Abstract
Porphyromonas gingivalis as the keystone periodontopathogen plays a critical role in the pathogenesis of periodontitis, and crucially accounts for inflammatory comorbidities such as cardiovascular disease and Alzheimer′s disease. We recently identified the existence of P. gingivalis persisters and revealed the unforeseen perturbation of innate response in human gingival epithelial cells (HGECs) due to these noxious persisters. Herein, RNA sequencing revealed how P. gingivalis persisters affected the expression profile of cytokine genes and related signaling pathways in HGECs. Results showed that metronidazole-treated P. gingivalis persisters (M-PgPs) impaired the innate host defense of HGECs, in a similar fashion to P. gingivalis. Notably, over one thousand differentially expressed genes were identified in HGECs treated with M-PgPs or P. gingivalis with reference to the controls. Gene Ontology and KEGG pathway analysis demonstrated significantly enriched signaling pathways, such as FOXO. Importantly, the FOXO1 inhibitor rescued the M-PgP-induced disruption of cytokine expression. This study suggests that P. gingivalis persisters may perturb innate host defense, through the upregulation of the FOXO signaling pathway. Thus, the current findings could contribute to developing new approaches to tackling P. gingivalis persisters for the effective control of periodontitis and P. gingivalis-related inflammatory comorbidities.
1. Introduction
Periodontal diseases (gingivitis and periodontitis) are among the major global oral health burdens, with disastrous socioeconomic impacts and humongous healthcare costs [1,2,3,4,5], and yet periodontitis is intimately linked to over 50 systemic diseases and conditions, such as diabetes mellitus, cardiovascular disease, cancer and Alzheimer′s disease [6,7,8,9]. Severe periodontitis, affecting 11.2% of the entire global population, was ranked as the 6th most prevalent disease in humans in the first Global Burden of Disease (GBD) study [4,5,8,10]. If uncontrolled, it may eventually lead to severe tooth loss and edentulous in adults. Porphyromonas gingivalis, the major periodontopathogen, has been recognized as the ‘keystone’ pathogen for periodontitis [11]. It enables the disturbance of microbe–host homeostasis by shifting microbial symbiosis/eubiosis to dysbiosis, even at a low abundance, leading to dysregulated immuno-inflammatory response and irreversible periodontal destruction [12,13]. Increasing evidence has linked periodontitis with other inflammatory comorbidities, and indeed, P. gingivalis plays multiple essential roles in this process [14,15]. Currently, it remains a challenge in clinical practice to precisely eliminate P. gingivalis cells in their biofilm and intracellular modes. This ‘keystone’ periodontopathogen develops tough intrinsic strategies to disrupt host immunity and survive under harsh environments [16,17], notably by forming persister cells as one of the key approaches [18,19].
Microbial persisters are a unique subpopulation of microorganisms with specific strategies to survive under the strain of a lethal dosage from multiple antimicrobials [20]. The first description of persisters can be dated back to 1944 [21] when Joseph W. Bigger found that penicillin failed to eliminate all of the cultured Staphylococcus. In fact, the remaining survivors, so-called ‘persisters’, are present in a dormant and non-dividing state, like bacteria in their stationary phase. After being overlooked for a half-century, there has been a rapid increase in interest and investigations on persisters over the past two decades. To date, persisters have been reported in almost all microorganisms tested [22], and several clinical symptoms, such as cystic fibrosis [23], oral carriage [24], urinary tract infections [25] and tuberculosis [26], have been closely associated with persisters. It is worth noting that it has been demonstrated that Salmonella persisters can dampen innate immune response and induce anti-inflammatory macrophage polarization, as well as enhance antibiotic resistance in the intestine [27,28]. Given the essential role that P. gingivalis plays in the relapse of periodontitis, our group has provided the first evidence of the presence of metronidazole-tolerant P. gingivalis persister cells [18], and further demonstrated that these metronidazole-treated P. gingivalis persisters (M-PgPs) enabled the suppression of the immuno-inflammatory response in human gingival epithelial cells (HGECs) [19], whereas the underlying mechanisms and related signaling pathways need further investigation.
HGECs as the frontline defense of periodontal tissues function as both physical [29] and biological barriers for maintaining periodontal homeostasis and health [30]. When challenged by pathogenic bacteria, HGECs are able to secrete pro-inflammatory cytokines and chemokines, such as C-X-C motif chemokine 5 (CXCL5), Interleukin (IL)-6 and IL-8 [31,32], which exhibit the potent effects on the recruitment of neutrophils [33], thereby playing a critical role in maintaining periodontal health. The inhibitory effects of M-PgPs on the expression of CXCL5, IL-6 and IL-8 demonstrated by our group show that even when treated with a lethal concentration of antibiotics, M-PgPs still disrupt the innate host responses of HGECs. Thus, targeting this inhibitory activity of M-PgPs could serve as a potential strategy to control periodontitis and P. gingivalis-related inflammatory comorbidities.
Here, the present study further investigated how these M-PgPs interacted with HGECs and affected the innate host defense. It was hypothesized that certain signaling pathways could be critically involved in the M-PgP-induced disruption of immuno-inflammatory responses in HGECs, and thus, this RNA sequencing (RNA-seq) study aimed to identify candidate genes and underlying signaling pathways.
2. Results
2.1. M-PgPs Inhibited the Innate Host Defense in HGECs
The previous study of our group showed that cytokine (IL-6, IL-8, and CXCL5) expression levels in HGECs were remarkably suppressed by M-PgPs [19]. Herein, we further investigated the relative mRNA expression levels of pro-inflammatory (IL-1β and TNF) and anti-inflammatory cytokines (IL-10 and TGF-β2) in M-PgP-treated HGECs. IL-1β (1 ng mL−1) was used for the pretreatment of HGECs to mimic the inflammatory condition. Notably, IL-1β and TNF were downregulated, while IL-10 and TGF-β2 were upregulated by M-PgPs, with reference to the untreated group, whether they were pre-treated with IL-1β or not (Figure 1). These findings showed that M-PgPs enabled the inhibition of the innate host defense in HGECs.
