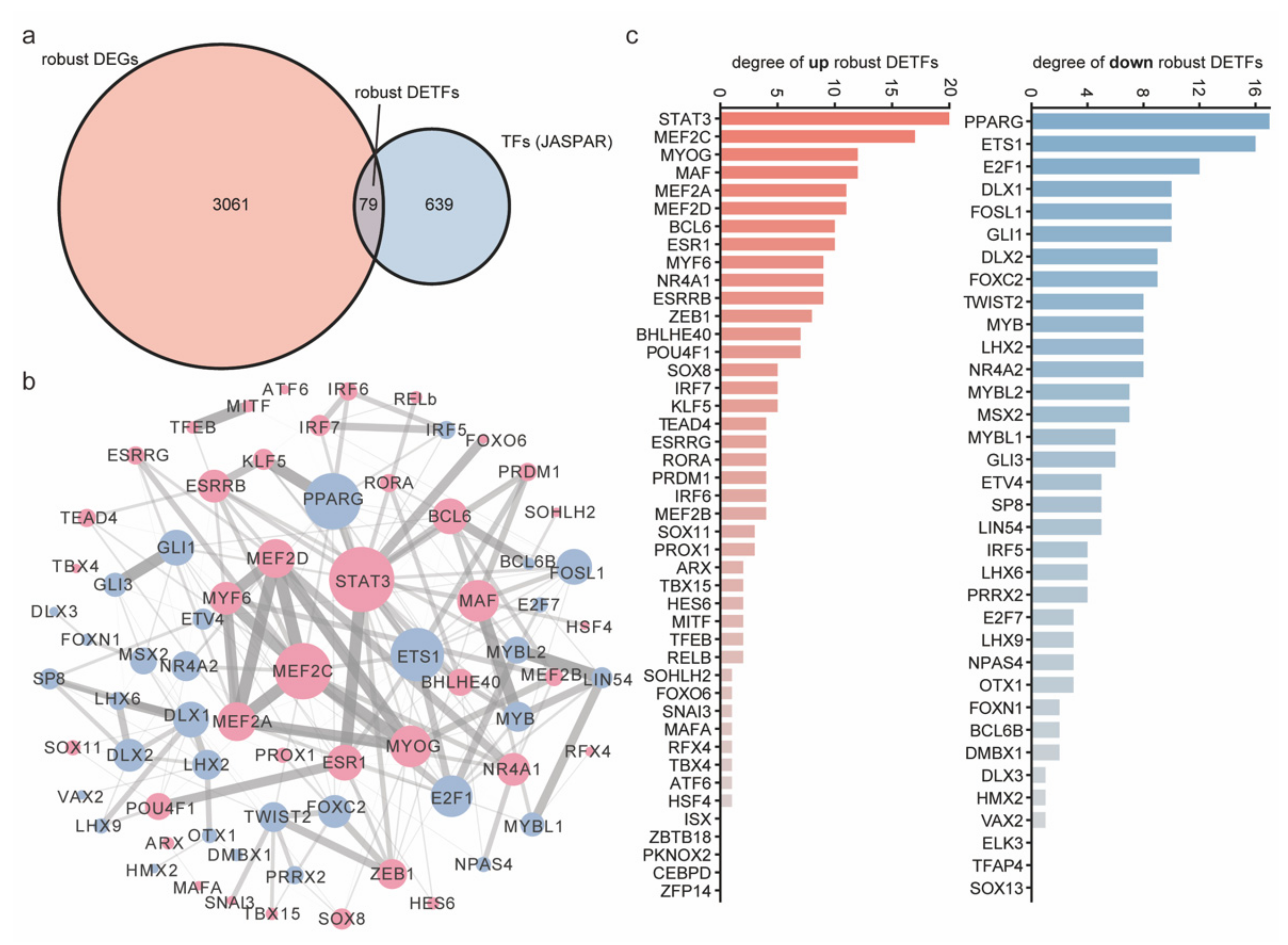
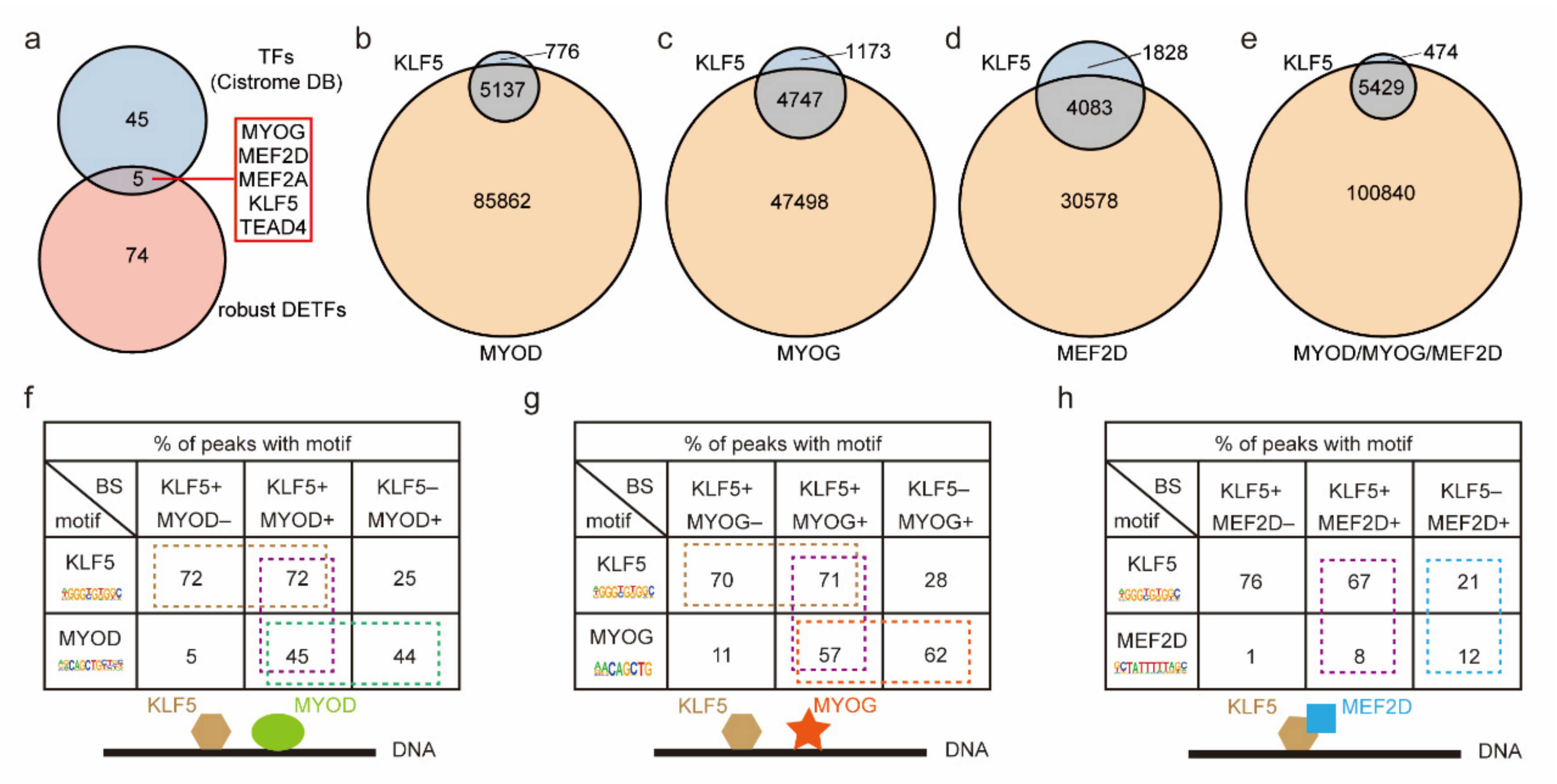
2.5. The Core Robust DETFs Were Screened by the PPI Network
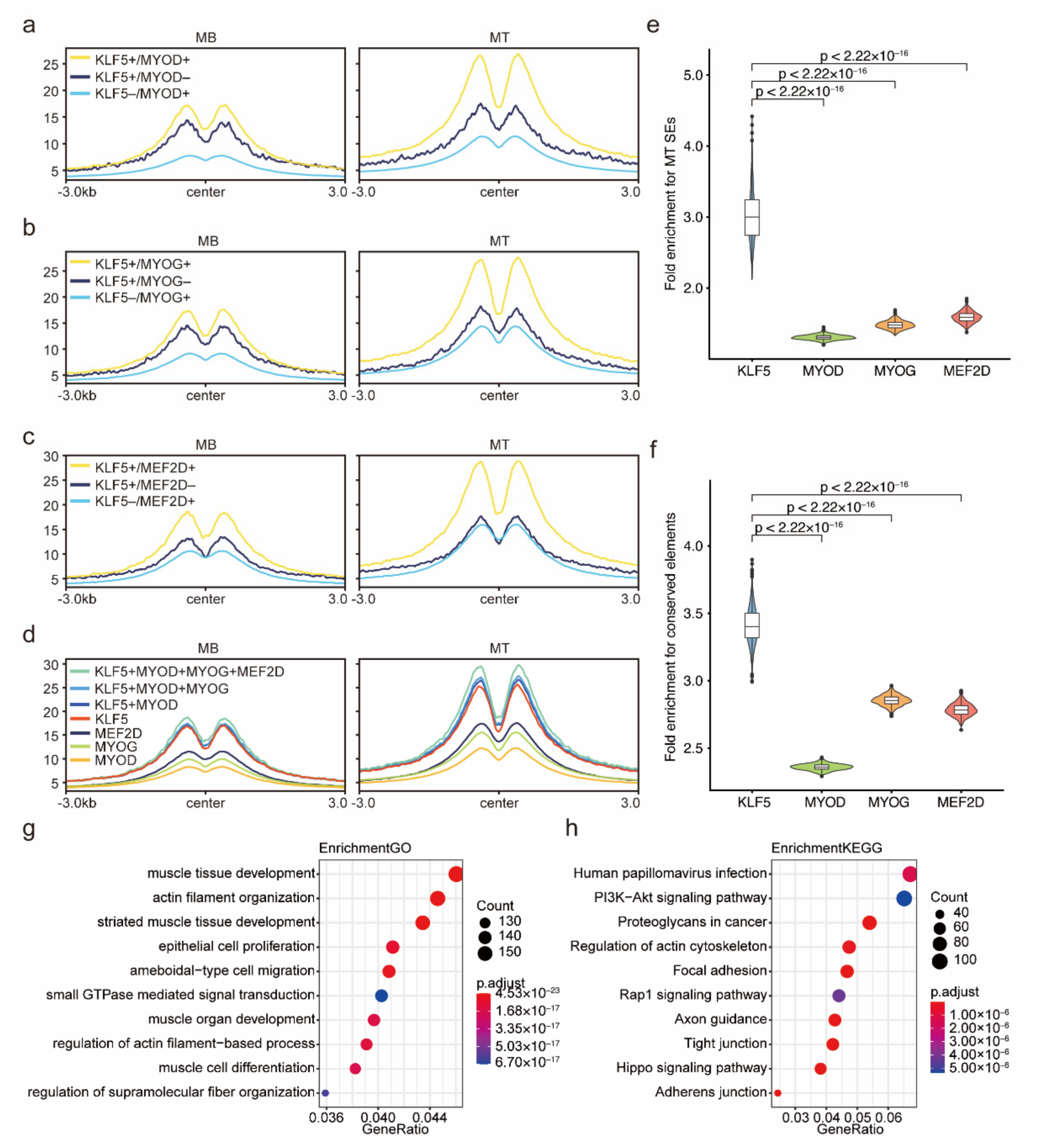
2.6. The Cooperative Binding Modes of KLF5 and Three Key Myogenic Factors Were Revealed
2.7. KLF5-Binding Sites Could Mark Key Enhancers for Myogenesis
3. Discussion
4. Materials and Methods
4.1. Dataset Collection
4.2. DEG Identification and RRA Integration
4.3. ChIP-Seq Data Analysis
4.4. GO/KEGG Analysis and PPI Network Construction
4.5. Cooperative Binding Analysis of TFs
4.6. Enrichment for MT SEs of Genomic Regions
4.7. Conservative Analysis of Genomic Regions
4.8. Functional Annotation of Genomic Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Janssen, I.; Heymsfield, S.B.; Wang, Z.; Ross, R. Skeletal Muscle Mass and Distribution in 468 Men and Women Aged 18–88 Yr. J. Appl. Physiol. 2000, 89, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, R.R. The Underappreciated Role of Muscle in Health and Disease. Am. J. Clin. Nutr. 2006, 84, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Ornatsky, O.I.; Andreucci, J.J.; McDermott, J.C. A Dominant-Negative Form of Transcription Factor MEF2 Inhibits Myogenesis. J. Biol. Chem. 1997, 272, 33271–33278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sincennes, M.-C.; Brun, C.E.; Rudnicki, M.A. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. Stem Cells Transl. Med. 2016, 5, 282–290. [Google Scholar] [CrossRef]
- Ferri, P.; Barbieri, E.; Burattini, S.; Guescini, M.; D’Emilio, A.; Biagiotti, L.; Del Grande, P.; De Luca, A.; Stocchi, V.; Falcieri, E. Expression and Subcellular Localization of Myogenic Regulatory Factors during the Differentiation of Skeletal Muscle C2C12 Myoblasts. J. Cell. Biochem. 2009, 108, 1302–1317. [Google Scholar] [CrossRef]
- Lu, J.; McKinsey, T.A.; Zhang, C.-L.; Olson, E.N. Regulation of Skeletal Myogenesis by Association of the MEF2 Transcription Factor with Class II Histone Deacetylases. Mol. Cell 2000, 6, 233–244. [Google Scholar] [CrossRef]
- Edmondson, D.G.; Lyons, G.E.; Martin, J.F.; Olson, E.N. Mef2 Gene Expression Marks the Cardiac and Skeletal Muscle Lineages during Mouse Embryogenesis. Development 1994, 120, 1251–1263. [Google Scholar] [CrossRef]
