
Development and Characterization of a Factor V-Deficient CRISPR Cell Model for the Correction of Mutations

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
2.1. NHEJ-PD Generates an FV KO Cellular Model for the HepG2 Cell Line
2.2. Generation of an FV TT Cellular Model Derived from the KO HepG2 Cell Line
2.3. Comparison and Characterization of the Three Cell Lines Obtained: WT HepG2, KO HepG2 and TT HepG2
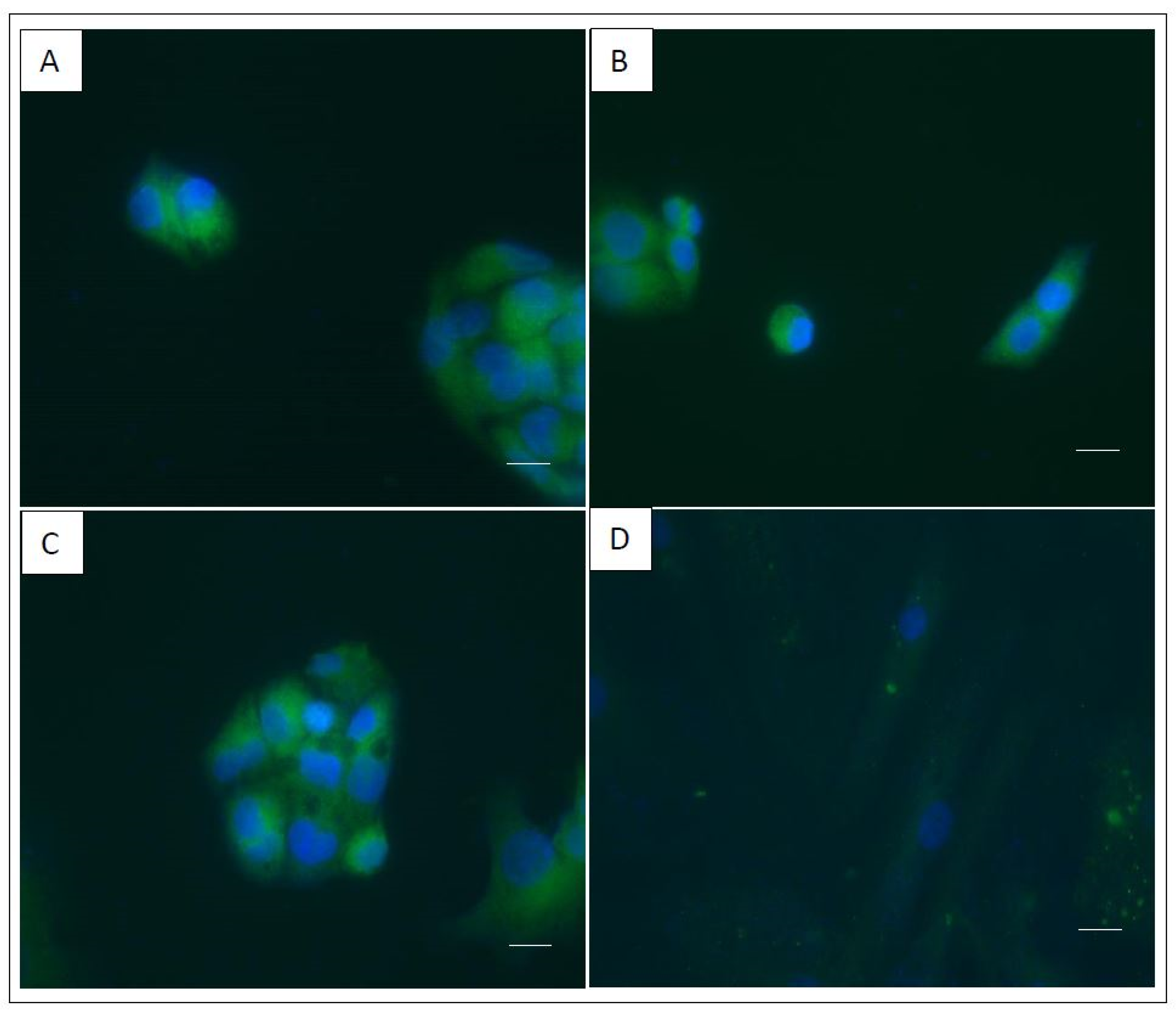
2.4. Functional FV Synthesized by the Three Cell Lines
2.5. Analysis of Off-Target TT Guides
3. Discussion
4. Materials and Methods
4.1. Clinical Characteristics of the Patient
4.2. Mutational Analysis
4.3. Cell Lines
4.4. Design and Generation of CRISPR-Cas9 Tools
4.5. Electroporation of the HepG2 Cell Line with RNPs
4.6. Sequencing Analysis
4.7. Characterization of HepG2 Cell Lines
4.8. Factor V Analysis and Quantification
4.9. Off-Target Analysis
4.10. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Segers, K.; Dahlbäck, B.; Nicolaes, G.A.F. Coagulation factor V and thrombophilia: Background and mechanisms. Thromb. Haemost. 2007, 98, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, S.; Reglińska-Matveyev, N.; Gierula, M.; Camire, R.M.; Crawley, J.T.B.; Lane, B.A.; Ahnström, J. Factor V has an anticoagulant cofactor activity that targets the early phase of coagulation. J. Biol. Chem. 2017, 292, 9335–9344. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, P.; Rosing, J.; Duckers, C.; Hackeng, T.M.; Simioni, P.; Castoldi, E. Factor V Has Anticoagulant Activity in Plasma in the Presence of TFPIα: Difference between FV1 and FV2. Thromb. Haemost. 2018, 118, 1194–1202. [Google Scholar] [CrossRef]