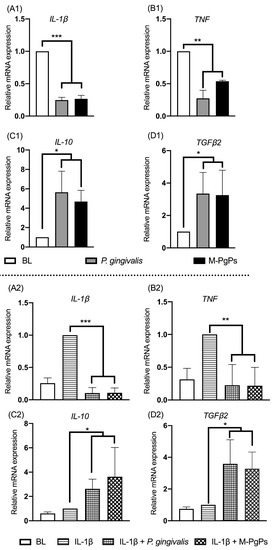
Figure 1.
M-PgPs inhibit the innate host defense of HGECs. Relative mRNA expression levels of IL-1β (A1,A2), TNF (B1,B2), IL-10 (C1,C2), and TGF-β2 (D1,D2)in HGECs after the treatment of M-PgPs (1:100) for 24 h were analyzed using RT-qPCR. (A1–D1) Under normal condition without IL-1β pre-treatment. (A2–D2) Under inflammatory condition with IL-1β pre-treatment. HGECs without any treatment served as blank control (BL). HGECs treated with P. gingivalis (1:100, 24 h) served as positive control. IL-1β (1 ng mL−1) was added 6 h before M-PgPs and P. gingivalis treatment. * p < 0.05; ** p < 0.01; *** p < 0.001. Data represent the mean ± SD of three independent experiments.
2.2. Differential Gene Expression
To verify the underlying mechanisms behind the inhibitory effects of M-PgPs on the immuno-inflammatory response of HGECs, RNA-seq was conducted to determine the possible genes and signaling pathways that could be involved in this process. Samples from six groups were compared: HGECs only (blank group, BL), M-PgP-treated HGECs, P. gingivalis-treated HGECs, IL-1β-treated HGECs (negative control group, NC), IL-1β + M-PgP-treated HGECs and IL-1β + P. gingivalis-treated HGECs. After sequencing, a differentially expressed gene (DEG) analysis was performed to identify gene expression changes among these groups.
Under the normal condition without IL-1β pretreatment, 1237 DEGs (p < 0.05, |Log2 fold change| ≥ 1) were identified in M-PgP-treated HGECs as compared with BL (Figure S1A), including 689 upregulated genes (Figure S2A) and 548 downregulated ones (Figure S2B). Moreover, there were totally 1452 DEGs in P. gingivalis-treated HGECs with reference to BL (Figure S1B), including 778 upregulated genes (Figure S2A) and 674 downregulated genes (Figure S2B). Under inflammatory condition with IL-1β pretreatment, a total of 1135 DEGs were identified in IL-1β + M-PgP-treated HGECs compared with NC (Figure S1C), which included 724 upregulated genes (Figure S2A) and 411 downregulated genes (Figure S2B). Furthermore, there were totally 1393 DEGs in IL-1β + P. gingivalis-treated HGECs referred to NC (Figure S1D), including 876 upregulated genes (Figure S2A) and 517 downregulated ones (Figure S2B).
Notably, some genes were similarly identified as DEGs in all groups, among which the top 20 DEGs are shown in Table S1. Importantly, no DEGs were identified between M-PgP- and P. gingivalis-treated HGECs, whether they were pre-treated with IL-1β or not.
2.3. Gene Ontology (GO) Functional Annotation and Enrichment Analyses
To generally describe the functions of the DEGs obtained from RNA-Seq, functional annotation was performed by comparing the sequences with the GO databases. DEGs annotated by the GO databases were then classified into three categories, including the cellular component (CC), biological progress (BP) and molecular function (MF). In total, 52 terms were assigned from the DEGs, and the top 10 terms of each category based on the number of DEGs assigned are shown in Figure 2.
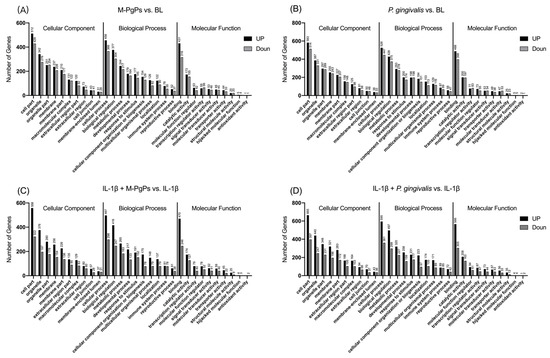
Figure 2.
Gene Ontology functional annotation of DEGs in different comparison groups. The top 10 terms of GO functional annotation assigned from the DEGs in different comparison groups are shown. M-PgPs vs. BL (A); P. gingivalis vs. BL (B); IL-1β + M-PgPs vs. IL-1β (C); and IL-1β + P. gingivalis vs. IL-1β (D). GO terms are shown on the x axis. Numbers of DEGs enriched in the terms are shown above the bar. Black bar: upregulated genes. Gray bar: downregulated genes. Samples for RNA-Seq were obtained from three independent experiments.
Enrichment analyses of GO terms were performed for DEGs using GOatools. GO terms with q < 0.05 were identified as significantly enriched. According to the functional enrichment results, several terms were significantly enriched within each category (CC, BP and MF) (Table S2). The top 20 terms of each category based on the Rich Ratio are shown in Table S3.
2.4. KEGG Pathway Enrichment Analysis
This analysis was performed to explore the enriched pathways for DEGs. As compared to the BL group, 31 and 28 KEGG pathways were identified as significantly enriched in the M-PgP-treated and P. gingivalis-treated HGECs, respectively (q < 0.05). It is worth noting that the most enriched pathway in both groups was Cytokine-cytokine receptor interaction. Additionally, the specific pathways of MAPK, TNF and FOXO were also significantly enriched in both groups (Figure 3A,B).