- Hayashi, S.; Manabe, I.; Suzuki, Y.; Relaix, F.; Oishi, Y. Klf5 Regulates Muscle Differentiation by Directly Targeting Muscle-Specific Genes in Cooperation with MyoD in Mice. Elife 2016, 5, e17462. [Google Scholar] [CrossRef]
- Sala, D.; Cunningham, T.J.; Stec, M.J.; Etxaniz, U.; Nicoletti, C.; Dall’Agnese, A.; Puri, P.L.; Duester, G.; Latella, L.; Sacco, A. The Stat3-Fam3a Axis Promotes Muscle Stem Cell Myogenic Lineage Progression by Inducing Mitochondrial Respiration. Nat. Commun. 2019, 10, 1796. [Google Scholar] [CrossRef]
- Pan, X.; Liu, B.; Chen, S.; Ding, H.; Yao, X.; Cheng, Y.; Xu, D.; Yin, Y.; Dai, X.; Sun, J. Nr4a1 as a Myogenic Factor Is Upregulated in Satellite Cells/Myoblast under Proliferation and Differentiation State. Biochem. Biophys. Res. Commun. 2019, 513, 573–581. [Google Scholar] [CrossRef]
- Robson, M.I.; Jose, I.; Czapiewski, R.; Lê Thành, P.; Booth, D.G.; Kelly, D.A.; Webb, S.; Kerr, A.R.W.; Schirmer, E.C. Tissue-Specific Gene Repositioning by Muscle Nuclear Membrane Proteins Enhances Repression of Critical Developmental Genes during Myogenesis. Mol. Cell 2016, 62, 834–847. [Google Scholar] [CrossRef] [Green Version]
- Frangini, M.; Franzolin, E.; Chemello, F.; Laveder, P.; Romualdi, C.; Bianchi, V.; Rampazzo, C. Synthesis of Mitochondrial DNA Precursors during Myogenesis, an Analysis in Purified C2C12 Myotubes*[S]. J. Biol. Chem. 2013, 288, 5624–5635. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.; Dang, H.C.P.; Djambazian, H.; Zuzan, H.; Fedyshyn, Y.; Ketela, T.; Moffat, J.; Hudson, T.J.; Sladek, R. Analysis of Early C2C12 Myogenesis Identifies Stably and Differentially Expressed Transcriptional Regulators Whose Knock-down Inhibits Myoblast Differentiation. Physiol. Genom. 2012, 44, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Chirn, G.-W.; Szustakowski, J.D.; Bakhtiarova, A.; Kosinski, P.A.; Kemp, D.; Nirmala, N. Uncovering Mechanisms of Transcriptional Regulations by Systematic Mining of Cis Regulatory Elements with Gene Expression Profiles. BioData Min. 2008, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q. Comprehensive Genomic Characterization of Long Non-Coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular RNA Is Enriched and Stable in Exosomes: A Promising Biomarker for Cancer Diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S. The Landscape