
Monoubiquitination in Homeostasis and Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Protein Substrates of Monoubiquitination
2.1. Histones H2A and H2B
2.2. Transcription Factors and Other Transcriptional Regulators
2.3. Enzymes
2.4. Membrane Proteins
3. Cellular Functions of Monoubiquitination
3.1. Transcription
3.2. Endocytosis and Degradation
3.3. Signal Transduction
3.4. Cell Death
3.5. DNA Damage Response (DDR)
4. Diseases Associated with Dysregulated Monoubiquitination
4.1. Fanconi Anemia
4.2. Parkinson’s Disease
5. Roles of Monoubiquitination in Cancer
5.1. Tumor Cell Proliferation and Death
5.2. Tumor Cell Migration, Invasion, and Metastasis
5.3. Cancer Therapy Response
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, L.; Luo, Z.Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Leighton, J.S.; Wang, Y.; Peng, X.; Chen, Z.; Chen, H.; Sun, Y.; Yao, F.; Li, J.; Zhang, H.; et al. Integrated Genomic Analysis of the Ubiquitin Pathway across Cancer Types. Cell Rep. 2018, 23, 213–226.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Zhang, P.; Ma, L. The role of deubiquitinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 589–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Urbe, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chenova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef]
- Komander, D.; Lord, C.J.; Scheel, H.; Swift, S.; Hofmann, K.; Ashworth, A.; Barford, D. The structure of the CYLD USP domain explains its specificity for Lys63-linked polyubiquitin and reveals a B box module. Mol. Cell 2008, 29, 451–464. [Google Scholar] [CrossRef]
- Lin, S.C.; Chung, J.Y.; Lamothe, B.; Rajashankar, K.; Lu, M.; Lo, Y.C.; Lam, A.Y.; Darnay, B.G.; Wu, H. Molecular basis for the unique deubiquitinating activity of the NF-kappaB inhibitor A20. J. Mol. Biol. 2008, 376, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Al-Hakim, A.K.; Zagorska, A.; Chapman, L.; Deak, M.; Peggie, M.; Alessi, D.R. Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem. J. 2008, 411, 249–260. [Google Scholar] [CrossRef] [Green Version]
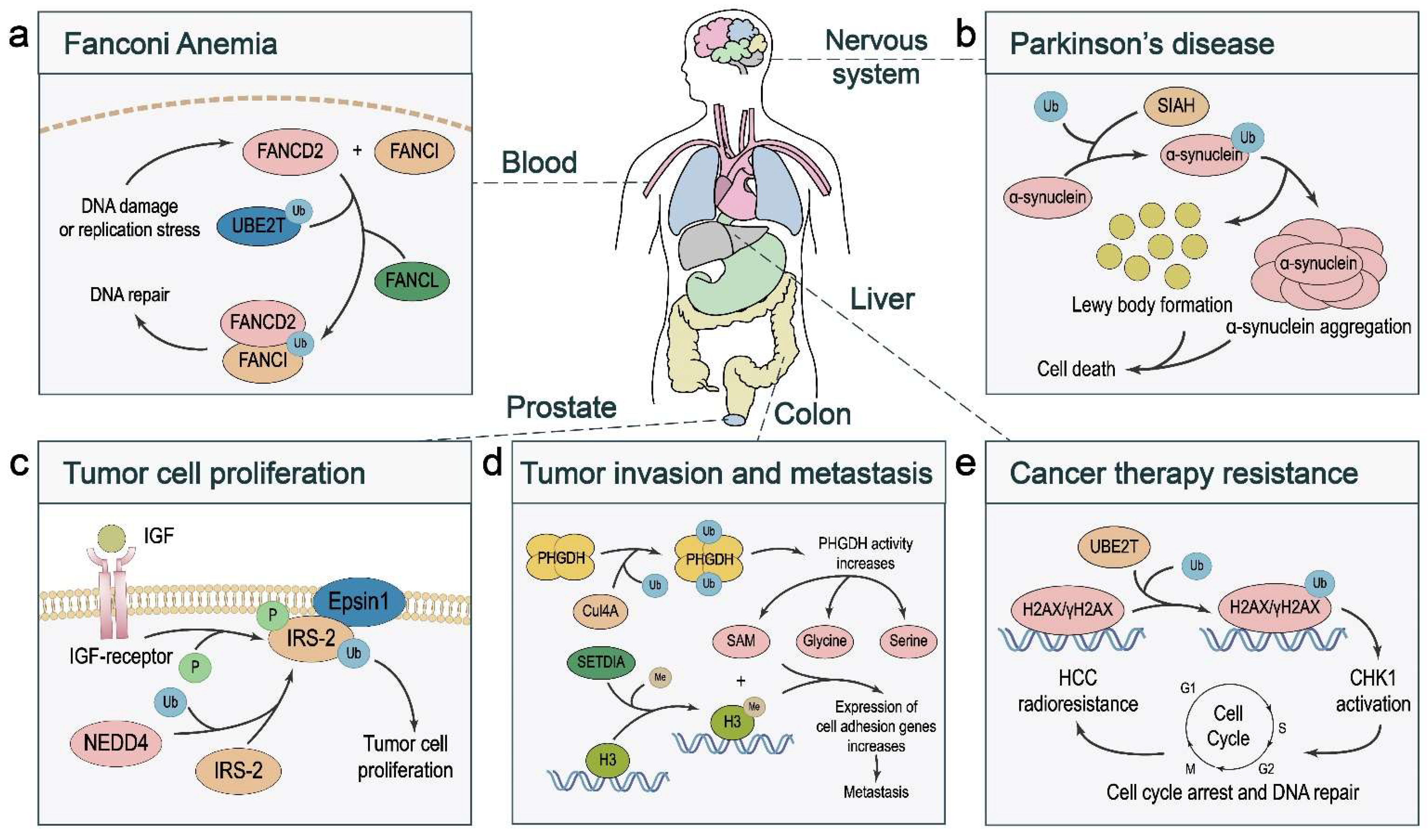
- Zhang, Y.; Yu, H.; Zhang, J.; Gao, H.; Wang, S.; Li, S.; Wei, P.; Liang, J.; Yu, G.; Wang, X.; et al. Cul4A-DDB1-mediated monoubiquitination of phosphoglycerate dehydrogenase promotes colorectal cancer metastasis via increased S-adenosylmethionine. J. Clin. Investig. 2021, 131, e146187. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tokheim, C.; Gu, S.S.; Wang, B.; Tang, Q.; Li, Y.; Traugh, N.; Zeng, Z.; Zhang, Y.; Li, Z.; et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell 2021, 184, 5357–5374.e22. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Schiapparelli, P.; Nguyen, H.N.; Igarashi, A.; Zhang, Q.; Abbadi, S.; Amzel, L.M.; Sesaki, H.; Quinones-Hinojosa, A.; Iijima, M. Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization. Oncogene 2017, 36, 3673–3685. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Yao, F.; Zhou, Z.; Kim, J.; Hang, Q.; Xiao, Z.; Ton, B.N.; Chang, L.; Liu, N.; Zeng, L.; Wang, W.; et al. SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat. Commun. 2018, 9, 2269. [Google Scholar] [CrossRef]
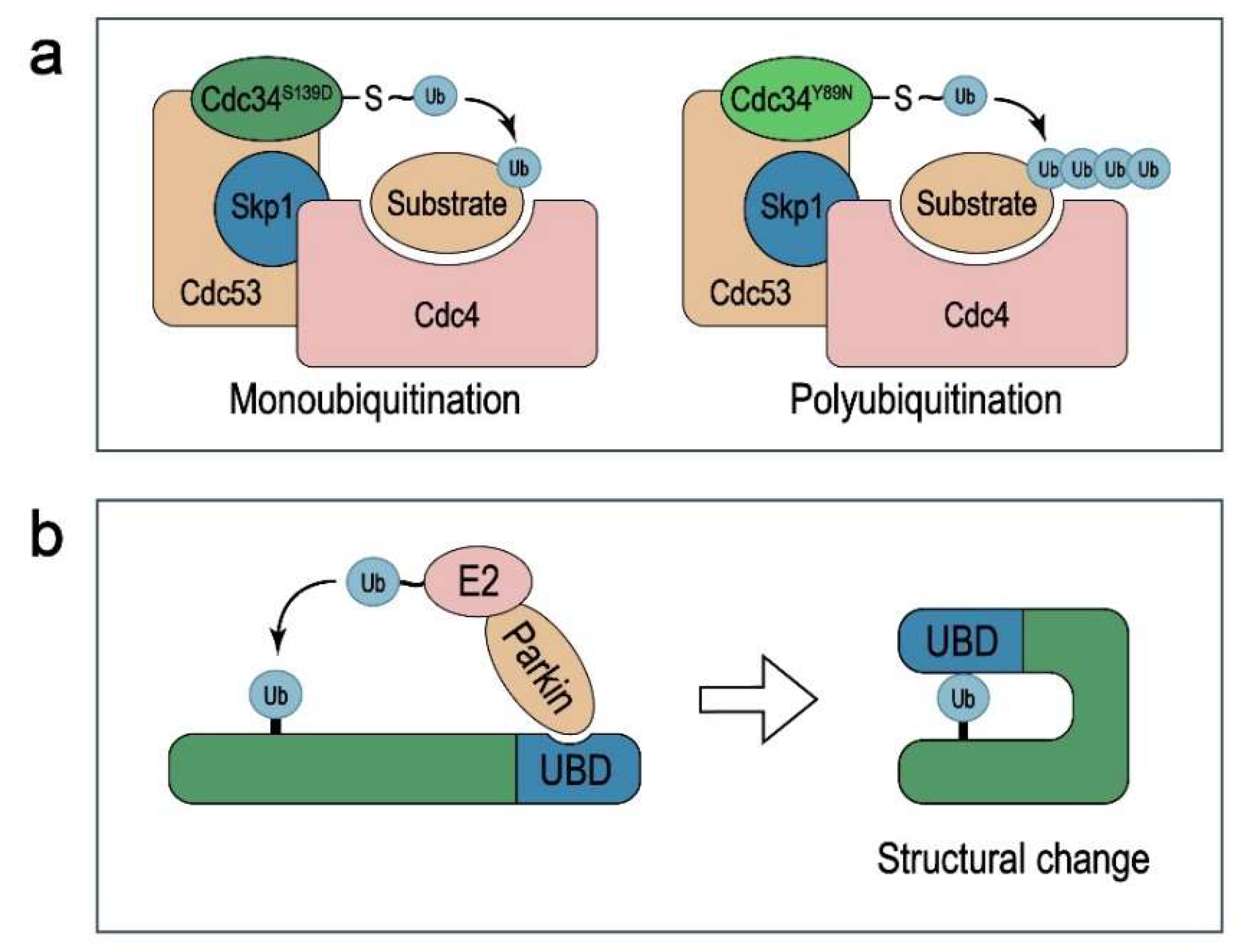