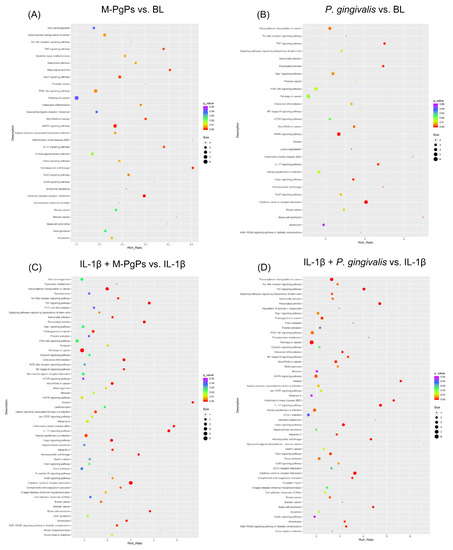
Figure 3.
KEGG pathway enrichment analysis of DEGs in different comparison groups. M-PgPs vs. BL (A); P. gingivalis vs. BL (B); IL-1β + M-PgPs vs. IL-1β (C); and IL-1β + P. gingivalis vs. IL-1β (D). Rich Ratio is shown on the x axis. KEGG pathway names are shown on the y axis. Size of the dots represents numbers of genes enriched in the term. Color of the dots represents q value. Samples for RNA-Seq were obtained from three independent experiments.
With reference to the NC group, 55 and 53 KEGG pathways were identified as significantly enriched in the IL-1β + M-PgP-treated and IL-1β + P. gingivalis-treated HGECs, respectively (q < 0.05). The most enriched pathway in both groups was the cytokine–cytokine receptor interaction. Additionally, TNF and IL-17 signaling pathways, as well as pathways in cancer and transcriptional misregulation in cancer, were also significantly enriched in both groups (Figure 3C, D).
Several signaling pathways (e.g., cytokine–cytokine receptor interaction, TNF signaling, IL-17 signaling, FOXO signaling, pathways in cancer, PI3K-Akt signaling and Toll-like receptor signaling) were identified as significantly enriched in all comparison groups (Figure 3). The numbers of genes mapped onto these signaling pathways are shown in Table S4.
2.5. FOXO Signaling Pathway Was Involved in the Inhibitory Effects of M-PgPs on the Innate Host Defense in HGECs
Since the FOXO signaling pathway was identified as significantly enriched in all groups, further verification assay was conducted. The relative mRNA expression levels of FOXO1 and several downstream genes (e.g., BCL2L11, BCL6, S1PR1, TRAIL and KLF2) were analyzed using RT-qPCR, thereby demonstrating that these gene expression levels were upregulated in both P. gingivalis- and M-PgP-treated HGECs as compared to the BL/NC groups (Figure 4). Then, the FOXO1 inhibitor (Fi) was used to block the FOXO signaling pathway. The preliminary results showed that the most appropriate concentration of Fi was 1 μM, and it could downregulate the relative mRNA expression levels of FOXO1, KLF2 and TRAIL (Figure S3). It is worth noting that higher Fi concentrations (≥2 μM) could lead to the death of HGECs, resulting in a cell viability of lower than 80% (Figure S3).
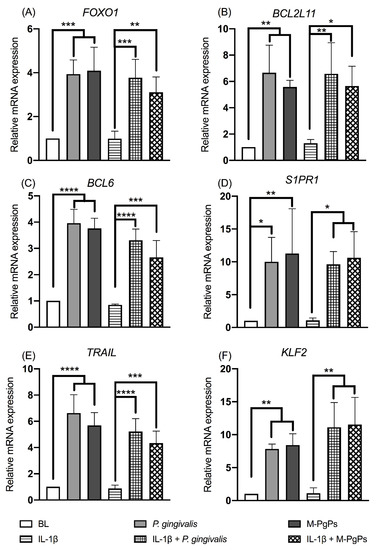
Figure 4.
FOXO signaling pathway in HGECs was upregulated by M-PgPs. Relative mRNA expression levels of FOXO1 (A), BCL2L11 (B), BCL6 (C), S1PR1 (D), TRAIL (E) and KLF2 (F) in HGECs after the treatment of M-PgPs (1:100) for 24 h were analyzed using RT-qPCR. HGECs without any treatment served as blank control (BL). HGECs treated with P. gingivalis (1:100, 24 h) served as positive control. IL-1β (1 ng mL−1) was added 6 h before M-PgPs and P. gingivalis treatment. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. Data represent the mean ± SD of three independent experiments.
Under the normal condition, Fi reversed the relative expression levels of FOXO1 mRNAs and the downstream genes (BCL6 and KLF2) affected by M-PgPs (Figure 5A–C). In parallel, the downregulated IL-1β and TNF as well as the upregulated IL-10 and TGFβ2 due to M-PgPs were completely or partly reversed by Fi (Figure 5D–G). Moreover, the downregulated cytokines (IL-6 and CXCL5) were also fully or partly reversed by Fi (Figure 5H and I), whereas under the inflammatory condition, Fi did not affect the FOXO1 mRNA expression level (Figure S4A) but reversed downstream genes such as KLF2 (Figure S4B). Meanwhile, the altered mRNA expression levels of IL-1β, TNF, IL-10, TGFβ2, IL-6, IL-8 and CXCL5 were partly reversed by Fi (Figure S4C–I).
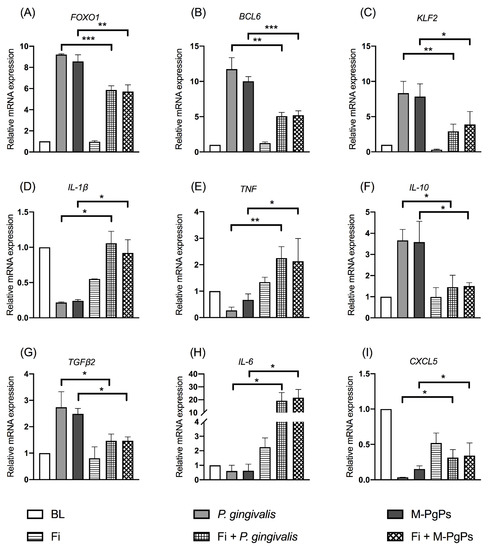
Figure 5.