of Long Noncoding RNAs in the Human Transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M. MapSplice: Accurate Mapping of RNA-Seq Reads for Splice Junction Discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, Z.; Figler, B.; Zhang, H.; Cheng, F. Introduction to RNA-Seq and Its Applications to Drug Discovery and Development. Drug Dev. Res. 2014, 75, 324–330. [Google Scholar] [CrossRef]
- Zhang, T.; Guan, X.; Choi, U.L.; Dong, Q.; Lam, M.M.T.; Zeng, J.; Xiong, J.; Wang, X.; Poon, T.C.W.; Zhang, H. Phosphorylation of TET2 by AMPK Is Indispensable in Myogenic Differentiation. Epigenet. Chromatin 2019, 12, 32. [Google Scholar] [CrossRef]
- Doynova, M.D.; Markworth, J.F.; Cameron-Smith, D.; Vickers, M.H.; O’Sullivan, J.M. Linkages between Changes in the 3D Organization of the Genome and Transcription during Myotube Differentiation in Vitro. Skelet. Muscle 2017, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, I.; Caccia, R.; Garcia-Manteiga, J.M.; Ferri, G.; Caretti, G.; Molineris, I.; Nishioka, K.; Gabellini, D. The Trithorax Protein Ash1L Promotes Myoblast Fusion by Activating Cdon Expression. Nat. Commun. 2018, 9, 5026. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Wu, G.; Li, W.-X.; Guan, D.; Lv, W.; Gong, M.; Ye, S.; Lu, A. A Transcriptomic Study of Myogenic Differentiation under the Overexpression of PPARγ by RNA-Seq. Sci. Rep. 2017, 7, 15308. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L. Histone Demethylase KDM6A Directly Senses Oxygen to Control Chromatin and Cell Fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M. Circ-ZNF609 Is a Circular RNA That Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Martone, J.; Briganti, F.; Legnini, I.; Morlando, M.; Picillo, E.; Sthandier, O.; Politano, L.; Bozzoni, I. The Lack of the Celf2a Splicing Factor Converts a Duchenne Genotype into a Becker Phenotype. Nat. Commun. 2016, 7, 10488. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lan, B.; Yi, X.; He, C.; Dang, L.; Zhou, X.; Lu, Y.; Sun, Y.; Liu, Z.; Bai, X. HRP2–DPF3a–BAF Complex Coordinates Histone Modification and Chromatin Remodeling to Regulate Myogenic Gene Transcription. Nucleic Acids Res. 2020, 48, 6563–6582. [Google Scholar] [CrossRef]