- Sadowski, M.; Sarcevic, B. Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the E2 catalytic core and amino acid residues proximal to the lysine. Cell Div. 2010, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, M.; Suryadinata, R.; Lai, X.; Heierhorst, J.; Sarcevic, B. Molecular basis for lysine specificity in the yeast ubiquitin-conjugating enzyme Cdc34. Mol. Cell Biol. 2010, 30, 2316–2329. [Google Scholar] [CrossRef] [Green Version]
- Fallon, L.; Belanger, C.M.; Corera, A.T.; Kontogiannea, M.; Regan-Klapisz, E.; Moreau, F.; Voortman, J.; Haber, M.; Rouleau, G.; Thorarinsdottir, T.; et al. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat. Cell Biol. 2006, 8, 834–842. [Google Scholar] [CrossRef]
- Fukushima, T.; Yoshihara, H.; Furuta, H.; Kamei, H.; Hakuno, F.; Luan, J.; Duan, C.; Saeki, Y.; Tanaka, K.; Iemura, S.; et al. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat. Commun. 2015, 6, 6780. [Google Scholar] [CrossRef] [Green Version]
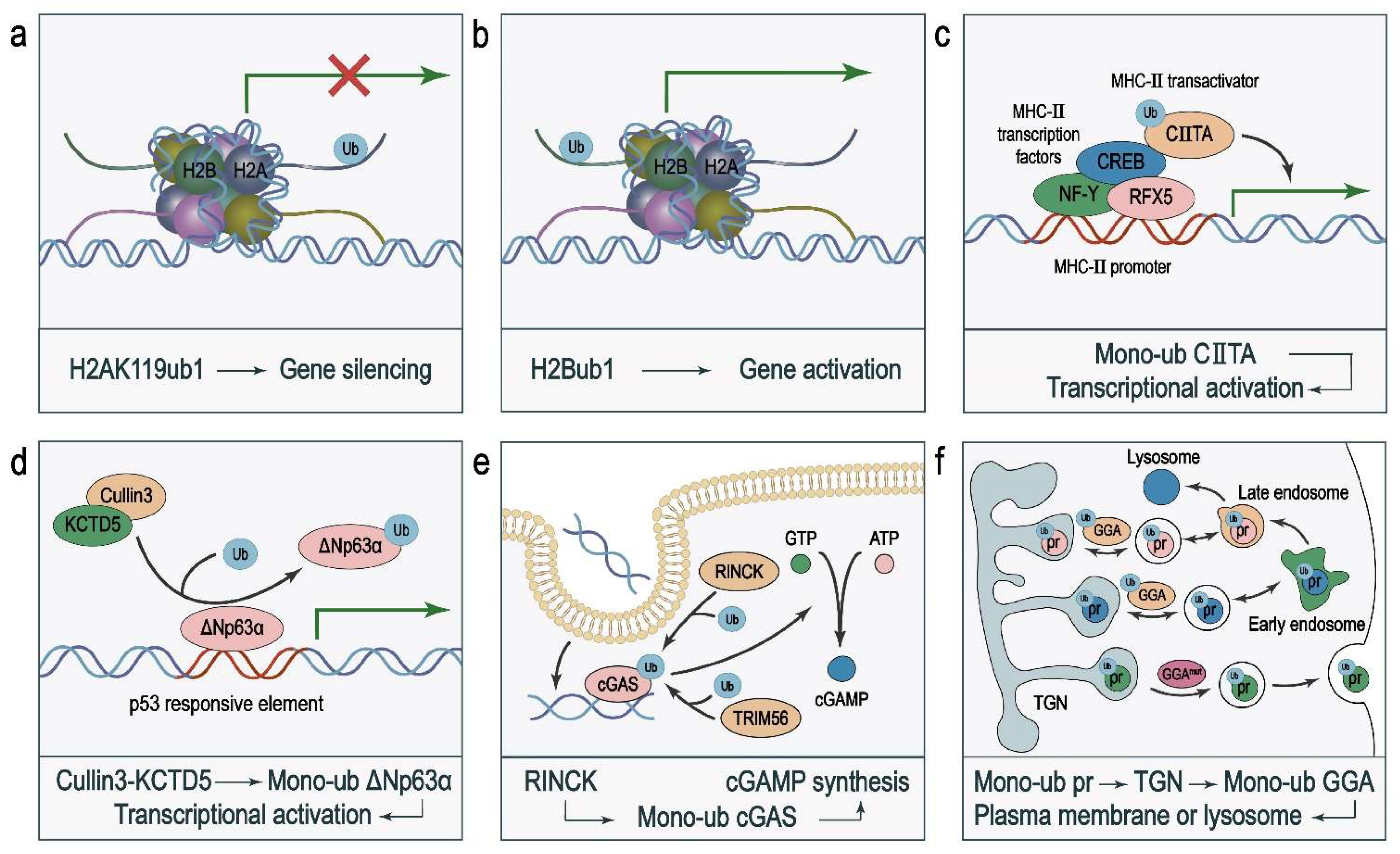
- Greer, S.F.; Zika, E.; Conti, B.; Zhu, X.S.; Ting, J.P. Enhancement of CIITA transcriptional function by ubiquitin. Nat. Immunol. 2003, 4, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; Di Fiore, P.P.; Dikic, I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 2003, 5, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Isasa, M.; Katz, E.J.; Kim, W.; Yugo, V.; Gonzalez, S.; Kirkpatrick, D.S.; Thomson, T.M.; Finley, D.; Gygi, S.P.; Crosas, B. Monoubiquitination of RPN10 regulates substrate recruitment to the proteasome. Mol. Cell 2010, 38, 733–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelzer, C.; Cabalzar, K.; Wolf, A.; Gonzalez, M.; Lenz, G.; Thome, M. The protease activity of the paracaspase MALT1 is controlled by monoubiquitination. Nat. Immunol. 2013, 14, 337–345. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Xiao, Z.; Lei, G.; Lee, H.; Koppula, P.; Cheng, W.; Mao, C.; Zhuang, L.; et al. H2A Monoubiquitination Links Glucose Availability to Epigenetic Regulation of the Endoplasmic Reticulum Stress Response and Cancer Cell Death. Cancer Res. 2020, 80, 2243–2256. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563. [Google Scholar] [CrossRef]
- Zhou, F.; Xie, F.; Jin, K.; Zhang, Z.; Clerici, M.; Gao, R.; van Dinther, M.; Sixma, T.K.; Huang, H.; Zhang, L.; et al. USP4 inhibits SMAD4 monoubiquitination and promotes activin and BMP signaling. EMBO J. 2017, 36, 1623–1639. [Google Scholar] [CrossRef]
- Ishiai, M. Regulation of the Fanconi Anemia DNA Repair Pathway by Phosphorylation and Monoubiquitination. Genes 2021, 12, 1763. [Google Scholar] [CrossRef]
- Li, L.; Tan, W.; Deans, A.J. Structural insight into FANCI-FANCD2 monoubiquitination. Essays Biochem. 2020, 64, 807–817. [Google Scholar]
- Gregory, R.C.; Taniguchi, T.; D’Andrea, A.D. Regulation of the Fanconi anemia pathway by monoubiquitination. Semin. Cancer Biol. 2003, 13, 77–82. [Google Scholar] [CrossRef]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Woelk, T.; Oldrini, B.; Maspero, E.; Confalonieri, S.; Cavallaro, E.; Di Fiore, P.P.; Polo, S. Molecular mechanisms of coupled monoubiquitination. Nat. Cell Biol. 2006, 8, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.E.; Riley, B.E.; Shaler, T.A.; Trevino, R.S.; Becker, C.H.; Schulman, H.; Kopito, R.R. Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods 2011, 8, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Talbert, P.B.; Henikoff, S. Histone variants at a glance. J. Cell Sci. 2021, 134, jcs244749. [Google Scholar] [CrossRef]