- Tabibian, S.; Shiravand, Y.; Shams, M.; Safa, M.; Gholami, M.S.; Heydari, F.; Ahmadi, A.; Rashidpanah, J.; Dorgalaleh, A. A Comprehensive Overview of Coagulation Factor V and Congenital Factor V Deficiency. Semin. Thromb. Hemost. 2019, 45, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Stoilova-McPhie, S.; Parmenter, C.D.J.; Segers, K.; Villoutreix, B.O.; Nicolaes, G.A.F. Defining the structure of membrane-bound human blood coagulation factor Va. J. Thromb. Haemost. 2008, 6, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Ruben, E.A.; Rau, M.J.; Fitzpatrick, J.A.J.; Di Cera, E. Cryo-EM structures of human coagulation factors V and Va. Blood 2021, 137, 3137–3144. [Google Scholar] [CrossRef]
- Lippi, G.; Favaloro, E.J.; Montagnana, M.; Manzato, F.; Guidi, G.C.; Franchini, M. Inherited and acquired factor V deficiency. Blood Coagul. Fibrinolysis 2011, 22, 160–166. [Google Scholar] [CrossRef]
- Thalji, N.; Camire, R. Parahemophilia: New Insights into Factor V Deficiency. Semin. Thromb. Hemost. 2013, 39, 607–612. [Google Scholar] [CrossRef]
- Congenital Factor V Deficiency. The Portal for Rare Diseases and Orphan Drugs Orphanet. Available online: https://www.orpha.net/consor/cgi-bin/index.php?lng=EN (accessed on 26 March 2022).