- Dell’Orso, S.; Wang, A.H.; Shih, H.-Y.; Saso, K.; Berghella, L.; Gutierrez-Cruz, G.; Ladurner, A.G.; O’Shea, J.J.; Sartorelli, V.; Zare, H. The Histone Variant MacroH2A1. 2 Is Necessary for the Activation of Muscle Enhancers and Recruitment of the Transcription Factor Pbx1. Cell Rep. 2016, 14, 1156–1168. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK Enables Robust Identification of Essential Genes from Genome-Scale CRISPR/Cas9 Knockout Screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust Rank Aggregation for Gene List Integration and Meta-Analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, L.; Li, S.; Zhang, W.; Ma, X.; Cheng, G.; Yang, W.; Zan, L. Identification of Potential Key Genes Associated with Adipogenesis through Integrated Analysis of Five Mouse Transcriptome Datasets. Int. J. Mol. Sci. 2018, 19, 3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Chao, F.; Zhuo, Z.; Ma, Z.; Li, W.; Chen, G. Identification of Hub Genes in Prostate Cancer Using Robust Rank Aggregation and Weighted Gene Co-Expression Network Analysis. Aging 2019, 11, 4736. [Google Scholar] [CrossRef] [PubMed]
- Schöneich, C.; Dremina, E.; Galeva, N.; Sharov, V. Apoptosis in Differentiating C2C12 Muscle Cells Selectively Targets Bcl-2-Deficient Myotubes. Apoptosis 2014, 19, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, M.N.; Luxford, C.; Skibsted, L.H.; Davies, M.J. Oxidation of Myosin by Haem Proteins Generates Myosin Radicals and Protein Cross-Links. Biochem. J. 2008, 410, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Rossi, A.C.; Smerdu, V.; Leinwand, L.A.; Reggiani, C. Developmental Myosins: Expression Patterns and Functional Significance. Skelet. Muscle 2015, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Guo, Q.; Zhang, G.; Cao, X.; Chen, W.; Li, Y.; Guan, M.; Yu, J.; Wang, X.; Yan, Y. High Myosin Binding Protein H Expression Predicts Poor Prognosis in Glioma Patients. Sci. Rep. 2022, 12, 1525. [Google Scholar] [CrossRef]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common Markers of Proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef]
- Peng, X.L.; So, K.K.; He, L.; Zhao, Y.; Zhou, J.; Li, Y.; Yao, M.; Xu, B.; Zhang, S.; Yao, H.; et al. MyoD- and FoxO3-Mediated Hotspot Interaction Orchestrates Super-Enhancer Activity during Myogenic Differentiation. Nucleic Acids Res. 2017, 45, 8785–8805. [Google Scholar] [CrossRef] [Green Version]