FOXO1 inhibitor (Fi) rescued the inhibitory effects of M-PgPs on the immuno-inflammatory response of HGECs under normal condition. Relative mRNA expression levels of FOXO1 (A), BCL6 (B), KLF2 (C), IL-1β (D), TNF (E), IL-10 (F), TGF-β2 (G), IL-6 (H) and CXCL5 (I) in HGECs after the treatment of M-PgPs (1:100) for 24 h were analyzed using RT-qPCR. HGECs without any treatment served as blank control (BL). HGECs treated with P. gingivalis (1:100, 24 h) served as positive control. Fi (1 μM) was added 2 h before M-PgPs and P. gingivalis treatment. * p < 0.05; ** p < 0.01; *** p < 0.001. Data represent the mean ± SD of three independent experiments.
3. Discussion
In the present study, RNA-seq analysis was undertaken to identify the potential mechanisms (e.g., the candidate genes and signaling pathways) underlying the P. gingivalis persister-induced disruption of immuno-inflammatory responses in HGECs. The findings indicated that the FOXO signaling pathway was crucially involved in this process. Interestingly, the rescue experiment verified that the FOXO1 inhibitor could reverse the cytokine expression levels altered by M-PgPs. Our current work demonstrates that M-PgPs can impair innate host defense through the upregulation of the FOXO signaling pathway. This study may contribute to developing novel approaches to tackling P. gingivalis persisters for effectively controlling periodontitis and P. gingivalis-related inflammatory comorbidities.
As the first line of defense in the periodontium, gingival epithelial cells play essential roles in maintaining periodontal homeostasis and health. Thus, numerous studies have investigated the immuno-inflammatory responses in HGECs challenged by pathogenic bacteria such as P. gingivalis [34,35,36]. Our group has recently demonstrated, for the first time, that M-PgPs perturb the innate immune responses in HGECs [19] in an identical fashion to P. gingivalis. Similar results were found in the present study, showing that the pro-inflammatory cytokine genes (IL-1β and TNF) were downregulated, while the anti-inflammatory cytokine genes (IL-10 and TGF-β2) were upregulated by M-PgPs, with reference to the untreated group. However, the underlying mechanisms involved in the M-PgP-induced disruption of innate host defense and the relevant biological implications remain unclear.
In this study, RNA sequencing was performed in HGECs after treatments of M-PgPs and P. gingivalis, and over one thousand DEGs were identified among the groups. Interestingly, no notable inter-group difference was identified under both normal and inflammatory conditions. This finding indicates that M-PgPs maintain a similar capability to P. gingivalis in suppressing immuno-inflammatory responses in HGECs in accordance with our previous study [19]. Further study is required to confirm this point, and explore the molecular signature of P. gingivalis persister pathogenicity.
Next, GO term assignment and enrichment analyses showed that multiple GO terms were significantly enriched in all groups (e.g., cell junction, metabolic process, response to stimulus, transcription regulator activity, translation regulator activity and antioxidant activity). These annotations and classifications provide us with plenty of resources and information for illustrating the underlying processes, functions and pathways related to the actions of HGECs in response to certain pathogens and their persisters.
Moreover, KEGG pathway enrichment analyses demonstrated that the FOXO signaling pathway was significantly enriched in M-PgP-treated HGECs. It is well known that the FOXO transcription family consists of four members (FOXO1, FOXO3, FOXO4 and FOXO6) in mammals and acts as a critical signaling mediator in various cell biological processes, such as oxidative stress responses, apoptosis, proliferation, energy metabolism and inflammation [37]. Particularly, FOXO plays critical roles in the homeostasis of immune-relevant cells [38,39]. As the representative and the best studied member of the FOXO family [40], FOXO1 has been investigated in various cell types and genetically modified mice models [41,42,43,44], while only a few studies have explored its functions in HGECs. Further investigation on the modulation of FOXO in HGECs would enable us to gain a better understanding of the initiation and progression of periodontitis. Our study revealed that the mRNA levels of FOXO1 and its downstream genes were upregulated by M-PgPs and P. gingivalis. This finding is consistent with a previous study in which P. gingivalis could induce the activation of FOXO1 in gingival epithelial cells [45]. Considering the finding that M-PgPs suppressed the immuno-inflammatory response in HGECs, the FOXO signaling pathway may be involved in this notable observation.
In order to confirm this hypothesis, FOXO1 inhibitor (Fi) was applied, as previously described [46], to perturb activation by M-PgPs. It is worth noting that Fi totally or partly rescued the relative mRNA expression levels of those genes affected by M-PgPs under normal and inflammatory conditions. Thus, M-PgPs could perturb the innate response of HGECs, at least partly, through the upregulation of the FOXO signaling pathway, and blocking the FOXO signaling pathway may possibly disrupt this process.
The Fi (AS1842856) was first discovered by Nagashima and co-workers for treating type 2 diabetes mellitus [46], and indeed, it has been widely used for the management of diabetes in animal models [47,48,49]. In the present study, Fi was used to treat HGECs for the first time, and surprisingly, it could to some extent rescue the M-PgP-induced disruption of immuno-inflammatory responses. Notably, Fi is a specific and powerful inhibitor of FOXO1, and the current finding confirms the essential regulatory effects of the FOXO signaling pathway in the M-PgP-dysregulated innate response in HGECs. Further studies are warranted to determine the potential usage of Fi in other FOXO-related research in connection to the pathogenicity of P. gingivalis [45] and M-PgPs.
Besides FOXO, this study also identified other signaling pathways that were highly enriched in all groups, such as cytokine–cytokine receptor interaction, the TNF signaling pathway and the IL-17 signaling pathway. Cytokines as a broad category of small proteins play essential roles in immunoregulatory and inflammatory processes. The resultant actions need to be exerted through the interaction with specific receptors. Thus, these cytokine-related signaling pathways critically account for microbe-induced inflammatory responses. Remarkably, the majority of the DEGs enriched in these pathways (e.g., TNF, CCL2, CCL20, FOS, IL-1 and TNFAIP3) revealed a downregulated trend, which is in accordance with the observation of the inhibition of the innate host defense to M-PgPs and P. gingivalis. It is noted that multiple signaling pathways could be involved in this process. Thus, further investigations are needed to better illustrate the regulatory mechanisms and develop more effective solutions.
