
Modulation of ODH Propane Selectivity by Zeolite Support Desilication: Vanadium Species Anchored to Al-Rich Shell as Crucial Active Sites

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
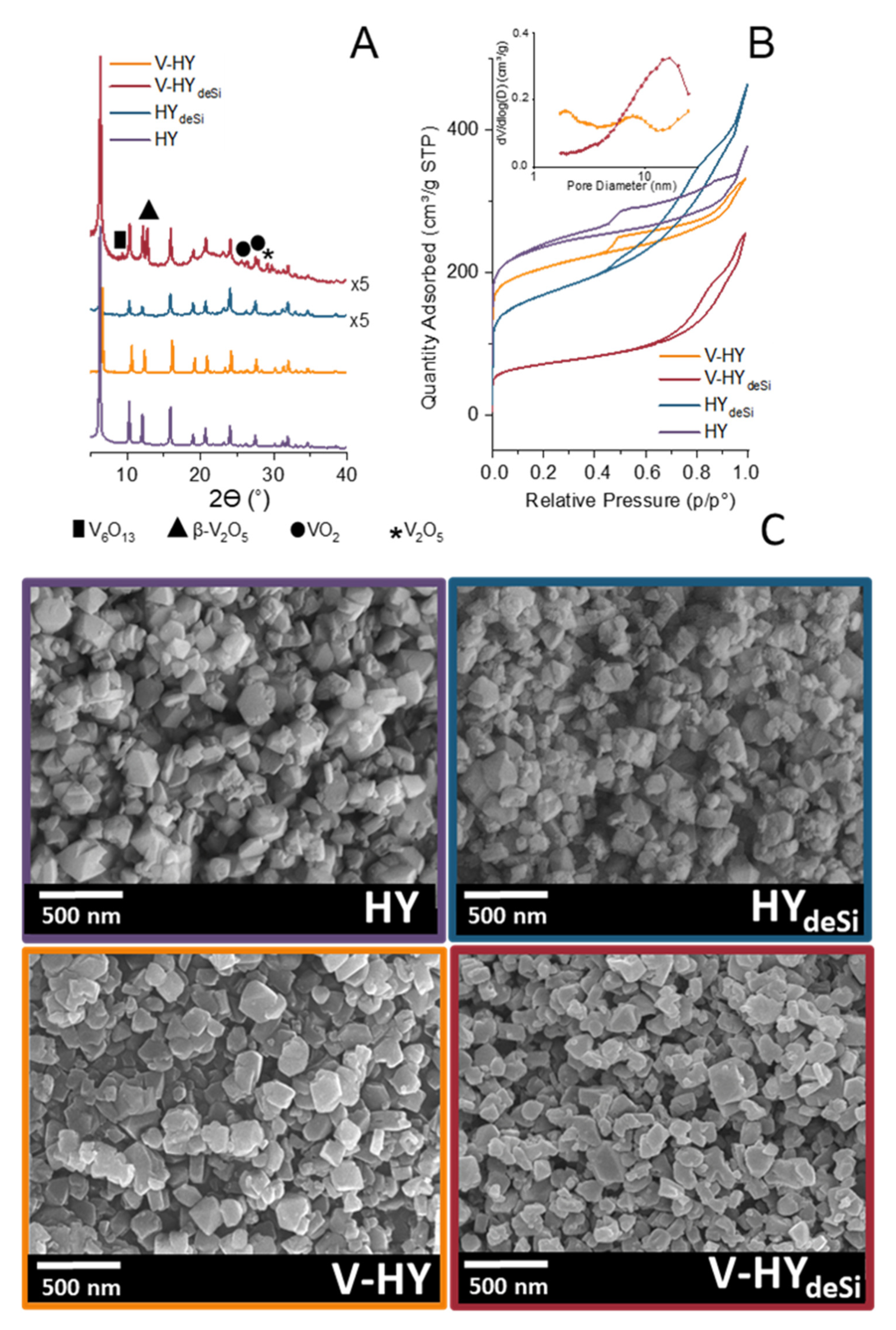
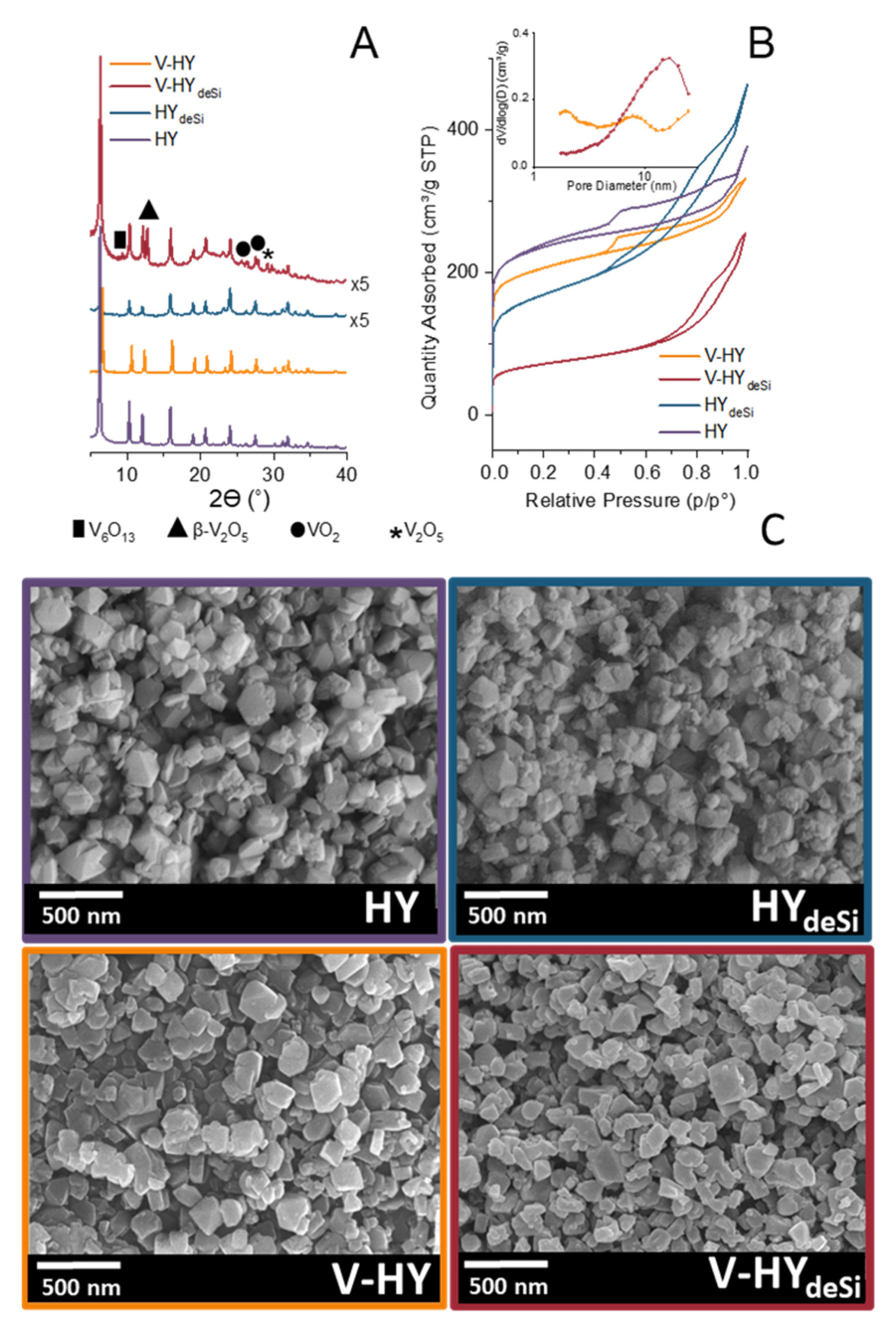
2.1. Structure, Morphology, and Textural Properties
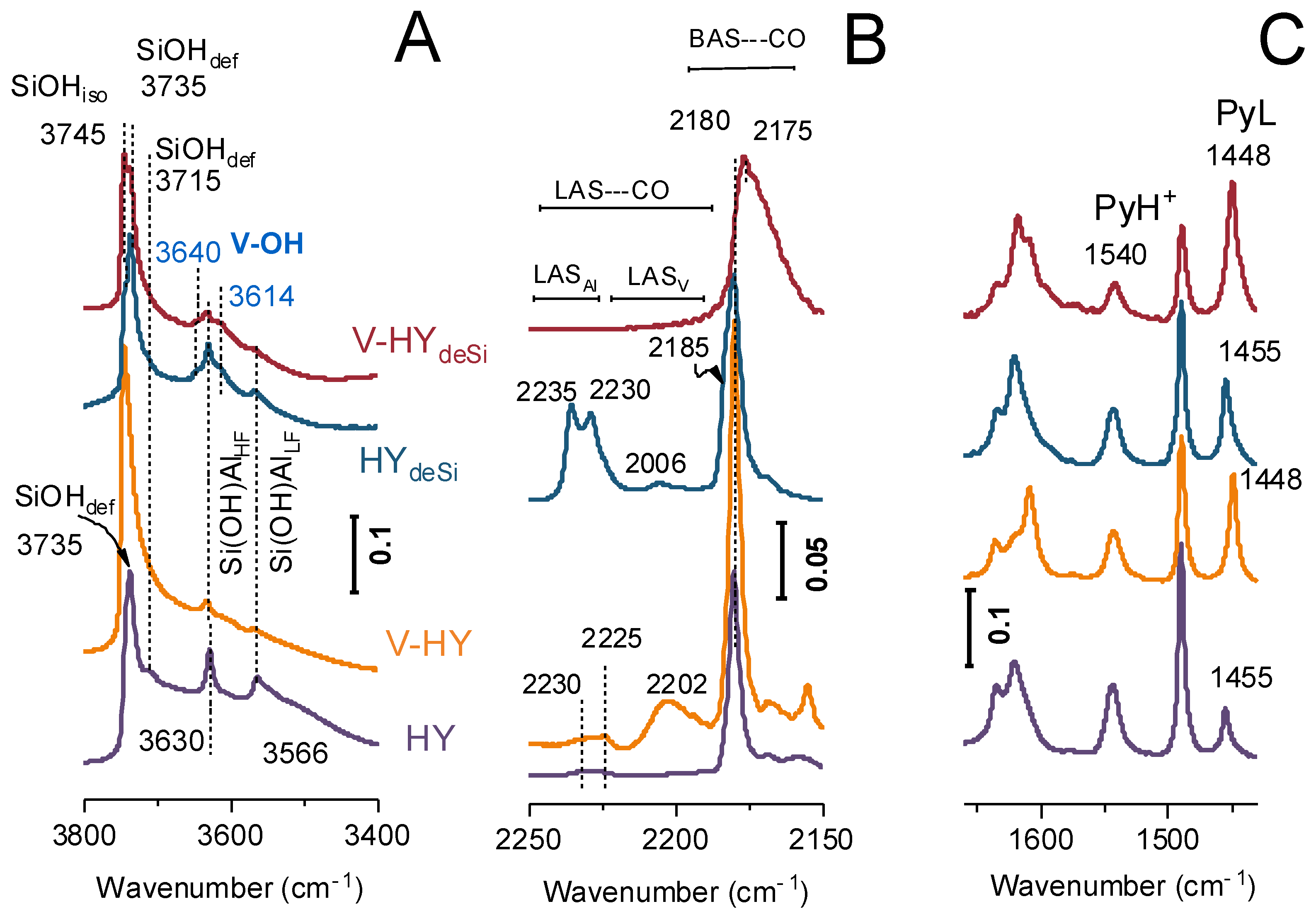