- Peyvandi, F.; Di Michele, D.; Bolton-Maggs, P.H.B.; Lee, C.A.; Tripodi, A.; Srivastava, A. Project on Consensus Definitions in Rare Bleeeding Disorders of the Factor VIII/Factor IX Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Classification of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity: Classification of rare bleeding disorders. J. Thromb. Haemost. 2012, 10, 1938–1943. [Google Scholar] [CrossRef]
- Jain, S.; Acharya, S.S. Management of rare coagulation disorders in 2018. Transfus. Apher. Sci. 2018, 57, 705–712. [Google Scholar] [CrossRef]
- Asselta, R.; Peyvandi, F. Factor V Deficiency. Semin. Thromb. Hemost. 2009, 35, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Heger, A.; Svae, T.E.; Neisser-Svae, A.; Jordan, S.; Behizad, M.; Römisch, J. Biochemical quality of the pharmaceutically licensed plasma OctaplasLG® after implementation of a novel prion protein (PrPSc) removal technology and reduction of the solvent/detergent (S/D) process time. Vox Sang. 2009, 97, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Cushing, M.M.; Asmis, L.; Calabia, C.; Rand, J.H.; Haas, T. Efficacy of solvent/detergent plasma after storage at 2–8 °C for 5 days in comparison to other plasma products to improve factor V levels in factor V deficient plasma. Transfus. Apher. Sci. 2016, 55, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Spinella, P.C.; Borasino, S.; Alten, J. Solvent/Detergent-Treated Plasma in the Management of Pediatric Patients Who Require Replacement of Multiple Coagulation Factors: An Open-Label, Multicenter, Post-marketing Study. Front. Pediatr. 2020, 8, 572. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef]
- Bonafont, J.; Mencía, Á.; García, M.; Torres, R.; Rodríguez, S.; Carretero, M.; Chacón-Solano, E.; Modamio-Høybjør, S.; Marinas, L.; León, C.; et al. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 2019, 27, 986–998. [Google Scholar] [CrossRef]
- Canver, M.C.; Bauer, D.E.; Dass, A.; Yien, Y.Y.; Chung, J.; Masuda, T.; Maeda, T.; Paw, B.H.; Orkin, S.H. Characterization of Genomic Deletion Efficiency Mediated by Clustered Regularly Interspaced Palindromic Repeats (CRISPR)/Cas9 Nuclease System in Mammalian Cells. J. Biol. Chem. 2014, 289, 21312–21324. [Google Scholar] [CrossRef]
- Zheng, Q.; Cai, X.; Tan, M.H.; Schaffert, S.; Arnold, C.P.; Gong, X.; Chen, C.Z.; Huang, S. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 2014, 57, 115–124. [Google Scholar] [CrossRef]
- Geisinger, J.M.; Turan, S.; Hernandez, S.; Spector, L.P.; Calos, M.P. In vivo blunt-end cloning through CRISPR/Cas9-facilitated non-homologous end-joining. Nucleic Acids Res. 2016, 44, e76. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W. Development and Application of CRISPR-Cas Based Tools. Front. Cell Dev. Biol. 2022, 10, 834646. [Google Scholar] [CrossRef]
- López-Manzaneda, S.; Ojeda-Pérez, I.; Zabaleta, N.; García-Torralba, A.; Alberquilla, O.; Torres, R.; Sánchez-Domínguez, R.; Torella, L.; Olivier, E.; Mountford, J.; et al. In Vitro and In Vivo Genetic Disease Modeling via NHEJ-Precise Deletions Using CRISPR-Cas9. Mol. Ther. Methods Clin. Dev. 2020, 19, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, B.; Castoldi, E.; Mingozzi, F.; Bernardi, F.; Castaman, G. A Novel Factor V Null Mutation Detected in a Thrombophilic Patient With Pseudo-Homozygous APC Resistance and in an Asymptomatic Unrelated Subject. Blood 1998, 92, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.; Pelaez, I.; Alias, L.; Baena, M.; De Pablo-Moreno, J.A.; Serrano, L.J.; Camero, M.D.; Tizzano, E.F.; Berrueco, R.; Liras, A. High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies. Int. J. Mol. Sci. 2021, 22, 9705. [Google Scholar] [CrossRef] [PubMed]
- Patent “In Vitro Method to Recover the Expression of the F5 Gene Encoding Coagulation Factor V”. Invention Patent with Examination. Patent Recognition. UCM Office for the Transfer of Research Results. 2021. Available online: https://consultas2.oepm.es/pdf/ES/0000/000/02/78/53/ES-2785323_B2.pdf (accessed on 26 March 2022).