- Clayton, A.L.; Hazzalin, C.A.; Mahadevan, L.C. Enhanced histone acetylation and transcription: A dynamic perspective. Mol. Cell 2006, 23, 289–296. [Google Scholar] [CrossRef]
- Li, W.; Tian, W.; Yuan, G.; Deng, P.; Sengupta, D.; Cheng, Z.; Cao, Y.; Ren, J.; Qin, Y.; Zhou, Y.; et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 2021, 590, 498–503. [Google Scholar] [CrossRef]
- Armache, A.; Yang, S.; Martinez de Paz, A.; Robbins, L.E.; Durmaz, C.; Cheong, J.Q.; Ravishankar, A.; Daman, A.W.; Ahimovic, D.J.; Klevorn, T.; et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 2020, 583, 852–857. [Google Scholar] [CrossRef]
- Zhang, Y. Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev. 2003, 17, 2733–2740. [Google Scholar] [CrossRef] [Green Version]
- Mattiroli, F.; Penengo, L. Histone Ubiquitination: An Integrative Signaling Platform in Genome Stability. Trends Genet. 2021, 37, 566–581. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Gao, B.; Sun, Y.; Cao, L.; Genardi, S.M.; Wang, C.R.; Li, H.; Sun, Z.; Yang, Y.; et al. USP22 controls iNKT immunity through MED1 suppression of histone H2A monoubiquitination. J. Exp. Med. 2020, 217, e20182218. [Google Scholar] [CrossRef] [PubMed]
- Tamburri, S.; Lavarone, E.; Fernandez-Perez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackledge, N.P.; Fursova, N.A.; Kelley, J.R.; Huseyin, M.K.; Feldmann, A.; Klose, R.J. PRC1 Catalytic Activity Is Central to Polycomb System Function. Mol. Cell 2020, 77, 857–874.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fursova, N.A.; Turberfield, A.H.; Blackledge, N.P.; Findlater, E.L.; Lastuvkova, A.; Huseyin, M.K.; Dobrinic, P.; Klose, R.J. BAP1 constrains pervasive H2AK119ub1 to control the transcriptional potential of the genome. Genes Dev. 2021, 35, 749–770. [Google Scholar] [CrossRef]
- Kolovos, P.; Nishimura, K.; Sankar, A.; Sidoli, S.; Cloos, P.A.; Helin, K.; Christensen, J. PR-DUB maintains the expression of critical genes through FOXK1/2- and ASXL1/2/3-dependent recruitment to chromatin and H2AK119ub1 deubiquitination. Genome Res. 2020, 30, 1119–1130. [Google Scholar] [CrossRef]
- Fujino, T.; Goyama, S.; Sugiura, Y.; Inoue, D.; Asada, S.; Yamasaki, S.; Matsumoto, A.; Yamaguchi, K.; Isobe, Y.; Tsuchiya, A.; et al. Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway. Nat. Commun. 2021, 12, 1826. [Google Scholar] [CrossRef]
- Minsky, N.; Shema, E.; Field, Y.; Schuster, M.; Segal, E.; Oren, M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat. Cell Biol. 2008, 10, 483–488. [Google Scholar] [CrossRef]
- Xie, W.; Nagarajan, S.; Baumgart, S.J.; Kosinsky, R.L.; Najafova, Z.; Kari, V.; Hennion, M.; Indenbirken, D.; Bonn, S.; Grundhoff, A.; et al. RNF40 regulates gene expression in an epigenetic context-dependent manner. Genome Biol. 2017, 18, 32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wang, J.; Tong, T.R.; Wu, X.; Nelson, R.; Yuan, Y.C.; Reno, T.; Liu, Z.; Yun, X.; Kim, J.Y.; et al. Loss of H2B monoubiquitination is associated with poor-differentiation and enhanced malignancy of lung adenocarcinoma. Int. J. Cancer 2017, 141, 766–777. [Google Scholar] [CrossRef]