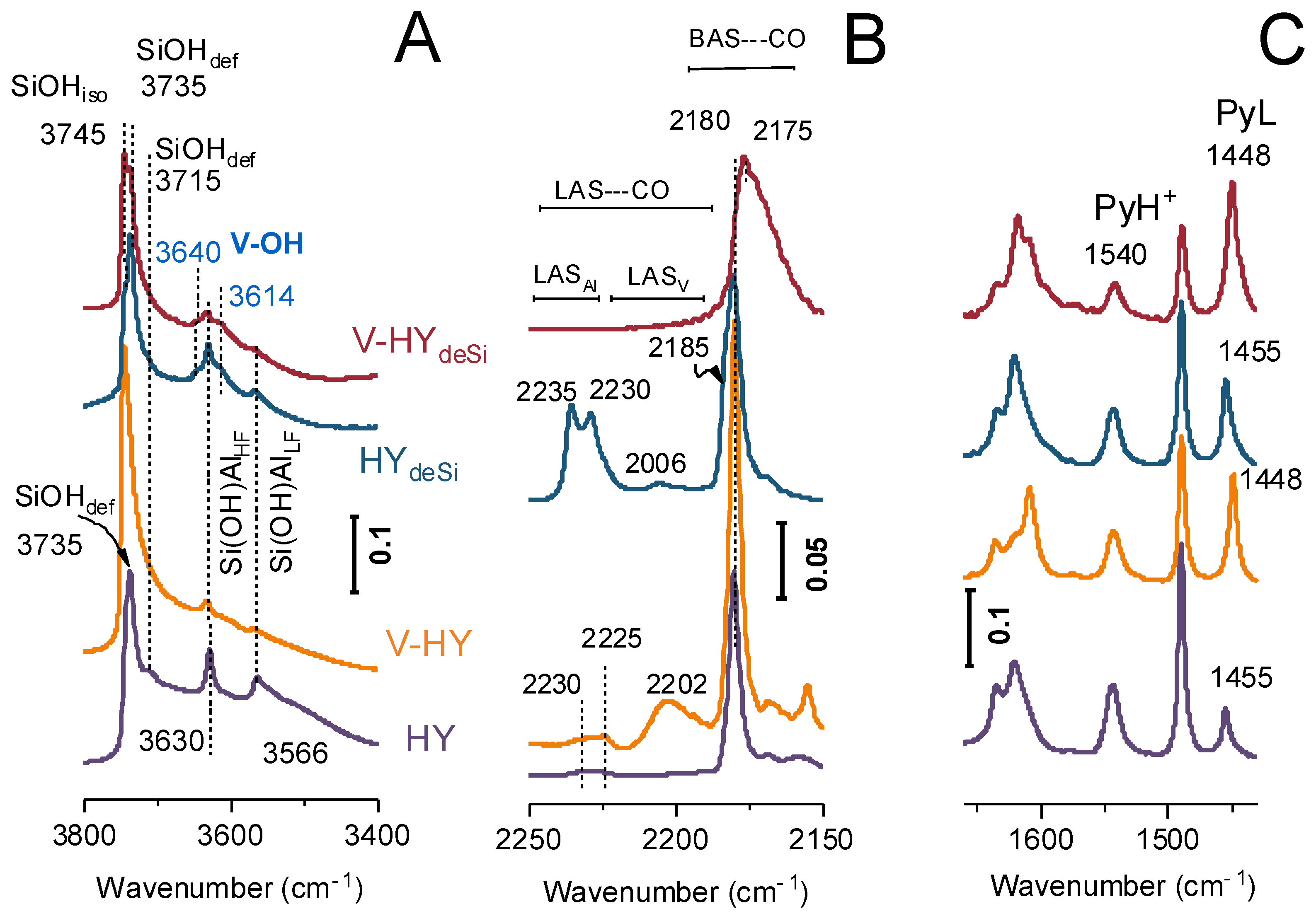
2.2. Acidity FT-IR Characterization with CO and Pyridine as the Probes
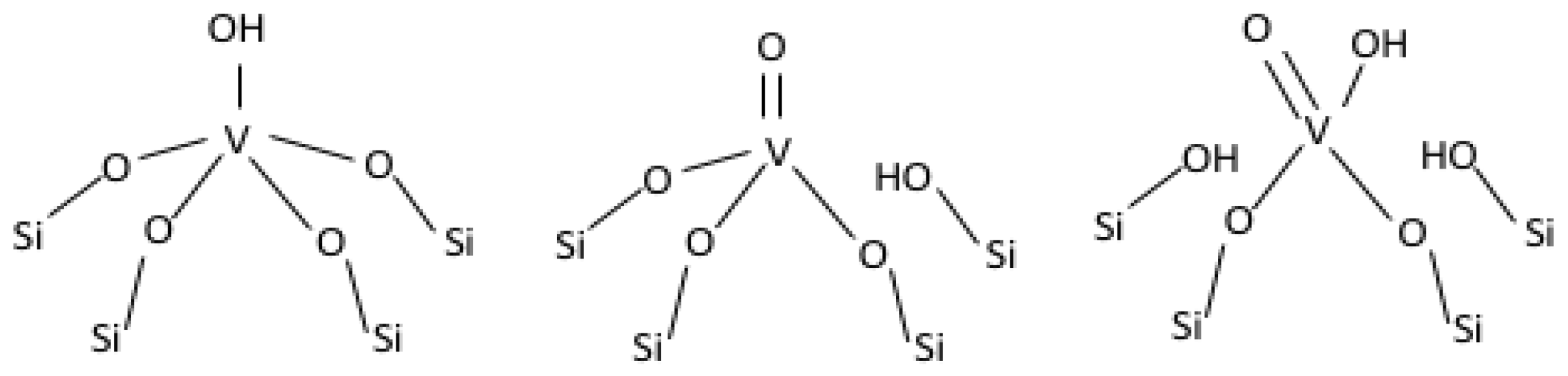
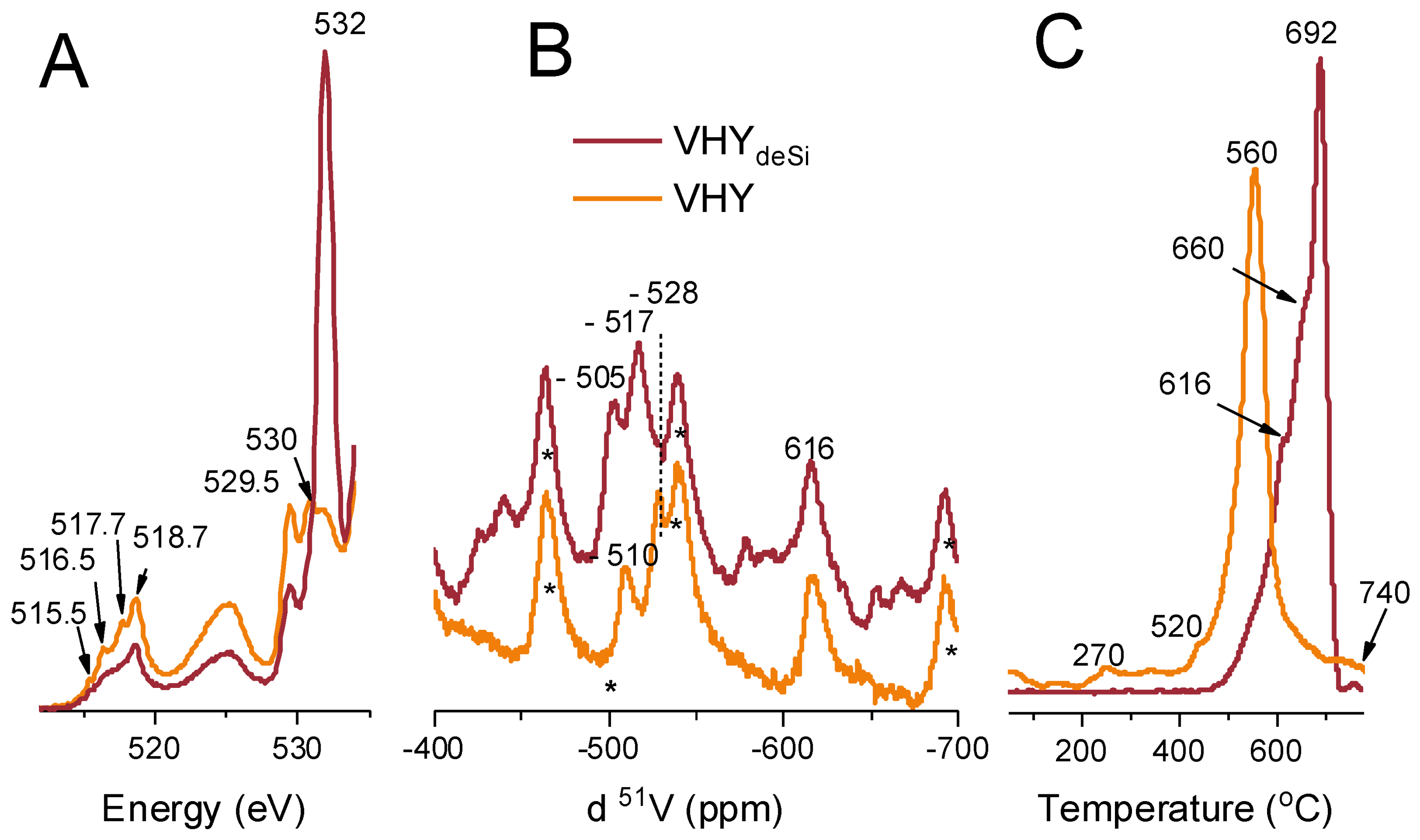
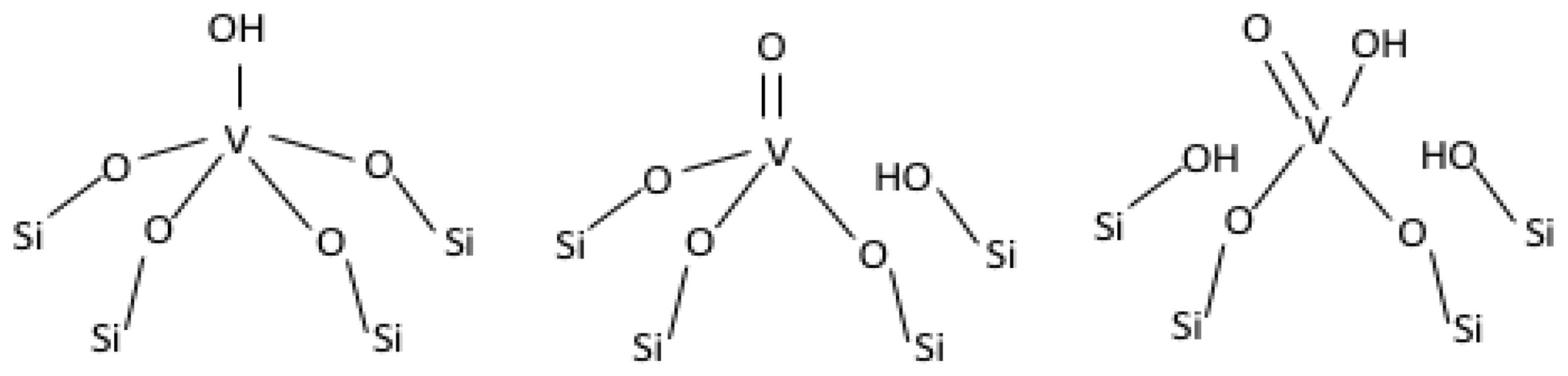
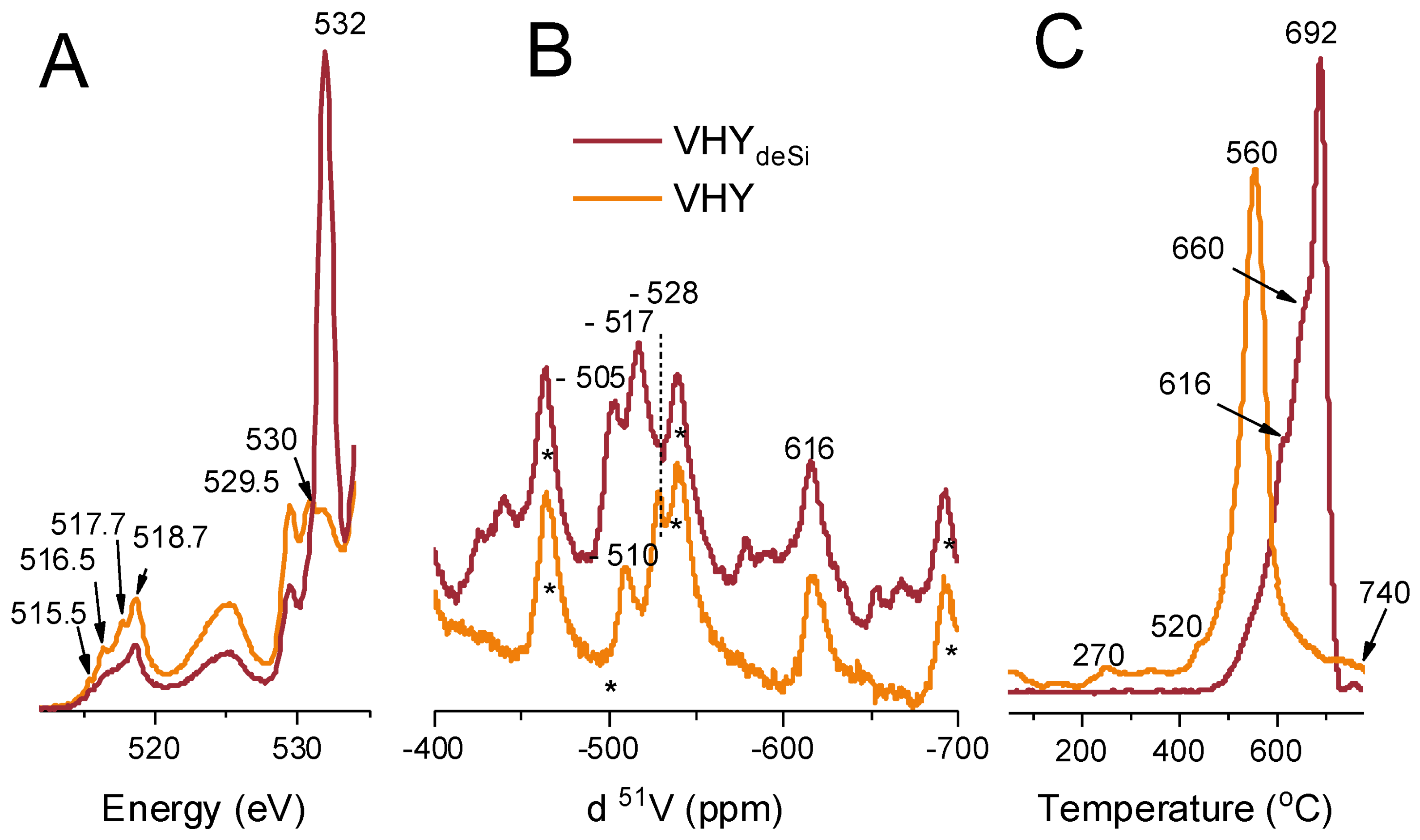
2.3. On the Nature of Vanadium Species