- In Vitro Method to Recover the Expression of the F5 Gene Encoding Coagulation Factor V. Transfer Catalog. UCM Office for the Transfer of Research Results. 2021. Available online: https://www.ucm.es/otri/complutransfer-metodo-in-vitro-para-recuperar-laexpresion-del-gen-f5-que-codifica-el-factor-v-de-la-coagulacion-1 (accessed on 26 March 2022).
- van Beek, D.; Funaki, B. Hemorrhage as a Complication of Percutaneous Liver Biopsy. Semin. Interv. Radiol 2013, 30, 413–416. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Midia, M.; Odedra, D.; Shuster, A.; Midia, R.; Muir, J. Predictors of bleeding complications following percutaneous image-guided liver biopsy: A scoping review. Diagn. Interv. Radiol. 2019, 25, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, J.; Patel, J.; Caldwell, H.; Davies, S.; Hebditch, V.; Hollywood, C.; Hubscher, S.; Karkhanis, S.; Lester, W.; Roslund, N.; et al. Guidelines on the use of liver biopsy in clinical practice from the British Society of Gastroenterology, the Royal College of Radiologists and the Royal College of Pathology. Gut 2020, 69, 1382–1403. [Google Scholar] [CrossRef]
- Wilson, D.B.; Salem, H.H.; Mruk, J.S.; Maruyama, I.; Majerus, P.W. Biosynthesis of coagulation Factor V by a human hepatocellular carcinoma cell line. J. Clin. Investig. 1984, 73, 654–658. [Google Scholar] [CrossRef]
- HepG2 (Liver Hepatocellular Carcinoma) Cell Line. 2021. Available online: https://www.hepg2.com/ (accessed on 26 March 2022).
- Shulman, M.; Nahmias, Y. Long-term culture and coculture of primary rat and human hepatocytes. Methods Mol. Biol. 2013, 945, 287–302. [Google Scholar] [CrossRef]
- Thompson, W.L.; Takebe, T. Human liver model systems in a dish. Dev. Growth Differ. 2021, 63, 47–58. [Google Scholar] [CrossRef]
- Mizoi, K.; Arakawa, H.; Yano, K.; Koyama, S.; Kojima, H.; Ogihara, T. Utility of Three-Dimensional Cultures of Primary Human Hepatocytes (Spheroids) as Pharmacokinetic Models. Biomedicines 2020, 8, 374. [Google Scholar] [CrossRef]
- Serrano, L.J.; de la Torre, P.; Liras, A.; Flores, A.I. Cell therapy for factor V deficiency: An approach based on human decidua mesenchymal stem cells. Biomed. Pharmacother. 2021, 142, 112059. [Google Scholar] [CrossRef] [PubMed]
- Pettinato, G.; Lehoux, S.; Ramanathan, R.; Salem, M.M.; He, L.X.; Muse, O.; Flaumenhaft, R.; Thompson, M.T.; Rouse, E.A.; Cummings, R.D.; et al. Generation of fully functional hepatocyte-like organoids from human induced pluripotent stem cells mixed with Endothelial Cells. Sci. Rep. 2019, 9, 8920. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, E.; Doudna, J.A. Rewriting a genome. Nature 2013, 495, 50–51. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhang, B. Combined deficiency of coagulation factors V and VIII: An update. Semin. Thromb. Hemost. 2013, 39, 613–620. [Google Scholar] [CrossRef]