- Jing, Y.Y.; Cai, F.F.; Zhang, L.; Han, J.; Yang, L.; Tang, F.; Li, Y.B.; Chang, J.F.; Sun, F.; Yang, X.M.; et al. Epigenetic regulation of the Warburg effect by H2B monoubiquitination. Cell Death Differ. 2020, 27, 1660–1676. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, K.E.; Ponnamperuma, R.M.; Yamashita, T.; Tokino, T.; Lee, L.A.; Young, M.F.; Weinberg, W.C. deltaNp63alpha functions as both a positive and a negative transcriptional regulator and blocks in vitro differentiation of murine keratinocytes. Oncogene 2003, 22, 3635–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Peng, Y.; Fan, S.; Chen, Y.; Zheng, X.; Li, C. Cullin3/KCTD5 induces monoubiquitination of DeltaNp63alpha and impairs its activity. FEBS Lett. 2018, 592, 2334–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadd, M.S.; Bulatov, E.; Ciulli, A. Serendipitous SAD Solution for DMSO-Soaked SOCS2-ElonginC-ElonginB Crystals Using Covalently Incorporated Dimethylarsenic: Insights into Substrate Receptor Conformational Flexibility in Cullin RING Ligases. PLoS ONE 2015, 10, e0131218. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, A.; de Vries-Smits, A.M.; Brenkman, A.B.; van Triest, M.H.; van den Broek, N.; Colland, F.; Maurice, M.M.; Burgering, B.M. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 2006, 8, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Juilland, M.; Thome, M. Holding All the CARDs: How MALT1 Controls CARMA/CARD-Dependent Signaling. Front. Immunol. 2018, 9, 1927. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, C.; Leder, L.; Blank, J.; Bernardi, A.; Melkko, S.; Decock, A.; D’Arcy, A.; Villard, F.; Erbel, P.; Hughes, N.; et al. Structural determinants of MALT1 protease activity. J. Mol. Biol. 2012, 419, 4–21. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Fang, J. E3-Independent Constitutive Monoubiquitination Complements Histone Methyltransferase Activity of SETDB1. Mol. Cell 2016, 62, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Kim, C.; Shin, W.J.; Sklan, E.H.; Eoh, H.; Jung, J.U. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 2018, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.S.; Zhang, Z.Y.; Cai, H.; Zhao, M.; Mao, J.; Dai, J.; Xia, T.; Zhang, X.M.; Li, T. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Liu, P. Cytosolic DNA sensing by cGAS: Regulation, function, and human diseases. Signal Transduct. Target. Ther. 2021, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Miller, L.D. RAS Mutations and Oncogenesis: Not all RAS Mutations are Created Equally. Front. Genet. 2011, 2, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, A.T.; Carracedo, A.; Locasale, J.W.; Anastasiou, D.; Takeuchi, K.; Kahoud, E.R.; Haviv, S.; Asara, J.M.; Pandolfi, P.P.; Cantley, L.C. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal 2011, 4, ra13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Azzo, A.; Bongiovanni, A.; Nastasi, T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 2005, 6, 429–441. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [Green Version]
- Bonifacino, J.S. The GGA proteins: Adaptors on the move. Nat. Rev. Mol. Cell Biol. 2004, 5, 23–32. [Google Scholar] [CrossRef]