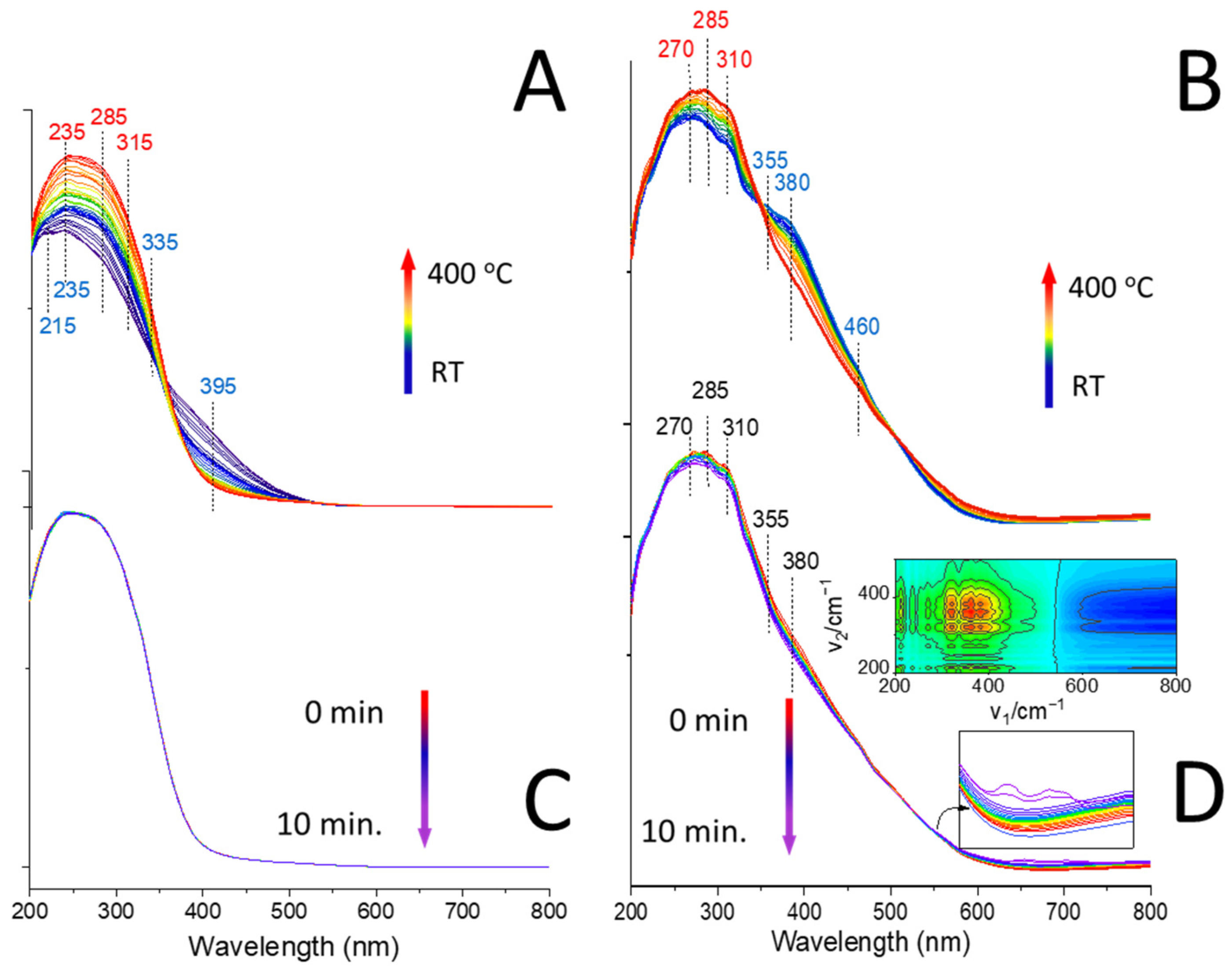
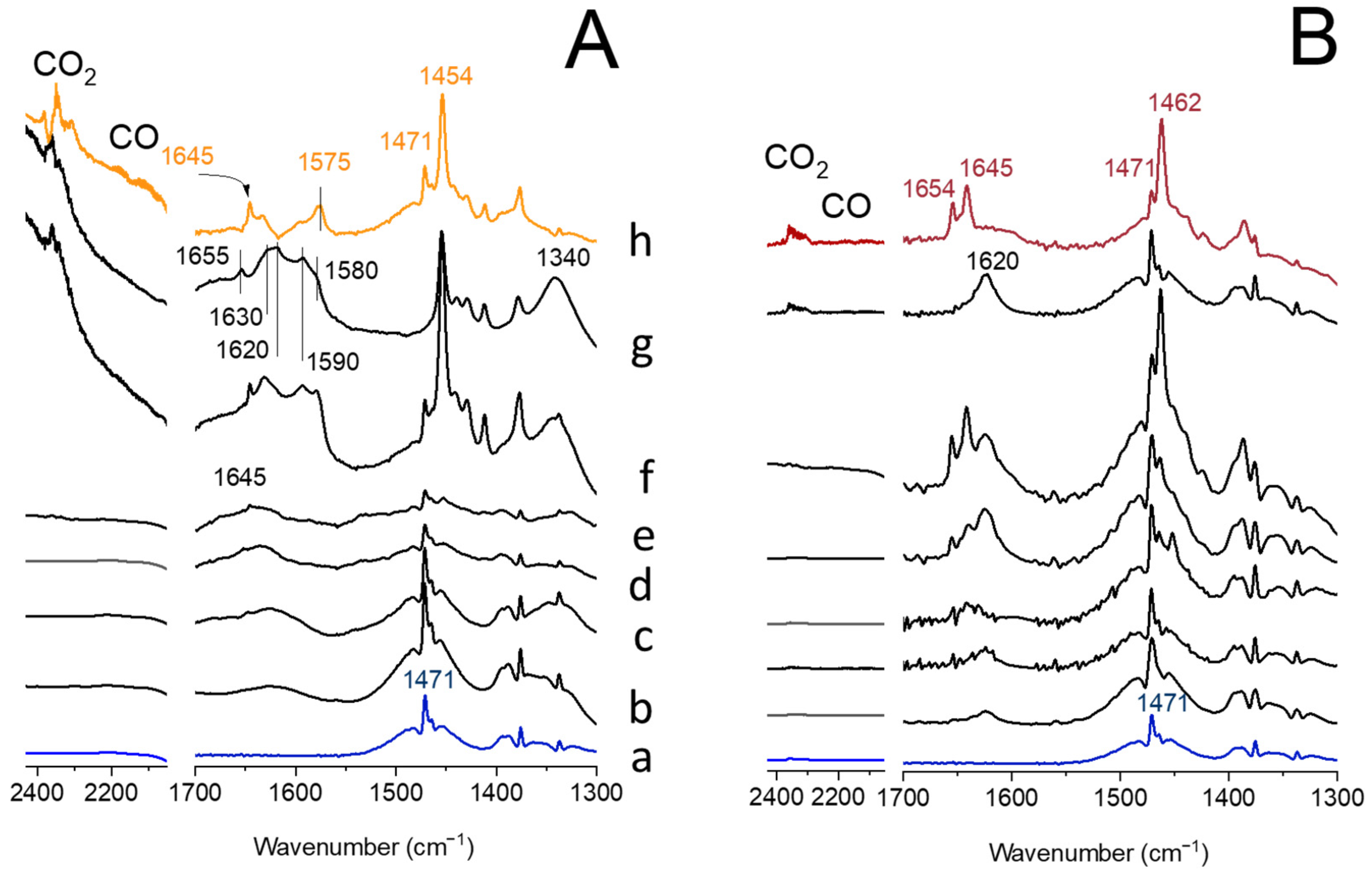
2.4. In Situ FT-IR Studies of ODH Process
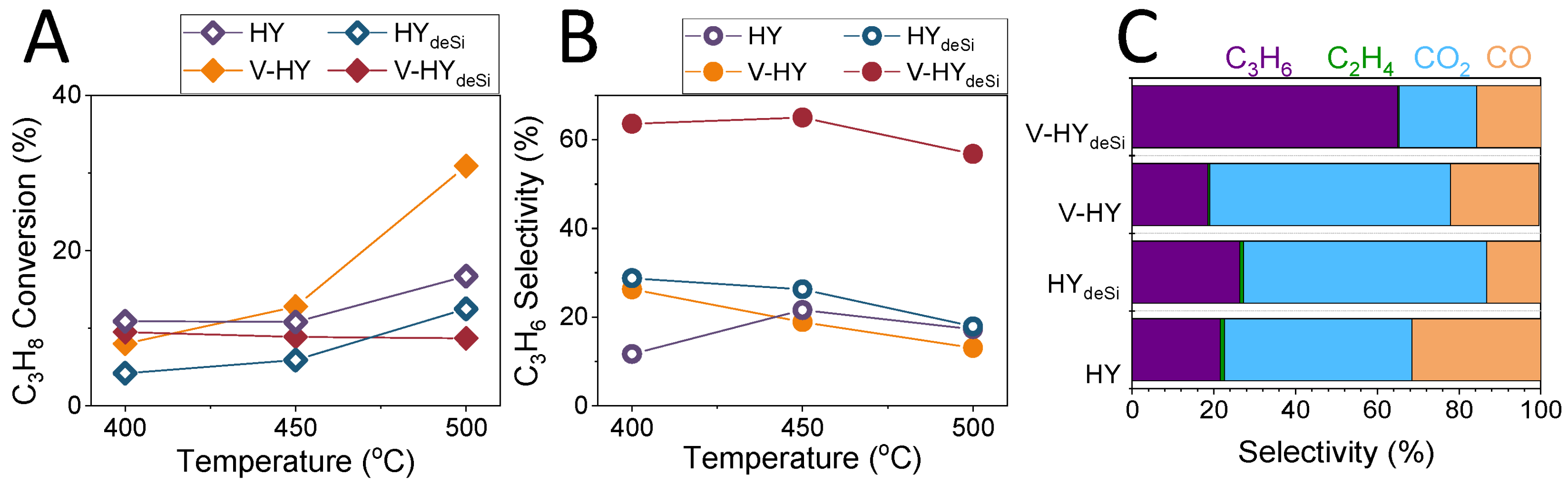
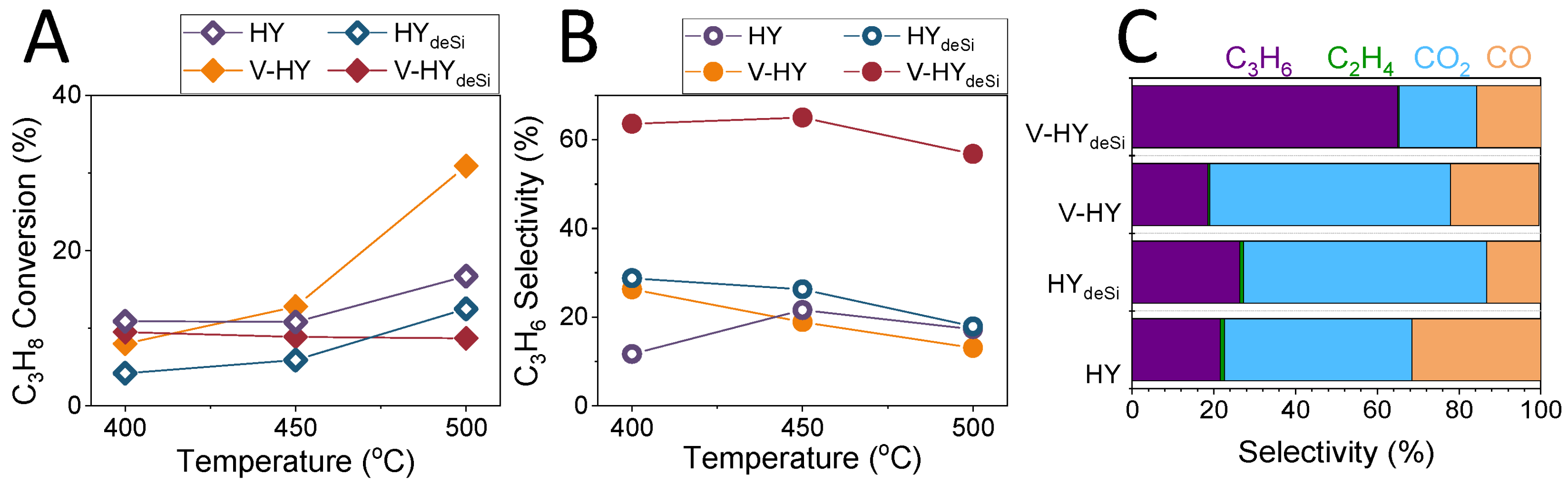
2.5. Catalytic Properties of the System
3. Materials and Methods
3.1. Synthesis
3.2. X-ray Diffraction (XRD)
3.3. X-ray Fluorescence (XRF)