- Gong, J.; Chung, T.H.; Zheng, J.; Zheng, H.; Chang, L.J. Transduction of modified factor VIII gene improves lentiviral gene therapy efficacy for hemophilia A. J. Biol. Chem. 2021, 297, 101397. [Google Scholar] [CrossRef]
- Zhou, B.; Ho, S.S.; Greer, S.U.; Spies, N.; Bell, J.M.; Zhang, X.; Zhu, X.; Arthur, J.G.; Byeon, S.; Pattni, R.; et al. Haplotype-resolved and integrated genome analysis of the cancer cell line HepG2. Nucleic Acids Res. 2019, 47, 3846–3861. [Google Scholar] [CrossRef]
- Ajzner, E.; Balogh, I.; Haramura, G.; Boda, Z.; Kalmár, K.; Pfliegler, G.; Dahlbäck, B.; Muszbek, L. Anti-factor V auto-antibody in the plasma and platelets of a patient with repeated gastrointestinal bleeding. J. Thromb. Haemost. 2003, 1, 943–949. [Google Scholar] [CrossRef]
- Su, K.; Wang, L.; Wang, M.; Wang, H. A novel mutation (Ser951LeufsTer8) in F5 gene leads to hereditary coagulation factor V deficiency. Blood Coagul. Fibrinolysis 2021, 32, 140–145. [Google Scholar] [CrossRef]
- Luo, S.; Liu, S.; Xu, M.; Li, X.; Zhang, H.; Jin, Y.; Yang, L.; Wang, M. Analysis of phenotype and genotype of a family with hereditary coagulation factor V deficiency caused by the compound heterozygous mutations. Blood Coagul. Fibrinolysis 2020, 31, 485–489. [Google Scholar] [CrossRef]
- Al-Numair, N.S.; Ramzan, K.; Saleh, M.; Alzahrani, H.; Tarawah, A.; Abu-Douleh, E.; Elbaik, L.; Imtiaz, F.; Owaidah, T.M. First description of the molecular and clinical characterization of hereditary factor V deficiency in Saudi Arabia: Report of four novel mutations. Blood Coagul. Fibrinolysis 2019, 30, 224–232. [Google Scholar] [CrossRef]
- Cui, J.; O’Shea, K.S.; Purkayastha, A.; Saunders, T.L.; Ginsburg, D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature 1996, 384, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Translational Animal Models in Drug Discovery and Development; Bentham Science Publishers: New York, NY, USA, 2018. [Google Scholar]
- Weyand, A.C.; Grzegorski, S.J.; Rost, M.S.; Lavik, K.I.; Ferguson, A.C.; Menegatti, M.; Richter, C.E.; Asselta, R.; Duga, S.; Peyvandi, F.; et al. Analysis of factor V in zebrafish demonstrates minimal levels needed for early hemostasis. Blood Adv. 2019, 3, 1670–1680. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TT gRNA 2 Off-Targets | |||
|---|---|---|---|
| Gene | Bp | Mismatches * | |
| EXON | SFTA3 | 409 | 5 |
| TFCP | 396 | 5 | |
| KIF9 | 387 | 5 | |
| ABLIM3 | 496 | 4 | |
| GAS7 | 464 | 5 | |
| INTRON | iGPC4 | 474 | 2 |
| iPRKCH | 382 | 3 | |
| iWDR4 | 492 | 3 | |
| iWDFY4 | 385 | 3 | |
| iZNRF3 | 403 | 3 | |
| iEPC1 | 500 | 13 | |
| iNRG3 | 423 | 4 | |