- Prag, G.; Lee, S.; Mattera, R.; Arighi, C.N.; Beach, B.M.; Bonifacino, J.S.; Hurley, J.H. Structural mechanism for ubiquitinated-cargo recognition by the Golgi-localized, gamma-ear-containing, ADP-ribosylation-factor-binding proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 2334–2339. [Google Scholar] [CrossRef] [Green Version]
- Scott, P.M.; Bilodeau, P.S.; Zhdankina, O.; Winistorfer, S.C.; Hauglund, M.J.; Allaman, M.M.; Kearney, W.R.; Robertson, A.D.; Boman, A.L.; Piper, R.C. GGA proteins bind ubiquitin to facilitate sorting at the trans-Golgi network. Nat. Cell Biol. 2004, 6, 252–259. [Google Scholar] [CrossRef]
- Li, D.; Zhou, W.; Pang, J.; Tang, Q.; Zhong, B.; Shen, C.; Xiao, L.; Hou, T. A magic drug target: Androgen receptor. Med. Res. Rev. 2019, 39, 1485–1514. [Google Scholar] [CrossRef]
- Burgdorf, S.; Leister, P.; Scheidtmann, K.H. TSG101 interacts with apoptosis-antagonizing transcription factor and enhances androgen receptor-mediated transcription by promoting its monoubiquitination. J. Biol. Chem. 2004, 279, 17524–17534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
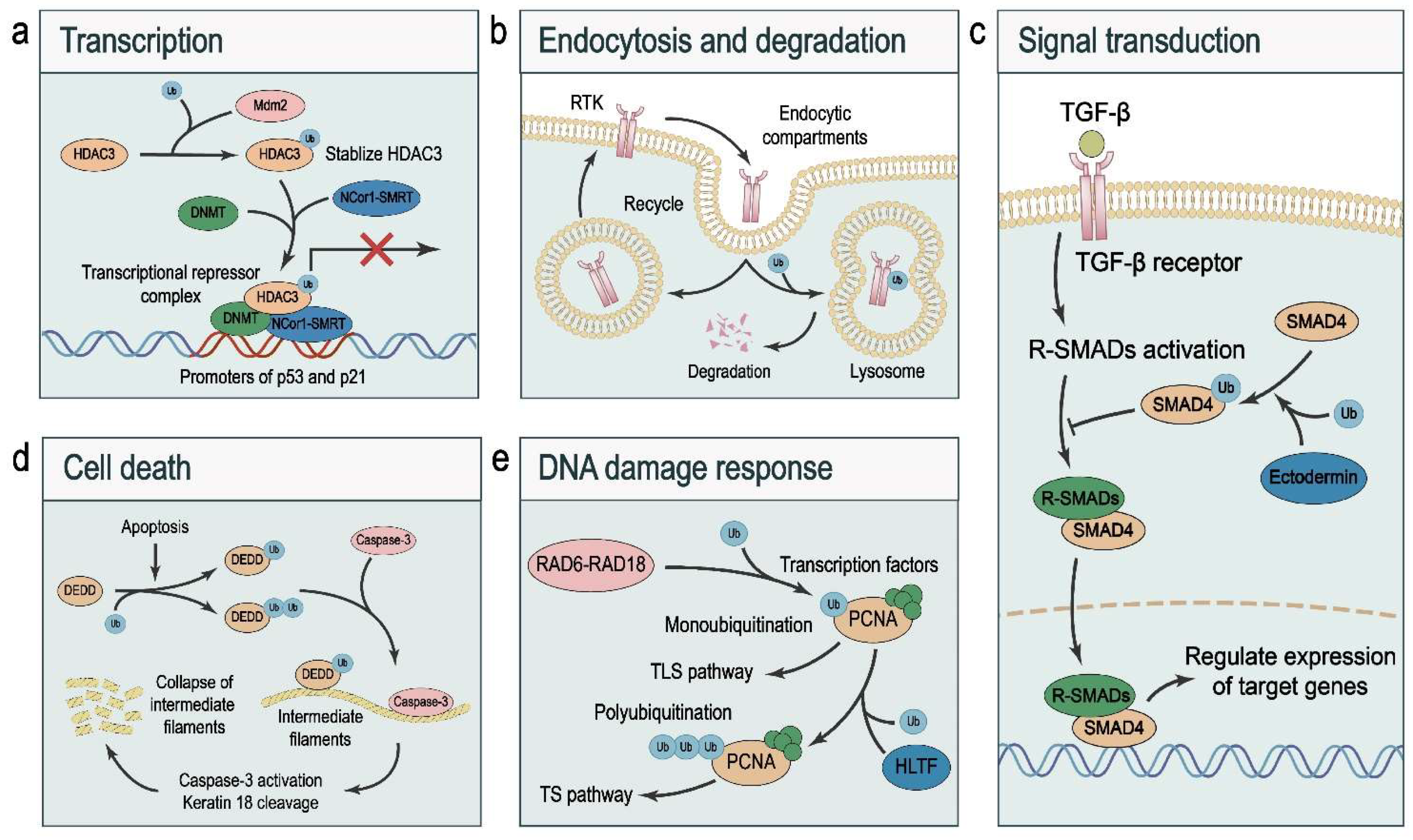
- Choi, Y.M.; An, S.; Bae, S.; Jung, J.H. Mdm2 is required for HDAC3 monoubiquitination and stability. Biochem. Biophys. Res. Commun. 2019, 517, 353–358. [Google Scholar] [CrossRef]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neben, C.L.; Lo, M.; Jura, N.; Klein, O.D. Feedback regulation of RTK signaling in development. Dev. Biol. 2019, 447, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Waterman, H.; Yarden, Y. Molecular mechanisms underlying endocytosis and sorting of ErbB receptor tyrosine kinases. FEBS Lett. 2001, 490, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Boudjadi, S.; Chatterjee, B.; Sun, W.; Vemu, P.; Barr, F.G. The expression and function of PAX3 in development and disease. Gene 2018, 666, 145–157. [Google Scholar] [CrossRef]
- Boutet, S.C.; Disatnik, M.H.; Chan, L.S.; Iori, K.; Rando, T.A. Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell 2007, 130, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Deveraux, Q.; Ustrell, V.; Pickart, C.; Rechsteiner, M. A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem. 1994, 269, 7059–7061. [Google Scholar] [CrossRef]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647. [Google Scholar] [CrossRef] [Green Version]
- Sakata, E.; Bohn, S.; Mihalache, O.; Kiss, P.; Beck, F.; Nagy, I.; Nickell, S.; Tanaka, K.; Saeki, Y.; Forster, F.; et al. Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 1479–1484. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Yu, X.; Yi, X.; Wu, K.; Dwabe, S.; Atefi, M.; Elshimali, Y.; Kemp, K.T., 2nd; Bhat, K.; Haro, J.; et al. Aberrant Phosphorylation of SMAD4 Thr277-Mediated USP9x-SMAD4 Interaction by Free Fatty Acids Promotes Breast Cancer Metastasis. Cancer Res. 2017, 77, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Zacchigna, L.; Cordenonsi, M.; Soligo, S.; Adorno, M.; Rugge, M.; Piccolo, S. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 2005, 121, 87–99. [Google Scholar] [CrossRef]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. Regulation of TGF-beta Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 2014, 3, 981–993. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Bauer, A.; Zhang, L.; Selinger, D.W.; Lu, C.X.; Ten Dijke, P. Fine-tuning BMP7 signalling in adipogenesis by UBE2O/E2-230K-mediated monoubiquitination of SMAD6. EMBO J. 2013, 32, 996–1007. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Parsons, R. Discovery of the PTEN Tumor Suppressor and Its Connection to the PI3K and AKT Oncogenes. Cold Spring Harb. Perspect. Med. 2020, 10, a036129. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Barriopedro, I.; Bosch-Presegue, L.; Marazuela-Duque, A.; de la Torre, C.; Colomer, C.; Vazquez, B.N.; Fuhrmann, T.; Martinez-Pastor, B.; Lu, W.; Braun, T.; et al. SIRT6-dependent cysteine monoubiquitination in the PRE-SET domain of Suv39h1 regulates the NF-kappaB pathway. Nat. Commun. 2018, 9, 101. [Google Scholar] [CrossRef]
- Fuseya, Y.; Fujita, H.; Kim, M.; Ohtake, F.; Nishide, A.; Sasaki, K.; Saeki, Y.; Tanaka, K.; Takahashi, R.; Iwai, K. The HOIL-1L ligase modulates immune signalling and cell death via monoubiquitination of LUBAC. Nat. Cell Biol. 2020, 22, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Schwartz, L.M. Autophagic Cell Death During Development—Ancient and Mysterious. Front. Cell Dev. Biol. 2021, 9, 656370. [Google Scholar] [CrossRef]
- Lee, J.C.; Peter, M.E. Regulation of apoptosis by ubiquitination. Immunol. Rev. 2003, 193, 39–47. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Kayastha, G.; McGovern, N.B.; Hwang, S.G.; Marcu, M.G.; Trepel, J.; Cai, S.Y.; Marchesi, V.T.; Neckers, L. Caspase-dependent deubiquitination of monoubiquitinated nucleosomal histone H2A induced by diverse apoptogenic stimuli. Cell Death Differ. 2001, 8, 1182–1196. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.C.; Schickling, O.; Stegh, A.H.; Oshima, R.G.; Dinsdale, D.; Cohen, G.M.; Peter, M.E. DEDD regulates degradation of intermediate filaments during apoptosis. J. Cell Biol. 2002, 158, 1051–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.M.; Martin, D.A.; Zheng, L.; Ng, S.Y.; Bertin, J.; Cohen, J.; Lenardo, M.J. Death-effector filaments: Novel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol. 1998, 141, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prenzel, T.; Begus-Nahrmann, Y.; Kramer, F.; Hennion, M.; Hsu, C.; Gorsler, T.; Hintermair, C.; Eick, D.; Kremmer, E.; Simons, M.; et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011, 71, 5739–5753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zhang, Y.; Redon, C.E.; Reinhold, W.C.; Chen, A.P.; Fogli, L.K.; Holbeck, S.L.; Parchment, R.E.; Hollingshead, M.; Tomaszewski, J.E.; et al. Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS ONE 2017, 12, e0171582. [Google Scholar] [CrossRef]
- Williamson, J.; Hughes, C.M.; Burke, G.; Davison, G.W. A combined gamma-H2AX and 53BP1 approach to determine the DNA damage-repair response to exercise in hypoxia. Free Radic. Biol. Med. 2020, 154, 9–17. [Google Scholar] [CrossRef]
- Luczak, M.W.; Zhitkovich, A. Monoubiquitinated gamma-H2AX: Abundant product and specific biomarker for non-apoptotic DNA double-strand breaks. Toxicol. Appl. Pharmacol. 2018, 355, 238–246. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair 2010, 9, 1219–1228. [Google Scholar] [CrossRef]
- Alcon, P.; Shakeel, S.; Chen, Z.A.; Rappsilber, J.; Patel, K.J.; Passmore, L.A. FANCD2-FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat. Struct. Mol. Biol. 2020, 27, 240–248. [Google Scholar] [CrossRef]