3.4. Scanning Electron Microscopy (SEM)
3.5. X-ray Absorption Spectroscopy (XAS)
3.6. Nuclear Magnetic Resonance (51V MAS NMR)
3.7. Temperature Programmed Reduction with Hydrogen (H2-TPR)
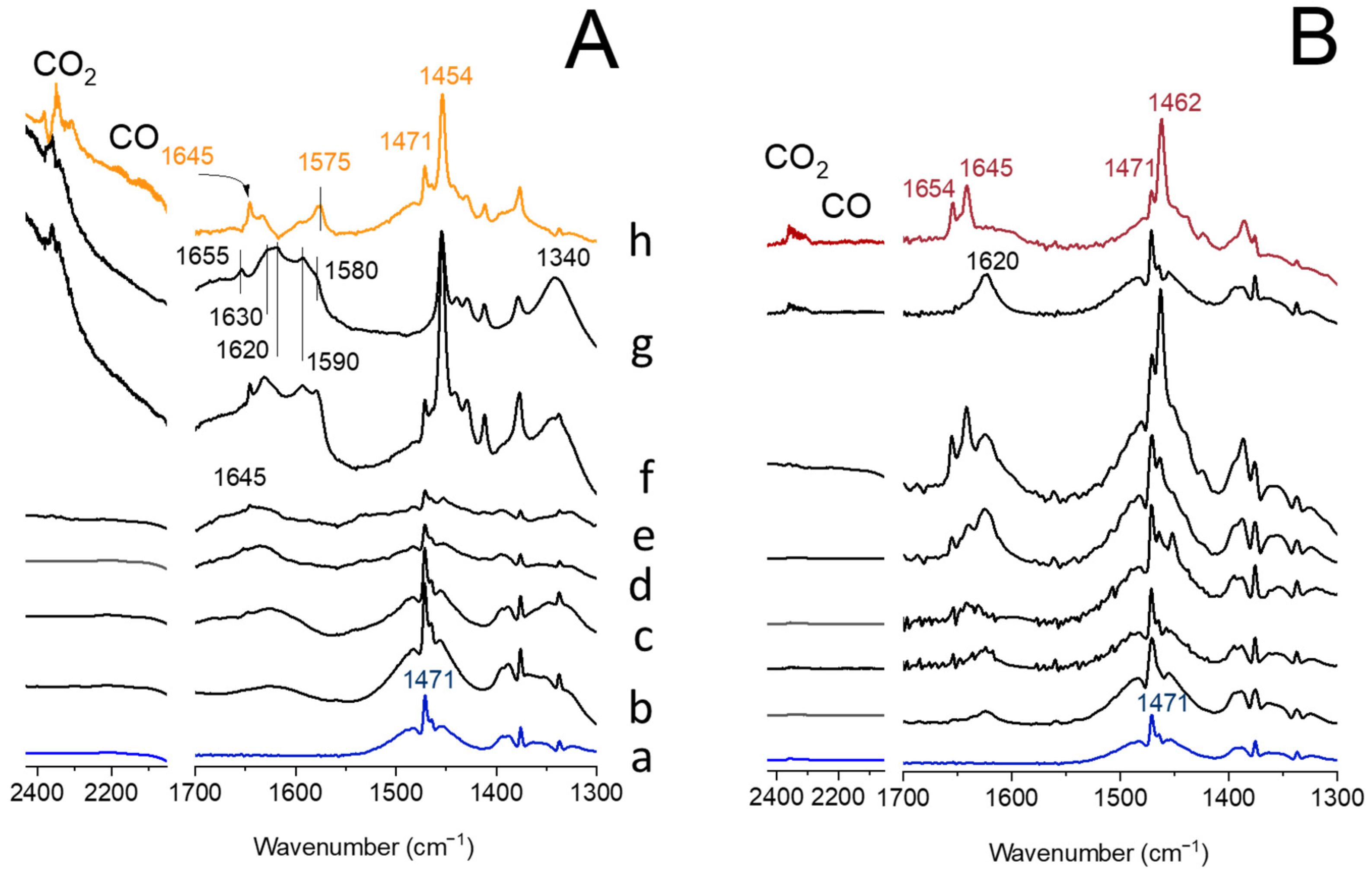
3.8. FT-IR Spectroscopy Studies of Probe Molecules Sorption
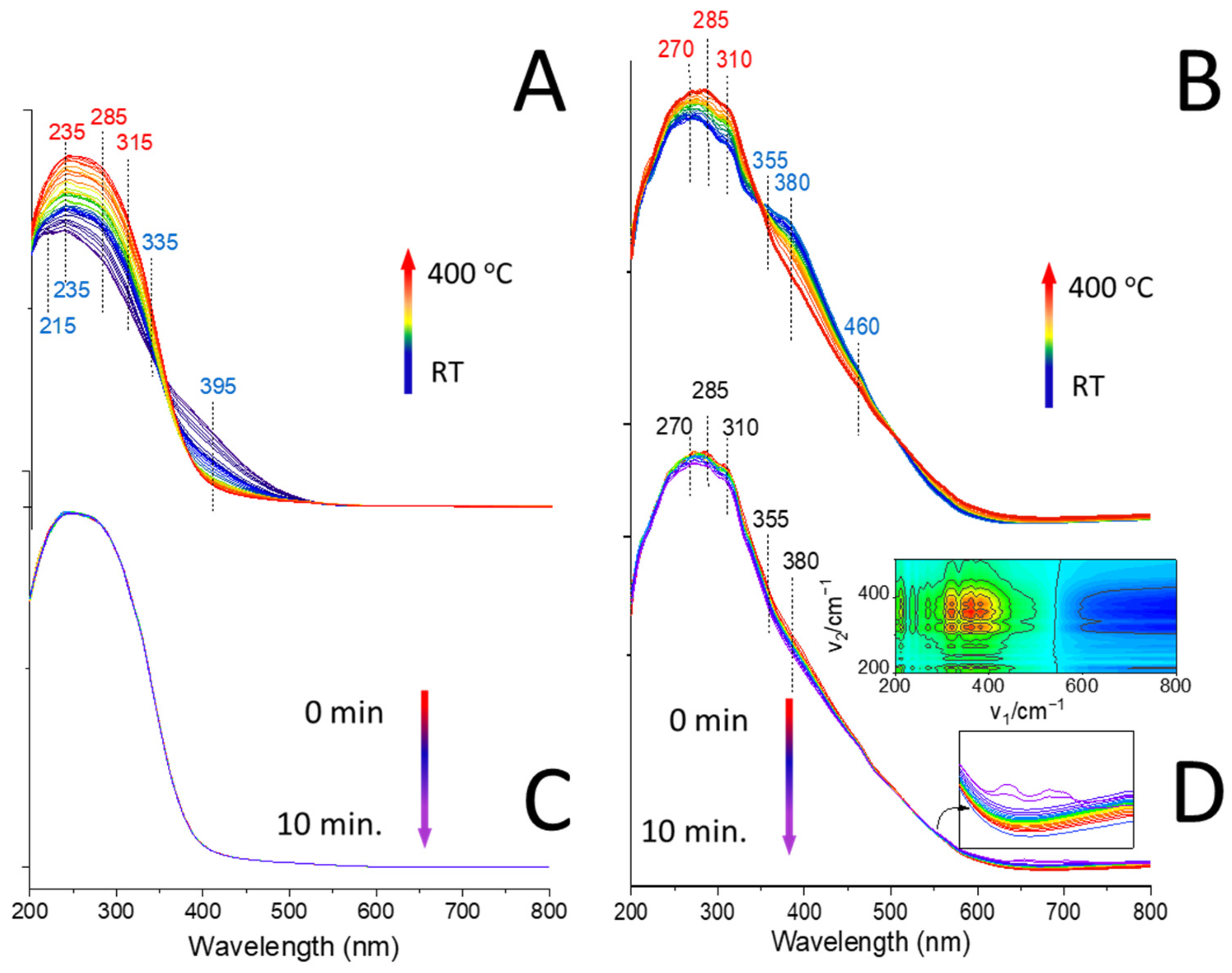
3.9. Operando DR UV-vis Spectroscopy Studies
3.10. Catalytic Tests
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dickson, D.; Hussain, A.; Kumpf, B. The Future of Petrochemicals: Growth Surrounded by Uncertainty. 2019. Available online: https://www2.deloitte.com/content/dam/Deloitte/us/Documents/energy-resources/the-future-of-petrochemicals.pdf (accessed on 2 December 2021).