It is worth noting that P. gingivalis persisters can maintain their pathogenicity even when treated with a lethal concentration of metronidazole [19]. This finding inspires us to reconsider the rationale and strategy of antibiotic usage in clinical practice. More studies are required to develop novel and precise approaches to tackling microbe-induced inflammatory diseases and systemic comorbidities. Indeed, we have recently demonstrated that bismuth drugs can rescue P. gingivalis-perturbed innate host responses [50], and P. gingivalis persisters could be effectively eliminated via the synergistic combination of bismuth drugs such as colloidal bismuth subcitrate (CBS) with metronidazole [51]. Further studies are required to develop new anti-persister drug delivery systems to effectively tackle bacterial persisters and modulate dysregulated immunoinflammatory responses to control P. gingivalis-related diseases.
Nevertheless, there are several limitations of this study. For instance, persister cells only take up a small proportion of the whole population, and it remains challenging to precisely isolate them from unlysed cells after antimicrobial treatments [52,53]. As such, in our previous work [19] and this study, we used the term ‘metronidazole-treated P. gingivalis persisters (M-PgPs)’, which reflected the components of P. gingivalis persisters and metronidazole-killed P. gingivalis cells. Moreover, we only verified the RNA-Seq results using RT-qPCR, but did not assess the protein expression levels. It is noted that protein expression is a complicated process, and it is affected by different microenvironmental conditions. Indeed, proinflammatory cytokines could be possibly degraded by P. gingivalis gingipains [54,55]. Further investigation must extend to FOXO transcription factors and proteins to clarify these points.
4. Materials and Methods
4.1. Culture of Cells
HGECs obtained from CELLnTEC (CELLnTEC, Berne, Switzerland) were used in this study as our established protocol [19]. They were cultured in epithelial culture medium (CnT-prime, CELLnTEC, Berne, Switzerland, changed every two days) in a humidified incubator (37 °C with 5% CO2). The 3rd to 5th passages of HGECs were used in all experiments.
4.2. Bacterial Culture and M-PgPs Formation
P. gingivalis (ATCC 33277) was employed and cultured following our established protocol [19]. Frozen stocked bacteria were first grown on blood agar plates (44 g L−1 BD Columbia agar base (Becton Dickinson GmbH, Heidelberg, Germany), 5% horse blood (Hemostat, Dixon, CA, USA), 5 mg L−1 hemin (Sigma-Aldrich, St. Louis, MO, USA), 1 mg L−1 vitamin K1 (Sigma-Aldrich, St. Louis, MO, USA)) in an anaerobic atmosphere at 37 °C (10% H2, 5% CO2 and 85% N2). After one week, a single colony was picked and placed in liquid Trypticase soy broth (30 g L−1 TSB; Becton Dickinson GmbH, Heidelberg, Germany) supplemented with 5 g L−1 yeast extract, 5 mg L−1 hemin and 1 mg L−1 vitamin K1; it was then cultured under the same anaerobic conditions.
M-PgPs formation was performed following our previous study [19]. In brief, P. gingivalis was cultured in broth for 48 h and re-suspended in fresh broth to an OD600 of 0.1. Subsequently, the bacteria were incubated in the stationary phase (72 h) and further treated with metronidazole (MTZ, Sigma-Aldrich, St. Louis, MO, USA) at 100 mg L−1 for 6 h. It was confirmed by our recent study that about 1% of the P. gingivalis cells remained viable after the 6 h MTZ treatment [51].
4.3. Infection of HGECs with M-PgPs
HGECs (4 × 105 cells) were seeded into 6-well plates. Following cellular adherence, the cells were pre-treated with or without IL-1β (1 ng ml−1) to mimick the inflammatory condition. Those without IL-1β treatment were defined as the blank control (BL). After 4 h, cells were treated with 1 μM FOXO1 inhibitor (Fi) AS1842856 [46] (Selleckchem, Houston, TX, USA) or not. After another 2 h, cells were infected with M-PgPs or P. gingivalis (MOI: 100) for 24 h, according to previous studies [19,56,57]. Total RNAs were collected for the following tests.
4.4. RT-qPCR
Total RNA (600 ng) was used to synthesize cDNA with the QuantiTect Reverse Transcription Kit (Qiagen, Benelux BV Qiagen GmbH, Hilden, Germany). RT-qPCR was undertaken using the ABI 7500 Real-time PCR System (Applied Biosystems, Carlsbad, CA, USA) with the QuantiNova SYBR Green PCR Kit (Qiagen, Benelux BV Qiagen GmbH, Hilden, Germany). The expression level of individual genes was normalized to β-actin using the comparative 2−ΔΔCT method. The primer sequences are listed in Table 1.

Table 1.
Primer sequences used for RT-qPCR.
4.5. Library Preparation and RNA Sequencing
Library preparation, RNA-seq and data analysis were performed at the Annoroad Gene Technology Corporation (Beijing, China). The samples were obtained from three independent experiments. Total RNA was purified using the RNeasy Plus Mini Kit (Qiagen, Benelux BV Qiagen GmbH, Hilden, Germany). Annoroad Gene Technology Corporation (Beijing, China) constructed cDNA libraries and performed sequencing using these RNA samples. In brief, after RNA quantity and quality detection and RNA fragment size analysis, mRNA was enriched from total RNA using oligo (dT) magnetic beads. Then, sequencing libraries were prepared using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (#E7530L, New England Biolabs, Inc, Ipswich, MA, USA), and index codes were added for attribute sequencing. The library RNA concentration was measured using the Qubit® RNA Assay Kit (Life technologies, Waltham, MA, USA) for initial quantification, and then adjusted to 1 ng μL−1. To make sure the insert size matched the valid library concentration (>10 nM), it was tested and accurately quantified using the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA) and the CFX96 RT-PCR System (Bio-Rad Laboratories, Hercules, CA, USA), respectively. The index-coded samples were clustered in a cBot cluster generation system using the HiSeq PE Cluster Kit v4-cBot-HS (Illumina, Santiago, CA, USA). Library sequencing was then undertaken using the NovaSeq 6000 platform (Illumina, Santiago, CA, USA) to generate 150 bp paired-end reads.