- Pradeepa, M.M. Causal Role of Histone Acetylations in Enhancer Function. Transcription 2017, 8, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Fei, W.; Liu, M.; Zhang, Y.; Cao, S.; Wang, X.; Xie, B.; Wang, J. Identification of Key Pathways and Hub Genes in the Myogenic Differentiation of Pluripotent Stem Cell: A Bioinformatics and Experimental Study. J. Orthop. Surg. Res. 2021, 16, 4. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, Q.; Shen, Y.; Liu, C.; Huang, T.; Huai, D.; Dai, J. LINC00704 Contributes to the Proliferation and Accelerates the Cell Cycle of Nasopharyngeal Carcinoma Cells via Regulating ETS1/CDK6 Axis. Kaohsiung J. Med. Sci. 2022, 38, 312–320. [Google Scholar] [CrossRef]
- Zhuang, R.; Zhang, X.; Lu, D.I.; Wang, J.; Zhuo, J.; Wei, X.; Ling, Q.; Xie, H.; Zheng, S.; Xu, X. LncRNA DRHC Inhibits Proliferation and Invasion in Hepatocellular Carcinoma via C-Myb-regulated MEK/ERK Signaling. Mol. Carcinog. 2019, 58, 366–375. [Google Scholar] [CrossRef]
- Qian, C.-S.; Li, L.-J.; Huang, H.-W.; Yang, H.-F.; Wu, D.-P. MYC-Regulated LncRNA NEAT1 Promotes B Cell Proliferation and Lymphomagenesis via the MiR-34b-5p-GLI1 Pathway in Diffuse Large B-Cell Lymphoma. Cancer Cell Int. 2020, 20, 87. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kim, B.G.; Jang, Y.; Kang, S.; Lee, J.H.; Cho, N.H. The Stromal Loss of MiR-4516 Promotes the FOSL1-Dependent Proliferation and Malignancy of Triple Negative Breast Cancer. Cancer Lett. 2020, 469, 256–265. [Google Scholar] [CrossRef]
- Judd, J.; Lovas, J.; Huang, G.N. Defined Factors to Reactivate Cell Cycle Activity in Adult Mouse Cardiomyocytes. Sci. Rep. 2019, 9, 18830. [Google Scholar] [CrossRef] [Green Version]
- Franco, H.L.; Casasnovas, J.; Rodríguez-Medina, J.R.; Cadilla, C.L. Redundant or Separate Entities?—Roles of Twist1 and Twist2 as Molecular Switches during Gene Transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, K.; Zhang, Y.; Barnes, S.D.; Jaichander, P.; Zheng, Y.; Hassan, M.; Malladi, V.S.; Skapek, S.X.; Xu, L. Twist2 Amplification in Rhabdomyosarcoma Represses Myogenesis and Promotes Oncogenesis by Redirecting MyoD DNA Binding. Genes Dev. 2019, 33, 626–640. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Ahmad, K.; Ramachandran, S. Cooperative Binding between Distant Transcription Factors Is a Hallmark of Active Enhancers. Mol. Cell 2021, 81, 1651–1665. [Google Scholar] [CrossRef]