- Kaddouri, A. CH bond activation in the presence or absence of oxygen or nitrous oxide. React. Kinet. Mech. Catal. 2004, 82, 401–409. [Google Scholar] [CrossRef]
- Erdöhelyi, A.; Solymosi, F. Partial oxidation of ethane over supported vanadium pentoxide catalysts. J. Catal. 1990, 123, 31–42. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Grabowski, R. Kinetics of Oxidative Dehydrogenation of C2-C3 Alkanes on Oxide Catalysts. Catal. Rev. 2006, 48, 199–268. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Yang, D.; Wang, Z.; Yuan, Z.-Y. State-of-the-art catalysts for direct dehydrogenation of propane to propylene. Chin. J. Catal. 2019, 40, 1233–1254. [Google Scholar] [CrossRef]
- Langeslay, R.R.; Kaphan, D.M.; Marshall, C.L.; Stair, P.C.; Sattelberger, A.P.; Delferro, M. Catalytic Applications of Vanadium: A Mechanistic Perspective. Chem. Rev. 2019, 119, 2128–2191. [Google Scholar] [CrossRef]
- Blasco, T.; Nieto, J. Oxidative dyhydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl. Catal. A Gen. 1997, 157, 117–142. [Google Scholar] [CrossRef]
- Grasselli, R.K. Fundamental Principles of Selective Heterogeneous Oxidation Catalysis. Top. Catal. 2002, 21, 79–88. [Google Scholar] [CrossRef]
- Liu, J.; Mohamed, F.; Sauer, J. Selective oxidation of propene by vanadium oxide monomers supported on silica. J. Catal. 2014, 317, 75–82. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H. Insights into the structural requirements for oxidative dehydrogenation of propane on crystalline Mg-V-O catalysts. Appl. Catal. A Gen. 2018, 568, 1–10. [Google Scholar] [CrossRef]
- Cortes, I.; Rubio, O.; Herguido, J.; Menéndez, M. Kinetics under dynamic conditions of the oxidative dehydrogenation of butane with doped V/MgO. Catal. Today 2004, 91–92, 281–284. [Google Scholar] [CrossRef]
- Madeira, L.M.; Martın-Aranda, R.M.; Maldonado-Hódar, F.J.; Fierro, J.L.G.; Portela, M.F. Oxidative Dehydrogenation ofn-Butane over Alkali and Alkaline Earth-Promoted α-NiMoO4Catalysts. J. Catal. 1997, 169, 469–479. [Google Scholar] [CrossRef]
- Lisi, L.; Ruoppolo, G.; Casaletto, M.; Galli, P.; Massucci, M.; Patrono, P.; Pinzari, F. Vanadium-metal(IV)phosphates as catalysts for the oxidative dehydrogenation of ethane. J. Mol. Catal. A Chem. 2005, 232, 127–134. [Google Scholar] [CrossRef]
- Grant, J.T.; Venegas, J.M.; McDermott, W.; Hermans, I. Aerobic Oxidations of Light Alkanes over Solid Metal Oxide Catalysts. Chem. Rev. 2017, 118, 2769–2815. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, A.M.; Mason, O.M. Ethylene production via Oxidative Dehydrogenation of Ethane using M1 catalyst. Catal. Today 2017, 285, 159–165. [Google Scholar] [CrossRef]
- Venegas, J.M.; Grant, J.T.; McDermott, W.P.; Burt, S.P.; Micka, J.; Carrero, C.A.; Hermans, I. Selective Oxidation of n-Butane and Isobutane Catalyzed by Boron Nitride. ChemCatChem 2017, 9, 2118–2127. [Google Scholar] [CrossRef]
- Tian, J.; Tan, J.; Xu, M.; Zhang, Z.; Wan, S.; Wang, S.; Lin, J.; Wang, Y. Propane oxidative dehydrogenation over highly selective hexagonal boron nitride catalysts: The role of oxidative coupling of methyl. Sci. Adv. 2019, 5, eaav8063. [Google Scholar] [CrossRef] [Green Version]
- Carrero, C.A.; Schloegl, R.; Wachs, I.E.; Schomaecker, R. Critical Literature Review of the Kinetics for the Oxidative Dehydrogenation of Propane over Well-Defined Supported Vanadium Oxide Catalysts. ACS Catal. 2014, 4, 3357–3380. [Google Scholar] [CrossRef]
- Rasmussen, S.B.; Mikolajska, E.; Daturi, M.; Bañares, M.A. Structural characteristics of an amorphous VPO monolayer on alumina for propane ammoxidation. Catal. Today 2012, 192, 96–103. [Google Scholar] [CrossRef]
- Held, A.; Kowalska-Kuś, J.; Millot, Y.; Averseng, F.; Calers, C.; Valentin, L.; Dzwigaj, S. Influence of the Preparation Procedure of Vanadium-Containing SiBEA Zeolites on Their Catalytic Activity in Propene Epoxidation. J. Phys. Chem. C 2018, 122, 18570–18582. [Google Scholar] [CrossRef]
- Wang, W.; Gao, X.; Yang, Q.; Wang, X.; Song, F.; Zhang, Q.; Han, Y.; Tan, Y. Vanadium oxide modified H-beta zeolite for the synthesis of polyoxymethylene dimethyl ethers from dimethyl ether direct oxidation. Fuel 2019, 238, 289–297. [Google Scholar] [CrossRef]
- Floris, B.; Sabuzi, F.; Coletti, A.; Conte, V. Sustainable vanadium-catalyzed oxidation of organic substrates with H2O2. Catal. Today 2017, 285, 49–56. [Google Scholar] [CrossRef]
- Grzybowska-Świerkosz, B. Thirty years in selective oxidation on oxides: What have we learned? Top. Catal. 2000, 11, 23–42. [Google Scholar] [CrossRef]
- Held, A.; Kowalska-Kuś, J.; Nowińska, K.; Góra-Marek, K. Potassium-modified silica-supported vanadium oxide catalysts applied for propene epoxidation. J. Catal. 2017, 347, 21–35. [Google Scholar] [CrossRef]
- Lv, G.; Zhang, L.; Zhai, Y.; Zhang, X.; Wang, F.; Jiang, T.; Shen, Y. Preparation of vanadium-MFI zeolite for oxidative desulfurization with the aid of ammonium carbonate. Appl. Catal. A Gen. 2020, 607, 117862. [Google Scholar] [CrossRef]
- Huang, C.; Wu, P.; Guo, Y.; Guo, Y. Facile synthesis of mesoporous kaolin catalyst carrier and its application in deep oxidative desulfurization. Microporous Mesoporous Mater. 2020, 306, 110415. [Google Scholar] [CrossRef]
- Yoon, T.-U.; Ahn, S.; Kim, A.R.; Notestein, J.M.; Farha, O.K.; Bae, Y.S. Cyclohexene epoxidation with H2O2 in the vapor and liquid phases over a vanadium-based metal–organic framework. Catal. Sci. Technol. 2020, 10, 4580–4585. [Google Scholar] [CrossRef]
- Liu, X.; Yang, F.; Gao, S.; Shao, B.; Zhou, S.; Kong, Y. Preparation of ZSM-5 containing vanadium and Brønsted acid sites with high promoting of styrene oxidation using 30% H2O2. Chin. J. Chem. Eng. 2020, 28, 1302–1310. [Google Scholar] [CrossRef]
- Guerrero-Pérez, M.O. Supported, bulk and bulk-supported vanadium oxide catalysts: A short review with an historical perspective. Catal. Today 2017, 285, 226–233. [Google Scholar] [CrossRef]
- Held, A.; Kowalska-Kuś, J.; Nowińska, K. Epoxidation of propene on vanadium species supported on silica supports of different structure. Catal. Commun. 2012, 17, 108–113. [Google Scholar] [CrossRef]
- Centi, G.; Trifiro, F. Catalytic behavior of V-containing zeolites in the transformation of propane in the presence of oxygen. Appl. Catal. A Gen. 1996, 143, 3–16. [Google Scholar] [CrossRef]
- Chalupka, K.; Thomas, C.; Millot, Y.; Averseng, F.; Dzwigaj, S. Mononuclear pseudo-tetrahedral V species of VSiBEA zeolite as the active sites of the selective oxidative dehydrogenation of propane. J. Catal. 2013, 305, 46–55. [Google Scholar] [CrossRef]
- Dźwigaj, S.; Gressel, I.; Grzybowska, B.; Samson, K. Oxidative dehydrogenation of propane on VSiβ catalysts. Catal. Today 2006, 114, 237–241. [Google Scholar] [CrossRef]
- Smoliło, M.; Samson, K.; Zhou, T.; Duraczyńska, D.; Ruggiero-Mikołajczyk, M.; Drzewiecka-Matuszek, A.; Rutkowska-Zbik, D. Oxidative Dehydrogenation of Propane over Vanadium-Containing Faujasite Zeolite. Molecules 2020, 25, 1961. [Google Scholar] [CrossRef] [Green Version]
- Christensen, C.H.; Johannsen, K.; Tornqvist, E.; Schmidt, I.; Topsoe, H. Mesoporous zeolite single crystal catalysts: Diffusion and catalysis in hierarchical zeolites. Catal. Today 2007, 128, 117–122. [Google Scholar] [CrossRef]