- Alvarez, V.; Frattini, C.; Sacristan, M.P.; Gallego-Sanchez, A.; Bermejo, R.; Bueno, A. PCNA Deubiquitylases Control DNA Damage Bypass at Replication Forks. Cell Rep. 2019, 29, 1323–1335 e5. [Google Scholar] [CrossRef] [Green Version]
- Leung, W.; Baxley, R.M.; Moldovan, G.L.; Bielinsky, A.K. Mechanisms of DNA Damage Tolerance: Post-Translational Regulation of PCNA. Genes 2018, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, Y.; Mitsuyuki, S.; Kanao, R.; Hishiki, A.; Hashimoto, H.; Masutani, C. Regulation of HLTF-mediated PCNA polyubiquitination by RFC and PCNA monoubiquitination levels determines choice of damage tolerance pathway. Nucleic Acids Res. 2018, 46, 11340–11356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.T.; Nijman, S.M.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [CrossRef]
- Wu, L.; Crawley, C.D.; Garofalo, A.; Nichols, J.W.; Campbell, P.A.; Khramtsova, G.F.; Olopade, O.I.; Weichselbaum, R.R.; Yamini, B. p50 mono-ubiquitination and interaction with BARD1 regulates cell cycle progression and maintains genome stability. Nat. Commun. 2020, 11, 5007. [Google Scholar] [CrossRef] [PubMed]
- Tsui, V.; Crismani, W. The Fanconi Anemia Pathway and Fertility. Trends Genet. 2019, 35, 199–214. [Google Scholar] [CrossRef] [PubMed]
- van Twest, S.; Murphy, V.J.; Hodson, C.; Tan, W.; Swuec, P.; O’Rourke, J.J.; Heierhorst, J.; Crismani, W.; Deans, A.J. Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol. Cell 2017, 65, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; van Twest, S.; Leis, A.; Bythell-Douglas, R.; Murphy, V.J.; Sharp, M.; Parker, M.W.; Crismani, W.; Deans, A.J. Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. Elife 2020, 9, e54128. [Google Scholar] [CrossRef]
- Liang, F.; Miller, A.S.; Longerich, S.; Tang, C.; Maranon, D.; Williamson, E.A.; Hromas, R.; Wiese, C.; Kupfer, G.M.; Sung, P. DNA requirement in FANCD2 deubiquitination by USP1-UAF1-RAD51AP1 in the Fanconi anemia DNA damage response. Nat. Commun. 2019, 10, 2849. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Yang, M.; Xu, Y.; Heisner, J.S.; Sun, J.; Stowe, D.F.; Kwok, W.M.; Camara, A.K.S. Peroxynitrite nitrates adenine nucleotide translocase and voltage-dependent anion channel 1 and alters their interactions and association with hexokinase II in mitochondria. Mitochondrion 2019, 46, 380–392. [Google Scholar] [CrossRef]
- Ham, S.J.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef]
- Sutherland, R.L.; Prall, O.W.; Watts, C.K.; Musgrove, E.A. Estrogen and progestin regulation of cell cycle progression. J. Mammary Gland Biol. Neoplasia 1998, 3, 63–72. [Google Scholar] [CrossRef]
- Wang, S.; Luo, H.; Wang, C.; Sun, H.; Sun, G.; Sun, N.; Zeng, K.; Song, H.; Zou, R.; Zhou, T.; et al. RNF8 identified as a co-activator of estrogen receptor alpha promotes cell growth in breast cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Yi, F.; Wang, Z.; Guo, Q.; Liu, J.; Bai, N.; Li, X.; Dong, X.; Ren, L.; Cao, L.; et al. The Functions of DNA Damage Factor RNF8 in the Pathogenesis and Progression of Cancer. Int. J. Biol. Sci. 2019, 15, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhao, C.; Kharman-Biz, A.; Zhuang, T.; Jonsson, P.; Liang, N.; Williams, C.; Lin, C.Y.; Qiao, Y.; Zendehdel, K.; et al. The atypical ubiquitin ligase RNF31 stabilizes estrogen receptor alpha and modulates estrogen-stimulated breast cancer cell proliferation. Oncogene 2014, 33, 4340–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Wang, R.; Zhang, Y.; Li, X.; Gan, Y.; Gao, F.; Rong, P.; Wang, W.; Li, W. The role of ubiquitination and deubiquitination in tumor invasion and metastasis. Int. J. Biol. Sci. 2022, 18, 2292–2303. [Google Scholar] [CrossRef]