4.6. Data Filtering and Alignment
Raw reads generated from sequencing were filtered using Perl scripts [58] to (1) discard the reads if the length of the trimmed reads was lower than 30 bp after trimming the Smart-seq2 public primer sequence of the reads; (2) the contaminated reads were removed from adapters when the read bases contained more than 5 bp of adapter sequences; (3) the low-quality reads (carrying over 15% bases with a quality value ≤19) were removed; (4) reads that contained over 5% ambiguous nucleotides were removed. Following filtering, the clean reads were aligned to the reference genome (GRCh38.p13) using HISAT2 v2.1.0 (Baltimore, MD, USA) [59]. Herein, the reference and annotation files were obtained from the ENSEMBL browser (http://www.ensembl.org/index.html, accessed on 17 May 2022). A genome index was created using Bowtie2 v2.2.3 [60]. The multi-mapped and/or unmapped reads were excluded from the analysis.
4.7. Analysis of Differentially Expressed Genes
The transcript expression levels were determined using FPKM (Fragments per Kilobase per Million Mapped Fragments) with HTSeq v0.6.0 (California Institute of Technology, Pasadena, CA, USA) [61]. Correlation analysis was performed using the genes expressed in at least one sample. Hierarchical clustering was carried out via the Pearson correlation distance, and the expression levels of each gene in each sample were estimated with DESeq2 using linear regression. A p-value was calculated using the Wald test [62] and corrected using the Benjamini–Hochberg q-value (false discovery rate, FDR) [63]. The genes (q < 0.05 and |log2 ratio| ≥ 1) were then defined as differentially expressed genes (DEGs) [64].
4.8. Functional Annotation and Enrichment Analysis
Both GO (Gene Ontology, http://geneontology.org/, accessed on 17 May 2022) and KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.kegg.jp/, accessed on 17 May 2022) enrichments of DEGs were applied using the hypergeometric test. Fisher’s exact test and multiple comparisons were then performed to adjust the p-value as the q-value (false discovery rate, FDR). The genes in the whole genome served as the background of the datasets. They were determined to be significantly enriched, with q < 0.05 for the GO terms and KEGG pathways.
4.9. Cell Viability Test
HGECs (5 × 103 cells) were seeded into 96-well plates. After adhesion, cells were treated with different concentrations of FOXO1 inhibitor (Fi) AS1842856 (0 μM to 100 μM) for 24 h. The cell viability was evaluated using a testing kit (CyQUANT™ MTT Cell Proliferation Assay, Thermo Fisher Scientific Inc., Waltham, MA, USA).
4.10. Statistical Analysis
All results were presented as the mean ± standard deviation (SD). At least 3 independent repeats were conducted separately in each experiment. Statistical calculations were performed using GraphPad Prism 8. Inter-group differences were determined via one-way analysis of variance. p < 0.05 was considered as statistically significant.
5. Conclusions
Our current study demonstrates significant DEGs in M-PgP-treated HGECs. Functional enrichment analyses of the DEGs using GO and KEGG identified greatly enriched terms and pathways involved in the M-PgP-induced inhibition of innate host defense in HGECs. Further verification confirmed that the FOXO signaling pathway may be involved in this observed action. These findings enhance our understanding of the specific survival strategies of M-PgPs after invading host cells. Targeting the FOXO signaling pathway could be an alternative approach to tackling P. gingivalis persisters for the effective control of periodontitis and P. gingivalis-related inflammatory comorbidities.
Supplementary Materials
The Supplementary materials are available online at https://www.mdpi.com/article/10.3390/ijms23105728/s1.
Author Contributions
L.J. conceived the project. C.W. and L.J. designed the study. C.W., X.L., T.C. and L.W. performed the experiments, and collected and analyzed the data. All authors contributed to the interpretation of the results. C.W. drafted the manuscript. L.J. conducted a critical review of the manuscript. L.J. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Hong Kong Research Grants Council (GRF No. 17122918 and 17119819) and the Modern Dental Laboratory/HKU Endowment Fund to L.J. Jin.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The RNA-seq datasets generated in this study have been deposited in NCBI’s Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi, accessed on 17 May 2022) for access through the GEO series accession number GSE184777.
Acknowledgments
We are grateful to Edith Tong and Wendy Yu at the Central Research Laboratories, Faculty of Dentistry, The University of Hong Kong, for their technical assistance.
Conflicts of Interest
The authors declare that they have no conflicts of interest in connection with this article.
References
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Petersen, P.E.; Ogawa, H. The global burden of periodontal disease: Towards integration with chronic disease prevention and control. Periodontol. 2000 2012, 60, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.J.; Armitage, G.C.; Klinge, B.; Lang, N.P.; Tonetti, M.; Williams, R.C. Global oral health inequalities: Task group—Periodontal disease. Adv. Dent. Res. 2011, 23, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassebaum, N.J.; Bernabe, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.; Marcenes, W. Global burden of severe periodontitis in 1990–2010: A systematic review and meta-regression. J. Dent. Res. 2014, 93, 1045–1053. [Google Scholar] [CrossRef]
- Listl, S.; Galloway, J.; Mossey, P.A.; Marcenes, W. Global economic impact of dental diseases. J. Dent. Res. 2015, 94, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Herrera, D.; Meyle, J.; Renvert, S.; Jin, L. White Paper on Prevention and Management of Periodontal Diseases for Oral Health and General Health. Available online: https://www.fdiworlddental.org/sites/default/files/2020-11/gphp-2018-white_paper-en.pdf (accessed on 15 April 2022).