- Verboekend, D.; Pérez-Ramírez, J. Desilication Mechanism Revisited: Highly Mesoporous All-Silica Zeolites Enabled Through Pore-Directing Agents. Chem.—Eur. J. 2011, 17, 1137–1147. [Google Scholar] [CrossRef]
- Sadowska, K.; Góra-Marek, K.; Drozdek, M.; Kuśtrowski, P.; Datka, J.; Triguero, J.M.; Rey, F. Desilication of highly siliceous zeolite ZSM-5 with NaOH and NaOH/tetrabutylamine hydroxide. Microporous Mesoporous Mater. 2013, 168, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Batista, C.; Teixeira, V.; Carneiro, J. Structural and Morphological Characterization of Magnetron Sputtered Nanocrystalline Vanadium Oxide Films for Thermochromic Smart Surfaces. J. Nano Res. 2008, 2, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Góra-Marek, K.; Datka, J.; Dzwigaj, S.; Che, M. Influence of V Content on the Nature and Strength of Acidic Sites in VSiβ Zeolite Evidenced by IR Spectroscopy. J. Phys. Chem. B 2006, 110, 6763–6767. [Google Scholar] [CrossRef]
- Dzwigaj, S.; Massiani, P.; Davidson, A.; Che, M. Role of silanol groups in the incorporation of V in beta zeolite. J. Mol. Catal. A Chem. 2000, 155, 169–182. [Google Scholar] [CrossRef]
- Busca, G.; Marchetti, L.; Centi, G.; Trifirò, F. Surface characterization of a grafted vanadium–titanium dioxide catalyst. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1985, 81, 1003–1014. [Google Scholar] [CrossRef]
- Rajadhyaksha, R.A.; Knözinger, H. Ammonia adsorption on vanadia supported on titania—Silica catalyst: An infrared spectroscopic investigation. Appl. Catal. 1989, 51, 81–92. [Google Scholar] [CrossRef]
- Khatib, S.; Guil-Lopez, R.; Peña, M.A.; Fierro, J.; Bañares, M.A. Alumina-supported V–Mo–O mixed oxide catalysts, the formation of phases involving aluminum: AlVMoO7. Catal. Today 2006, 118, 353–359. [Google Scholar] [CrossRef]
- Drzewiecka-Matuszek, A.; Tokarz-Sobieraj, R.; Witko, M.; Rutkowska-Zbik, D. Comparison of Catalytic Properties of Vanadium Centers Introduced into BEA Zeolite and Present on (010) V2O5 Surface–DFT Studies. Catalysts 2020, 10, 1080. [Google Scholar] [CrossRef]
- Tranca, D.C.; Keil, F.J.; Tranca, I.; Calatayud, M.; Dzwigaj, S.; Trejda, M.; Tielens, F. Methanol Oxidation to Formaldehyde on VSiBEA Zeolite: A Combined DFT/vdW/Transition Path Sampling and Experimental Study. J. Phys. Chem. C 2015, 119, 13619–13631. [Google Scholar] [CrossRef]
- Tielens, F. Exploring the reactivity of intraframework vanadium, niobium and tantalum sites in zeolitic materials using the molecular electrostatic potential. J. Mol. Struct. THEOCHEM 2009, 903, 23–27. [Google Scholar] [CrossRef]
- Wojtaszek, A.; Ziolek, M.; Dzwigaj, S.; Tielens, F. Comparison of competition between T=O and T–OH groups in vanadium, niobium, tantalum BEA zeolite and SOD based zeolites. Chem. Phys. Lett. 2011, 514, 70–73. [Google Scholar] [CrossRef]
- Lu, Y.-R.; Hsu, H.-H.; Chen, J.-L.; Chang, H.-W.; Chen, C.-L.; Chou, W.-C.; Dong, C.-L. Atomic and electronic aspects of the coloration mechanism of gasochromic Pt/Mo-modified V2O5 smart films: An in situ X-ray spectroscopic study. Phys. Chem. Chem. Phys. 2016, 18, 5203–5210. [Google Scholar] [CrossRef]
- Jang, W.L.; Lu, Y.M.; Chen, C.L.; Lu, Y.R.; Dong, C.L.; Hsieh, P.H.; Hwang, W.-S.; Chen, J.-L.; Chen, J.-M.; Chan, T.-S.; et al. Local geometric and electronic structures of gasochromic VO(x) films. Phys. Chem. Chem. Phys. 2014, 16, 4699–4708. [Google Scholar] [CrossRef]
- Zou, C.W.; Yan, X.D.; Han, J.; Chen, R.Q.; Gao, W. Microstructures and optical properties of β-V2O5 nanorods prepared by magnetron sputtering. J. Phys. D Appl. Phys. 2009, 42, 145402. [Google Scholar] [CrossRef]
- Hävecker, M.; Düngen, P.; Buller, S.; Knop-Gericke, A.; Trunschke, A.; Schlögl, R. Restructuring of silica supported vanadia during propane oxidative dehydrogenation studied by combined synchrotron radiation based in situ soft X-ray absorption and photoemission. Catal. Struct. React. 2017, 3, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Song, G.Y.; Nandi, R.; Cho, J.Y.; Heo, J.; Cho, D.-Y. Phase identification of vanadium oxide thin films prepared by atomic layer deposition using X-ray absorption spectroscopy. RSC Adv. 2020, 10, 26588–26593. [Google Scholar] [CrossRef] [PubMed]
- Trejda, M.; Ziolek, M.; Millot, Y.; Chalupka, K.; Che, M.; Dzwigaj, S. Methanol oxidation on VSiBEA zeolites: Influence of V content on the catalytic properties. J. Catal. 2011, 281, 169–176. [Google Scholar] [CrossRef]
- Schwarz, O.; Habel, D.; Ovsitser, O.; Kondratenko, E.; Hess, C.; Schomäcker, R.; Schubert, H. Impact of preparation method on physico-chemical and catalytic properties of VOx/γ-Al2O3 materials. J. Mol. Catal. A Chem. 2008, 293, 45–52. [Google Scholar] [CrossRef]
- Kubacka, A.; Włoch, E.; Sulikowski, B.; Valenzuela, R.; Corberán, V.C. Oxidative dehydrogenation of propane on zeolite catalysts. Catal. Today 2000, 61, 343–352. [Google Scholar] [CrossRef]
- Sulikowski, B.; Olejniczak, Z.; Włoch, E.; Rakoczy, J.; Valenzuela, R.; Corberán, V.C. Oxidative dehydrogenation of isobutane on MCM-41 mesoporous molecular sieves. Appl. Catal. A Gen. 2002, 232, 189–202. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S.; Trifiro, F.; Aboukais, A.; Aissi, C.F.; Guelton, M. Physicochemical characterization of V-silicalite. J. Phys. Chem. 1992, 96, 2617–2629. [Google Scholar] [CrossRef]
- Grzybek, G.; Góra-Marek, K.; Tarach, K.; Pyra, K.; Patulski, P.; Greluk, M.; Słowik, G.; Rotko, M.; Kotarba, A. Tuning the properties of the cobalt-zeolite nanocomposite catalyst by potassium: Switching between dehydration and dehydrogenation of ethanol. J. Catal. 2022, 407, 364–380. [Google Scholar] [CrossRef]
- Bratan, V.; Chesler, P.; Hornoiu, C.; Scurtu, M.; Postole, G.; Pietrzyk, P.; Gervasini, A.; Auroux, A.; Ionescu, N.I. In situ electrical conductivity study of Pt-impregnated VOx/γ-Al2O3 catalysts in propene deep oxidation. J. Mater. Sci. 2020, 55, 10466–10481. [Google Scholar] [CrossRef]
- Klisińska, A.; Loridant, S.; Grzybowska, B.; Stoch, J.; Gressel, I. Effect of additives on properties of V2O5/SiO2 and V2O5/MgO catalysts: II. Structure and physicochemical properties of the catalysts and their correlations with oxidative dehydrogenation of propane and ethane. Appl. Catal. A Gen. 2006, 309, 17–27. [Google Scholar] [CrossRef]
- Gao, X.; Wachs, I.E. Investigation of Surface Structures of Supported Vanadium Oxide Catalysts by UV−vis−NIR Diffuse Reflectance Spectroscopy. J. Phys. Chem. B 2000, 104, 1261–1268. [Google Scholar] [CrossRef]
- Guo, B.; Zhu, L.; Hu, X.; Zhang, Q.; Tong, D.; Li, G.; Hu, C. Nature of vanadium species on vanadium silicalite-1 zeolite and their stability in hydroxylation reaction of benzene to phenol. Catal. Sci. Technol. 2011, 1, 1060–1067. [Google Scholar] [CrossRef]
- Zhang, Z.-G.; Xu, H.-G.; Kong, X.; Zheng, W. Anion Photoelectron Spectroscopy and Density Functional Study of Small Aluminum−Vanadium Oxide Clusters. J. Phys. Chem. A 2010, 115, 13–18. [Google Scholar] [CrossRef]
- Vieira, L.H.; Possato, L.G.; Chaves, T.F.; Pulcinelli, S.H.; Santilli, C.V.; Martins, L. Studies on dispersion and reactivity of vanadium oxides deposited on lamellar ferrierite zeolites for condensation of glycerol into bulky products. Mol. Catal. 2018, 458, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Noda, I. Generalized Two-Dimensional Correlation Method Applicable to Infrared, Raman, and other Types of Spectroscopy. Appl. Spectrosc. 1993, 47, 1329–1336. [Google Scholar] [CrossRef]