| TT gRNA 4 Off-Targets | |||
| Gene | Bp | Mismatches * | |
| EXON | ARID1A | 384 | 4 |
| RSPO2 | 350 | 5 | |
| TREML1 | 394 | 5 | |
| PPIL2 | 403 | 5 | |
| GPR114 | 456 | 5 | |
| LYPD6 | 405 | 4 | |
| SCL25A3 | 472 | 5 | |
| SKIL | 382 | 4 | |
| TENM4 | 440 | 4 | |
| INTRON | iRAD51B | 405 | 2 |
| iMYO18 | 439 | 4 | |
| iTEM8 | 388 | 4 | |
| iEEFSE | 411 | 4 | |
| iFOXP1 | 402 | 4 | |
| Guide | Sequence | PAM |
|---|---|---|
| KO Guide 5 | CAATCAGACATTGCCCTCTA | TGG |
| KO Guide 1 | GAGGAATTCTGATTATGGTC | AGG |
| TT Guide 4 | CAATGTCTGATTGAGGTCTG | TGG |
| TT Guide 2 | TTCCTCAAATGACACTGGTC | AGG |
| Primers | Bp Product | |||||
|---|---|---|---|---|---|---|
| Gene | Sequence 5′-3′ | WT | KO | TT | ||
| Factor V | Forward | GAGAAGCACACACACCATGC | 403 | 368 | 319 | |
| Reverse | CTGTGGGGAAGGACTTGTGA | |||||
| TT gRNA 2 off-targets | ||||||
| Gene | Primer 5′-3′ | bp | ||||
| EXON | SFTA3 | Forward | CCAACAAGGGTACACACTGC | 409 | ||
| Reverse | TCTCAGAATTCCTCCCTGCG | |||||
| TFCP | Forward | CAGCCTTCCTATGAGACAACCA | 396 | |||
| Reverse | GAACCAAAGGCAGCACACAG | |||||
| KIF9 | Forward | GCTCCTGTCTTGGGTAGGTGG | 387 | |||
| Reverse | CCAAAGCTCCCTAGCCCTCTT | |||||
| ABLIM3 | Forward | CAGGCTATGTAGTCCCTGAGC | 496 | |||
| Reverse | ATCACCTGCAGAGGACCCAA | |||||
| GAS7 | Forward | ATGCCTGGAGAGGTCAGGAT | 464 | |||
| Reverse | CAGCCCTGCCATACACAAGA | |||||
| INTRON | iGPC4 | Forward | GACCTTGACCCCAGACAACT | 474 | ||
| Reverse | TTTGAGGGGGAGGAAGTGTTC | |||||
| iPRKCH | Forward | CACTGATTTCACCCTTAGCAAACC | 382 | |||
| Reverse | TAGGGCGAAGCTTTACCTGG | |||||
| iWDR4 | Forward | AGGTTGGCCTTGACTTTGGG | 492 | |||
| Reverse | CCAGTGTCCGAAAGGCAAAATG | |||||
| iWDFY4 | Forward | ATAGAGCATGTGGAGCCCTTG | 385 | |||
| Reverse | GGGAAGGGAGAAGGGCATTG | |||||
| iZNRF3 | Forward | GGATCTCTTACCAGCCCCCT | 403 | |||
| Reverse | GATCCTCCACTTGCTGATGG | |||||
| iEPC1 | Forward | TCTTGGAAAGTGTACATCTGAGG | 500 | |||
| Reverse | CAGGCTTGAAGGTCTGTTTACC | |||||
| iNRG3 | Forward | TGATGTTGTAGCCTAGGGAAGT | 423 | |||
| Reverse | AGCAAACCTCTGCATCCATCT | |||||
| TT gRNA 4 off-targets | ||||||
| Gene | Primer 5′-3′ | bp | ||||
| EXON | ARID1A | Forward | TGGGAAAGGAGCAACTCTGC | 384 | ||
| Reverse | TGTCCTTGCCTGAAAAGGCT | |||||
| RSPO2 | Forward | AGCAATGGGCAGAGCCATTC | 350 | |||
| Reverse | ATGTGAACCACACCTAACCTGA | |||||
| TREML1 | Forward | GAGCCAGTGCCATTTCCTGA | 394 | |||
| Reverse | CCCCTTCGATTTAGTTTGGAGC | |||||
| PPIL2 | Forward | CTGTTGTGCCACAGATTGCG | 403 | |||
| Reverse | ATACAGCCCTCCCGGTGTAG | |||||
| GPR114 | Forward | CAAAGATTGGGAGGTTCCGC | 456 | |||
| Reverse | AATGGGTCACTCAGTAGGCG | |||||
| LYPD6 | Forward | AGGTCTGCACTTCTTGTTGTG | 405 | |||
| Reverse | TGGTATGGAACTTGGGCTGT | |||||
| SCL25A3 | Forward | CGCAGGCATTTTTCCCTTGC | 472 | |||