- Ohkawa, Y.; Marfella, C.G.A.; Imbalzano, A.N. Skeletal Muscle Specification by Myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 2006, 25, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Kumar, R.M.; Penn, B.H.; Berkes, C.A.; Kooperberg, C.; Boyer, L.A.; Young, R.A.; Tapscott, S.J. Global and Gene-specific Analyses Show Distinct Roles for Myod and Myog at a Common Set of Promoters. EMBO J. 2006, 25, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.; Kim, W.; Davie, J. Myogenin Is Required for Assembly of the Transcription Machinery on Muscle Genes during Skeletal Muscle Differentiation. PLoS ONE 2021, 16, e0245618. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, S.C.J.; Stitzel, M.L.; Taylor, D.L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; van Bueren, K.L.; Chines, P.S.; Narisu, N.; Black, B.L. Chromatin Stretch Enhancer States Drive Cell-Specific Gene Regulation and Harbor Human Disease Risk Variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934. [Google Scholar] [CrossRef] [Green Version]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT Improves Functional Interpretation of Cis-Regulatory Regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, J.; Hu, H.; Wang, H.; Wang, C.; Lin, H.; Zhao, X. Dynamic Transcriptome Profiles of Postnatal Porcine Skeletal Muscle Growth and Development. BMC Genom. Data 2021, 22, 32. [Google Scholar] [CrossRef]
- Briata, P.; Lin, W.-J.; Giovarelli, M.; Pasero, M.; Chou, C.-F.; Trabucchi, M.; Rosenfeld, M.G.; Chen, C.-Y.; Gherzi, R. PI3K/AKT Signaling Determines a Dynamic Switch between Distinct KSRP Functions Favoring Skeletal Myogenesis. Cell Death Differ. 2012, 19, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Kaya-Çopur, A.; Marchiano, F.; Hein, M.Y.; Alpern, D.; Russeil, J.; Luis, N.M.; Mann, M.; Deplancke, B.; Habermann, B.H.; Schnorrer, F. The Hippo Pathway Controls Myofibril Assembly and Muscle Fiber Growth by Regulating Sarcomeric Gene Expression. Elife 2021, 10, e63726. [Google Scholar] [CrossRef]
- Watt, K.I.; Goodman, C.A.; Hornberger, T.A.; Gregorevic, P. The Hippo Signaling Pathway in the Regulation of Skeletal Muscle Mass and Function. Exerc. Sport Sci. Rev. 2018, 46, 92. [Google Scholar] [CrossRef]