- Reid, M.A.; Allen, A.E.; Liu, S.; Liberti, M.V.; Liu, P.; Liu, X.; Dai, Z.; Gao, X.; Wang, Q.; Liu, Y.; et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun. 2018, 9, 5442. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Li, G.; Wen, C.; Zeng, T.; Fan, Y.; Liu, C.; Fu, G.F.; Xie, C.; Lin, Q.; Xie, L.; et al. Monoubiquitination of p120-catenin is essential for TGFbeta-induced epithelial-mesenchymal transition and tumor metastasis. Sci. Adv. 2020, 6, eaay9819. [Google Scholar] [CrossRef]
- Inui, M.; Manfrin, A.; Mamidi, A.; Martello, G.; Morsut, L.; Soligo, S.; Enzo, E.; Moro, S.; Polo, S.; Dupont, S.; et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat. Cell Biol. 2011, 13, 1368–1375. [Google Scholar] [CrossRef]
- Pstrag, N.; Ziemnicka, K.; Bluyssen, H.; Wesoly, J. Thyroid cancers of follicular origin in a genomic light: In-depth overview of common and unique molecular marker candidates. Mol. Cancer 2018, 17, 116. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Tian, Z.; Liao, X.; Wu, G. SOX13/TRIM11/YAP axis promotes the proliferation, migration and chemoresistance of anaplastic thyroid cancer. Int. J. Biol. Sci. 2021, 17, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhu, Z.; Li, W.; Shen, M.; Cao, C.; Sun, Q.; Guo, Z.; Liu, L.; Wu, D. UBE2T-regulated H2AX monoubiquitination induces hepatocellular carcinoma radioresistance by facilitating CHK1 activation. J. Exp. Clin. Cancer Res. 2020, 39, 222. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef]
- Lam, P.Y.; Kobayashi, T.; Soon, M.; Zeng, B.; Dolcetti, R.; Leggatt, G.; Thomas, R.; Mattarollo, S.R. NKT Cell-Driven Enhancement of Antitumor Immunity Induced by Clec9a-Targeted Tailorable Nanoemulsion. Cancer Immunol. Res. 2019, 7, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front. Immunol. 2018, 9, 1838. [Google Scholar] [CrossRef]
- Kojo, S.; Elly, C.; Harada, Y.; Langdon, W.Y.; Kronenberg, M.; Liu, Y.C. Mechanisms of NKT cell anergy induction involve Cbl-b-promoted monoubiquitination of CARMA1. Proc. Natl. Acad. Sci. USA 2009, 106, 17847–17851. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zhou, D.; Yao, Y.; Sun, Y.; Yao, F.; Ma, L. Monoubiquitination in Homeostasis and Cancer. Int. J. Mol. Sci. 2022, 23, 5925. https://doi.org/10.3390/ijms23115925
Chen Y, Zhou D, Yao Y, Sun Y, Yao F, Ma L. Monoubiquitination in Homeostasis and Cancer. International Journal of Molecular Sciences. 2022; 23(11):5925. https://doi.org/10.3390/ijms23115925
Chicago/Turabian StyleChen, Yujie, Dandan Zhou, Yinan Yao, Yutong Sun, Fan Yao, and Li Ma. 2022. "Monoubiquitination in Homeostasis and Cancer" International Journal of Molecular Sciences 23, no. 11: 5925. https://doi.org/10.3390/ijms23115925
APA StyleChen, Y., Zhou, D., Yao, Y., Sun, Y., Yao, F., & Ma, L. (2022). Monoubiquitination in Homeostasis and Cancer. International Journal of Molecular Sciences, 23(11), 5925. https://doi.org/10.3390/ijms23115925