- Tonetti, M.S.; Kornman, K.S. Special Issue: Periodontitis and Systemic Diseases—Proceedings of a workshop jointly held by the European Federation of Periodontology and American Academy of Periodontology. J. Clin. Periodontol. 2013, 40, S1–S214. [Google Scholar]
- Jin, L.J.; Lamster, I.B.; Greenspan, J.S.; Pitts, N.B.; Scully, C.; Warnakulasuriya, S. Global burden of oral diseases: Emerging concepts, management and interplay with systemic health. Oral. Dis. 2016, 22, 609–619. [Google Scholar] [CrossRef]
- Monsarrat, P.; Blaizot, A.; Kemoun, P.; Ravaud, P.; Nabet, C.; Sixou, M.; Vergnes, J.N. Clinical research activity in periodontal medicine: A systematic mapping of trial registers. J. Clin. Periodontol. 2016, 43, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, M.S.; Jepsen, S.; Jin, L.; Otomo-Corgel, J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J. Clin. Periodontol. 2017, 44, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef]
- Honda, K. Porphyromonas gingivalis sinks teeth into the oral microbiota and periodontal disease. Cell Host Microbe 2011, 10, 423–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; McIntosh, M.L.; Alsam, A.; Kirkwood, K.L.; Lambris, J.D.; et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Uehara, A.; Muramoto, K.; Imamura, T.; Nakayama, K.; Potempa, J.; Travis, J.; Sugawara, S.; Takada, H. Arginine-specific gingipains from Porphyromonas gingivalis stimulate production of hepatocyte growth factor (scatter factor) through protease-activated receptors in human gingival fibroblasts in culture. J. Immunol. 2005, 175, 6076–6084. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G.; Wang, M.; Liang, S.; Triantafilou, M.; Triantafilou, K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc. Natl. Acad. Sci. USA 2008, 105, 13532–13537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Fung, Y.E.; Yin, X.; Seneviratne, C.J.; Che, C.M.; Jin, L. Controlled cellular redox, repressive hemin utilization and adaptive stress responses are crucial to metronidazole tolerance of Porphyromonas gingivalis persisters. J. Clin. Periodontol. 2018, 45, 1211–1221. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, T.; Li, X.; Jin, L. Metronidazole-treated Porphyromonas gingivalis persisters invade human gingival epithelial cells and perturb innate responses. Antimicrob. Agents Chemother. 2020, 64, e02529-19. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Bigger, J. Treatment of Staphylococcal infections with penicillin by intermittent sterilisation. Lancet 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.R.; Burns, J.L.; Lory, S.; Lewis, K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 2010, 192, 6191–6199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafleur, M.D.; Qi, Q.; Lewis, K. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob. Agents Chemother. 2010, 54, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goneau, L.W.; Yeoh, N.S.; MacDonald, K.W.; Cadieux, P.A.; Burton, J.P.; Razvi, H.; Reid, G. Selective target inactivation rather than global metabolic dormancy causes antibiotic tolerance in uropathogens. Antimicrob. Agents Chemother. 2014, 58, 2089–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Weinrick, B.C.; Kalivoda, E.J.; Yang, H.; Munsamy, V.; Vilcheze, C.; Weisbrod, T.R.; Larsen, M.H.; O’Donnell, M.R.; Pym, A.; et al. Dual-reporter Mycobacteriophages (Phi2DRMs) reveal preexisting Mycobacterium tuberculosis persistent cells in human sputum. MBio 2016, 7, e01023-16. [Google Scholar] [CrossRef] [Green Version]
- Stapels, D.A.C.; Hill, P.W.S.; Westermann, A.J.; Fisher, R.A.; Thurston, T.L.; Saliba, A.E.; Blommestein, I.; Vogel, J.; Helaine, S. Salmonella persisters undermine host immune defenses during antibiotic treatment. Science 2018, 362, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Bakkeren, E.; Huisman, J.S.; Fattinger, S.A.; Hausmann, A.; Furter, M.; Egli, A.; Slack, E.; Sellin, M.E.; Bonhoeffer, S.; Regoes, R.R.; et al. Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature 2019, 573, 276–280. [Google Scholar] [CrossRef]
- Gunther, J.; Seyfert, H.M. The first line of defence: Insights into mechanisms and relevance of phagocytosis in epithelial cells. Semin. Immunopathol. 2018, 40, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Choi, Y. Innate immune response to oral bacteria and the immune evasive characteristics of periodontal pathogens. J. Periodontal. Implant Sci. 2013, 43, 3–11. [Google Scholar] [CrossRef]
- Ramage, G.; Lappin, D.F.; Millhouse, E.; Malcolm, J.; Jose, A.; Yang, J.; Bradshaw, D.J.; Pratten, J.R.; Culshaw, S. The epithelial cell response to health and disease associated oral biofilm models. J. Periodontal. Res. 2017, 52, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Rath-Deschner, B.; Memmert, S.; Damanaki, A.; de Molon, R.S.; Nokhbehsaim, M.; Eick, S.; Kirschneck, C.; Cirelli, J.A.; Deschner, J.; Jager, A.; et al. CXCL5, CXCL8, and CXCL10 regulation by bacteria and mechanical forces in periodontium. Ann. Anat. 2021, 234, 151648. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, X.; Xiang, Y.; Qu, X.; Liu, H.; Liu, C.; Tan, M.; Jiang, J.; Qin, X. Role of epithelial chemokines in the pathogenesis of airway inflammation in asthma (Review). Mol. Med. Rep. 2018, 17, 6935–6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkaim, R.; Bugueno-Valdebenito, I.M.; Benkirane-Jessel, N.; Tenenbaum, H. Porphyromonas gingivalis and its lipopolysaccharide differently modulate epidermal growth factor-dependent signaling in human gingival epithelial cells. J. Oral. Microbiol. 2017, 9, 1334503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque-Souza, E.; Balzarini, D.; Ando-Suguimoto, E.S.; Ishikawa, K.H.; Simionato, M.R.L.; Holzhausen, M.; Mayer, M.P.A. Probiotics alter the immune response of gingival epithelial cells challenged by Porphyromonas gingivalis. J. Periodontal. Res. 2019, 54, 115–127. [Google Scholar] [CrossRef]