- Weterman, M.A.J.; Barth, P.G.; van Spaendonck-Zwarts, K.Y.; Aronica, E.; Poll-The, B.-T.; Brouwer, O.F.; van Tintelen, J.P.; Qahar, Z.; Bradley, E.J.; de Wissel, M. Recessive MYL2 Mutations Cause Infantile Type I Muscle Fibre Disease and Cardiomyopathy. Brain 2013, 136, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Betz, R.C.; Schoser, B.G.H.; Kasper, D.; Ricker, K.; Ramírez, A.; Stein, V.; Torbergsen, T.; Lee, Y.; Nöthen, M.M.; Wienker, T.F. Mutations in CAV3 Cause Mechanical Hyperirritability of Skeletal Muscle in Rippling Muscle Disease. Nat. Genet. 2001, 28, 218–219. [Google Scholar] [CrossRef]
- Xu, X.; Qiu, H.; Du, Z.-Q.; Fan, B.; Rothschild, M.F.; Yuan, F.; Liu, B. Porcine CSRP3: Polymorphism and Association Analyses with Meat Quality Traits and Comparative Analyses with CSRP1 and CSRP2. Mol. Biol. Rep. 2010, 37, 451–459. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, Z. Multiple Functions of Maf in the Regulation of Cellular Development and Differentiation. Diabetes. Metab. Res. Rev. 2015, 31, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, T.; Miki, T.; Kikuchi, M.; Fukuda, T.; Miyasaka, N.; Kamiyama, R.; Hirosawa, S. The Proto-Oncogene Bcl6 Inhibits Apoptotic Cell Death in Differentiation-Induced Mouse Myogenic Cells. Oncogene 1999, 18, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; Hou, X.; Liu, X.; Wang, L.; Gao, H.; Zhao, F.; Shi, L.; Shi, L.; Yan, H.; Deng, T.; et al. The Landscape of Chromatin Accessibility in Skeletal Muscle during Embryonic Development in Pigs. J. Anim. Sci. Biotechnol. 2021, 12, 56. [Google Scholar] [CrossRef]
- Kumagai, H.; Miyamoto-Mikami, E.; Hirata, K.; Kikuchi, N.; Kamiya, N.; Hoshikawa, S.; Zempo, H.; Naito, H.; Miyamoto, N.; Fuku, N. ESR1 Rs2234693 Polymorphism Is Associated with Muscle Injury and Muscle Stiffness. Med. Sci. Sports Exerc. 2019, 51, 19. [Google Scholar] [CrossRef]
- Blais, A.; Tsikitis, M.; Acosta-Alvear, D.; Sharan, R.; Kluger, Y.; Dynlacht, B.D. An Initial Blueprint for Myogenic Differentiation. Genes Dev. 2005, 19, 553–569. [Google Scholar] [CrossRef] [Green Version]
- Molkentin, J.D.; Olson, E.N. Combinatorial Control of Muscle Development by Basic Helix-Loop-Helix and MADS-Box Transcription Factors. Proc. Natl. Acad. Sci. USA 1996, 93, 9366–9373. [Google Scholar] [CrossRef] [Green Version]