| Reverse | GTCTGACATTCGCTCTTTACATGG | |||||
| SKIL | Forward | AAGTAGTCAGAGCTCGCTGG | 382 | |||
| Reverse | TGTAATCAGCCCACAGGATGG | |||||
| TENM4 | Forward | GGCCCTTTTGACACTCACCA | 440 | |||
| Reverse | CACCTCCTGGCCATTCTCAC | |||||
| INTRON | iRAD51B | Forward | TCTGCCACTCCTGACTTCCT | 405 | ||
| Reverse | TCTTCCTAGTCACCTCGCCT | |||||
| iMYO18 | Forward | TAGGGCTCATTGCAAGGAAGAT | 439 | |||
| Reverse | CCCCAGCCTCCAATCTGAC | |||||
| iTEM8 | Forward | GCCAAGCGTGTAGCAGACT | 388 | |||
| Reverse | TGGATGTCTGCCAGCAAAGG | |||||
| iEEFSE | Forward | GAAGACCTGCCTCTTCCTGA | 411 | |||
| Reverse | CCCTCCCCTTACCTTTTCCAC | |||||
| iFOXP1 | Forward | TGATAGAACCTCGCTTGGGG | 402 | |||
| Reverse | TAGTCTCTGGACTGGTGTGGG | |||||
| Antibody | Manufacturer | Fluorochrome | Concentration | Ref. |
|---|---|---|---|---|
| CD90 | Biolegend | PE | 50 µg/mL | 328,110 |
| CD29 | Biolegend | PerCP-Cy5.5 | 400 µg/mL | 303,024 |
| CD73 | Biolegend | PE | 400 µg/mL | 344,004 |
| CD45 | Biolegend | FITC | 200 µg/mL | 304,006 |
| CD34 | Biolegend | APC | 50 µg/mL | 343,608 |
| HLADR | Biolegend | APC | 200 µg/mL | 307,658 |
| OV6 | Santa Cruz Bio. | PE | 200 µg/mL | SC-101863 |
| CD11b | Biolegend | PerCP-Cy5.5 | 200 µg/mL | 301,328 |
| CD105 | Biolegend | APC | 100 µg/mL | 323,208 |
| CD44 | Biolegend | PE/Cy7 | 100 µg/mL | 338,816 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano, L.J.; Garcia-Arranz, M.; De Pablo-Moreno, J.A.; Segovia, J.C.; Olivera-Salazar, R.; Garcia-Olmo, D.; Liras, A. Development and Characterization of a Factor V-Deficient CRISPR Cell Model for the Correction of Mutations. Int. J. Mol. Sci. 2022, 23, 5802. https://doi.org/10.3390/ijms23105802
Serrano LJ, Garcia-Arranz M, De Pablo-Moreno JA, Segovia JC, Olivera-Salazar R, Garcia-Olmo D, Liras A. Development and Characterization of a Factor V-Deficient CRISPR Cell Model for the Correction of Mutations. International Journal of Molecular Sciences. 2022; 23(10):5802. https://doi.org/10.3390/ijms23105802
Chicago/Turabian StyleSerrano, Luis Javier, Mariano Garcia-Arranz, Juan A. De Pablo-Moreno, José Carlos Segovia, Rocío Olivera-Salazar, Damián Garcia-Olmo, and Antonio Liras. 2022. "Development and Characterization of a Factor V-Deficient CRISPR Cell Model for the Correction of Mutations" International Journal of Molecular Sciences 23, no. 10: 5802. https://doi.org/10.3390/ijms23105802
APA StyleSerrano, L. J., Garcia-Arranz, M., De Pablo-Moreno, J. A., Segovia, J. C., Olivera-Salazar, R., Garcia-Olmo, D., & Liras, A. (2022). Development and Characterization of a Factor V-Deficient CRISPR Cell Model for the Correction of Mutations. International Journal of Molecular Sciences, 23(10), 5802. https://doi.org/10.3390/ijms23105802