- Lee, K.; Roberts, J.S.; Choi, C.H.; Atanasova, K.R.; Yilmaz, O. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence 2018, 9, 845–859. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Peng, S.L. Immune regulation by Foxo transcription factors. Autoimmunity 2007, 40, 462–469. [Google Scholar] [CrossRef]
- Peng, S.L. Foxo in the immune system. Oncogene 2008, 27, 2337–2344. [Google Scholar] [CrossRef] [Green Version]
- Kandula, V.; Kosuru, R.; Li, H.; Yan, D.; Zhu, Q.; Lian, Q.; Ge, R.S.; Xia, Z.; Irwin, M.G. Forkhead box transcription factor 1: Role in the pathogenesis of diabetic cardiomyopathy. Cardiovasc. Diabetol. 2016, 15, 44. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Song, L.; Tian, C.; Wang, Y.; Miao, F.; Zheng, J.; Lu, C.; Alsadun, S.; Graves, D.T. FOXO1 Regulates Bacteria-Induced Neutrophil Activity. Front. Immunol. 2017, 8, 1088. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.Q.; Li, A.; Yang, Y.; Li, X.X.; Zhang, L.N.; Guo, H.C. The regulation of FOXO1 and its role in disease progression. Life Sci. 2018, 193, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Milovanova, T.N. Mucosal Immunity and the FOXO1 Transcription Factors. Front. Immunol. 2019, 10, 2530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Graves, D.T. FOXO transcription factors: Their clinical significance and regulation. Biomed Res. Int. 2014, 2014, 925350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Sztukowska, M.; Ojo, A.; Scott, D.A.; Wang, H.; Lamont, R.J. FOXO responses to Porphyromonas gingivalis in epithelial cells. Cell. Microbiol. 2015, 17, 1605–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, T.; Shigematsu, N.; Maruki, R.; Urano, Y.; Tanaka, H.; Shimaya, A.; Shimokawa, T.; Shibasaki, M. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: Improvement of fasting glycemia in diabetic db/db mice. Mol. Pharmacol. 2010, 78, 961–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, K.; Al Batran, R.; Altamimi, T.R.; Greenwell, A.A.; Saed, C.T.; Tabatabaei Dakhili, S.A.; Dimaano, M.T.E.; Zhang, Y.; Eaton, F.; Sutendra, G.; et al. FoxO1 inhibition alleviates type 2 diabetes-related diastolic dysfunction by increasing myocardial pyruvate dehydrogenase activity. Cell Rep. 2021, 35, 108935. [Google Scholar] [CrossRef]
- Jeon, H.H.; Yu, Q.; Witek, L.; Lu, Y.; Zhang, T.; Stepanchenko, O.; Son, V.J.; Spencer, E.; Oshilaja, T.; Shin, M.K.; et al. Clinical application of a FOXO1 inhibitor improves connective tissue healing in a diabetic minipig model. Am. J. Transl. Res. 2021, 13, 781–791. [Google Scholar]
- Lee, Y.K.; Diaz, B.; Deroose, M.; Lee, S.X.; Belvedere, S.; Accili, D.; Leibel, R.L.; Lin, H.V. FOXO1 inhibition synergizes with FGF21 to normalize glucose control in diabetic mice. Mol. Metab. 2021, 49, 101187. [Google Scholar] [CrossRef]
- Cheng, T.; Lai, Y.T.; Wang, C.; Wang, Y.; Jiang, N.; Li, H.; Sun, H.; Jin, L. Bismuth drugs tackle Porphyromonas gingivalis and attune cytokine response in human cells. Metallomics 2019, 11, 1207–1218. [Google Scholar] [CrossRef]
- Wang, C.; Li, X.; Cheng, T.; Sun, H.; Jin, L. Eradication of Porphyromonas gingivalis persisters through colloidal bismuth subcitrate synergistically combined with metronidazole. Front. Microbiol. 2021, 12, 748121. [Google Scholar] [CrossRef]
- Van Acker, H.; Sass, A.; Bazzini, S.; De Roy, K.; Udine, C.; Messiaen, T.; Riccardi, G.; Boon, N.; Nelis, H.J.; Mahenthiralingam, E.; et al. Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding production of reactive oxygen species. PLoS ONE 2013, 8, e58943. [Google Scholar] [CrossRef] [Green Version]
- Helaine, S.; Kugelberg, E. Bacterial persisters: Formation, eradication, and experimental systems. Trends Microbiol. 2014, 22, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulou, P.G.; Benakanakere, M.R.; Galicia, J.C.; Kinane, D.F. The host cytokine response to Porphyromonas gingivalis is modified by gingipains. Oral Microbiol. Immunol. 2009, 24, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopoulou, P.G.; Galicia, J.C.; Benakanakere, M.R.; Garcia, C.A.; Potempa, J.; Kinane, D.F. Porphyromonas gingivalis induce apoptosis in human gingival epithelial cells through a gingipain-dependent mechanism. BMC Microbiol. 2009, 9, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeger, S.; Jarzina, F.; Domann, E.; Meyle, J. Porphyromonas gingivalis activates NFkappaB and MAPK pathways in human oral epithelial cells. BMC Immunol. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Roberts, J.S.; Atanasova, K.R.; Chowdhury, N.; Yilmaz, O. A novel kinase function of a nucleoside-diphosphate-kinase homologue in Porphyromonas gingivalis is critical in subversion of host cell apoptosis by targeting heat-shock protein 27. Cell. Microbiol. 2018, 20, e12825. [Google Scholar] [CrossRef]
- Miller, M.R.; Brunelli, J.P.; Wheeler, P.A.; Liu, S.; Rexroad, C.E., 3rd; Palti, Y.; Doe, C.Q.; Thorgaard, G.H. A conserved haplotype controls parallel adaptation in geographically distant salmonid populations. Mol. Ecol. 2012, 21, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Li, X.; Cooper, N.G.F.; O’Toole, T.E.; Rouchka, E.C. Choice of library size normalization and statistical methods for differential gene expression analysis in balanced two-group comparisons for RNA-seq studies. BMC Genom. 2020, 21, 75. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).