- Lyu, P.; Settlage, R.E.; Jiang, H. Genome-Wide Identification of Enhancers and Transcription Factors Regulating the Myogenic Differentiation of Bovine Satellite Cells. BMC Genom. 2021, 22, 901. [Google Scholar] [CrossRef]
- Joshi, S.; Davidson, G.; Le Gras, S.; Watanabe, S.; Braun, T.; Mengus, G.; Davidson, I. TEAD Transcription Factors Are Required for Normal Primary Myoblast Differentiation in Vitro and Muscle Regeneration in Vivo. PLoS Genet. 2017, 13, e1006600. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.I.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Langdon, W.B. Performance of Genetic Programming Optimised Bowtie2 on Genome Comparison and Analytic Testing (GCAT) Benchmarks. BioData Min. 2015, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. DeepTools: A Flexible Platform for Exploring Deep-Sequencing Data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N. The UCSC Genome Browser Database: 2019 Update. Nucleic Acids Res. 2019, 47, D853–D858. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Zhang, Y.; Chen, C.; Hu, Q.; Fu, Y.; Xu, L.; Wang, C.; Liu, Y. Identification of Robust and Key Differentially Expressed Genes during C2C12 Cell Myogenesis Based on Multiomics Data. Int. J. Mol. Sci. 2022, 23, 6002. https://doi.org/10.3390/ijms23116002
Zhang S, Zhang Y, Chen C, Hu Q, Fu Y, Xu L, Wang C, Liu Y. Identification of Robust and Key Differentially Expressed Genes during C2C12 Cell Myogenesis Based on Multiomics Data. International Journal of Molecular Sciences. 2022; 23(11):6002. https://doi.org/10.3390/ijms23116002
Chicago/Turabian StyleZhang, Song, Yuanyuan Zhang, Choulin Chen, Qingqing Hu, Yang Fu, Lingna Xu, Chao Wang, and Yuwen Liu. 2022. "Identification of Robust and Key Differentially Expressed Genes during C2C12 Cell Myogenesis Based on Multiomics Data" International Journal of Molecular Sciences 23, no. 11: 6002. https://doi.org/10.3390/ijms23116002
APA StyleZhang, S., Zhang, Y., Chen, C., Hu, Q., Fu, Y., Xu, L., Wang, C., & Liu, Y. (2022). Identification of Robust and Key Differentially Expressed Genes during C2C12 Cell Myogenesis Based on Multiomics Data. International Journal of Molecular Sciences, 23(11), 6002. https://doi.org/10.3390/ijms23116002