- Noda, I.; Ozaki, Y. Two-Dimensional Correlation Spectroscopy: Applications in Vibrational and Optical Spectroscopy. In Principle of Two-Dimensional Correlation Spectroscopy; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2004; pp. 15–38. [Google Scholar]
- Gołąbek, K.; Tarach, K.A.; Góra-Marek, K. Standard and rapid scan infrared spectroscopic studies of o-xylene transformations in terms of pore arrangement of 10-ring zeolites—2D COS analysis. Dalton Trans. 2017, 46, 9934–9950. [Google Scholar] [CrossRef]
- Ternero-Hidalgo, J.; Daturi, M.; Clet, G.; Bazin, P.; Bañares, M.; Portela, R.; Guerrero-Pérez, M.; Rodríguez-Mirasol, J.; Cordero, T. A simultaneous operando FTIR & Raman study of propane ODH mechanism over V-Zr-O catalysts. Catal. Today 2022, 387, 197–206. [Google Scholar] [CrossRef]
- Held, A.; Kowalska-Kuś, J.; Nowińska, K. Propane-to-propene oxide oxidation on silica-supported vanadium catalysts with N2O as an oxidant. J. Catal. 2016, 336, 23–32. [Google Scholar] [CrossRef]
- Rozanska, X.; Fortrie, R.; Sauer, J. Size-Dependent Catalytic Activity of Supported Vanadium Oxide Species: Oxidative Dehydrogenation of Propane. J. Am. Chem. Soc. 2014, 136, 7751–7761. [Google Scholar] [CrossRef]
- Klisińska, A.; Samson, K.; Gressel, I.; Grzybowska, B. Effect of additives on properties of V2O5/SiO2 and V2O5/MgO catalysts: I. Oxidative dehydrogenation of propane and ethane. Appl. Catal. A Gen. 2006, 309, 10–16. [Google Scholar] [CrossRef]
- Julbe, A.; Farrusseng, D.; Jalibert, J.; Mirodatos, C.; Guizard, C. Characteristics and performance in the oxidative dehydrogenation of propane of MFI and V-MFI zeolite membranes. Catal. Today 2000, 56, 199–209. [Google Scholar] [CrossRef]
- Solsona, B.; Nieto, J.M.L.; Díaz, U. Siliceous ITQ-6: A new support for vanadia in the oxidative dehydrogenation of propane. Microporous Mesoporous Mater. 2006, 94, 339–347. [Google Scholar] [CrossRef]
- Buchbinder, A.M.; Ray, N.A.; Lu, J.; Van Duyne, R.P.; Stair, P.C.; Weitz, E.; Geiger, F.M. Displacement of Hexanol by the Hexanoic Acid Overoxidation Product in Alcohol Oxidation on a Model Supported Palladium Nanoparticle Catalyst. J. Am. Chem. Soc. 2011, 133, 17816–17823. [Google Scholar] [CrossRef]
- Chen, K.; Bell, A.A.T.; Iglesia, E. Kinetics and Mechanism of Oxidative Dehydrogenation of Propane on Vanadium, Molybdenum, and Tungsten Oxides. J. Phys. Chem. B 2000, 104, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Khodakov, A.; Yang, J.; Bell, A.T.; Iglesia, E. Isotopic Tracer and Kinetic Studies of Oxidative Dehydrogenation Pathways on Vanadium Oxide Catalysts. J. Catal. 1999, 186, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, K.; Reyniers, M.-F.; Marin, G.B. Reaction path analysis of propane selective oxidation over V2O5 and V2O5/TiO2. J. Catal. 2012, 289, 127–139. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Kozłowska, A.; Tokarz-Sobieraj, R.; Witko, M. Interaction of oxygen with the surface of vanadia catalysts. J. Mol. Catal. A Chem. 2007, 277, 27–34. [Google Scholar] [CrossRef]
- Zając, M.; Giela, T.; Freindl, K.; Korecki, J.; Madej, E.; Sikora, M.; Spiridis, N.; Stankiewicz, M.; Stępień, J.; Szade, J.; et al. The soft X-rays spectroscopy beamline at the National Synchrotron Radiation Centre Solaris. Synchrotron Radiat. Nat. Sci. 2020, 19, 1–4. [Google Scholar] [CrossRef]
- Bare, S.R.; Ressler, T. Characterization of Catalysts in Reactive Atmospheres by X-ray Absorption Spectroscopy. In Advances in Catalysis; Academic Press: Cambridge, MA, USA, 2009; pp. 339–465. [Google Scholar]
- Sadowska, K.; Góra-Marek, K.; Datka, J. Hierarchic zeolites studied by IR spectroscopy: Acid properties of zeolite ZSM-5 desilicated with NaOH and NaOH/tetrabutylamine hydroxide. Vib. Spectrosc. 2012, 63, 418–425. [Google Scholar] [CrossRef]
- Góra-Marek, K.; Derewinski, M.; Sarv, P.; Datka, J. IR and NMR studies of mesoporous alumina and related aluminosilicates. Catal. Today 2005, 101, 131–138. [Google Scholar] [CrossRef]
- Van Oers, C.; Góra-Marek, K.; Sadowska, K.; Mertens, M.; Meynen, V.; Datka, J.; Cool, P. In situ IR spectroscopic study to reveal the impact of the synthesis conditions of zeolite β nanoparticles on the acidic properties of the resulting zeolite. Chem. Eng. J. 2014, 237, 372–379. [Google Scholar] [CrossRef]
- Pyra, K.; Tarach, K.A.; Góra-Marek, K. Towards a greater olefin share in polypropylene cracking—Amorphous mesoporous aluminosilicate competes with zeolites. Appl. Catal. B Environ. 2021, 297, 120408. [Google Scholar] [CrossRef]
- Pietrzyk, P.; Podolska-Serafin, K.; Gora-Marek, K.; Krasowska, A.; Sojka, Z. Redox states of nickel in zeolites and molecular account into binding of N2 to nickel(I) centers—IR, EPR and DFT study. Microporous Mesoporous Mater. 2020, 291, 109692. [Google Scholar] [CrossRef]
- Gołąbek, K.; Tarach, K.A.; Góra-Marek, K. 2D COS analysis of m-xylene transformation over medium-pore zeolites. Microporous Mesoporous Mater. 2018, 266, 90–101. [Google Scholar] [CrossRef]
- Kapteijn, F.; Moulijn, J.A. Laboratory catalytic reactors: Aspects of catalyst testing. In Handbook of Heterogeneous Catalysis; Ertl, G., Knoezinger, H., Weitkampf, J., Eds.; Wiley/VCH: Weinheim, Germany, 1997; p. 1365. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Si/Al a | SBET b | Smicro c | Smeso e | Vmicro c | Vmeso d | BAS f | LAS f | LASV | LASV/m2 | LASV/Vtotal | BAS Py450/Py170 g |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m2·g−1 | m2·g−1 | m2·g−1 | cm3·g−1 | cm3·g−1 | µmol·g−1 | µmol·g−1 | µmol·g−1 | µmol g−1·m−2 | µmol cm–3 | [-] | ||
| HY | 31 | 883 | 724 | 158 | 0.30 | 0.22 | 208 | 95 | - | - | - | 0.31 |
| HYdeSi | 18 | 688 | 265 | 423 | 0.12 | 0.53 | 170 | 180 | - | - | - | 0.20 |
| V-HY | 31 | 775 | 645 | 130 | 0.23 | 0.28 | 145 | 221 | 126 | 0.16 | 0.14 | 0.15 |
| V-HYdeSi | 18 | 319 | 190 | 129 | 0.05 | 0.35 | 80 | 285 | 105 | 0.30 | 0.12 | 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smoliło-Utrata, M.; Tarach, K.A.; Samson, K.; Gackowski, M.; Madej, E.; Korecki, J.; Mordarski, G.; Śliwa, M.; Jarczewski, S.; Podobiński, J.; et al. Modulation of ODH Propane Selectivity by Zeolite Support Desilication: Vanadium Species Anchored to Al-Rich Shell as Crucial Active Sites. Int. J. Mol. Sci. 2022, 23, 5584. https://doi.org/10.3390/ijms23105584
Smoliło-Utrata M, Tarach KA, Samson K, Gackowski M, Madej E, Korecki J, Mordarski G, Śliwa M, Jarczewski S, Podobiński J, et al. Modulation of ODH Propane Selectivity by Zeolite Support Desilication: Vanadium Species Anchored to Al-Rich Shell as Crucial Active Sites. International Journal of Molecular Sciences. 2022; 23(10):5584. https://doi.org/10.3390/ijms23105584
Chicago/Turabian StyleSmoliło-Utrata, Małgorzata, Karolina A. Tarach, Katarzyna Samson, Mariusz Gackowski, Ewa Madej, Józef Korecki, Grzegorz Mordarski, Michał Śliwa, Sebastian Jarczewski, Jerzy Podobiński, and et al. 2022. "Modulation of ODH Propane Selectivity by Zeolite Support Desilication: Vanadium Species Anchored to Al-Rich Shell as Crucial Active Sites" International Journal of Molecular Sciences 23, no. 10: 5584. https://doi.org/10.3390/ijms23105584
APA StyleSmoliło-Utrata, M., Tarach, K. A., Samson, K., Gackowski, M., Madej, E., Korecki, J., Mordarski, G., Śliwa, M., Jarczewski, S., Podobiński, J., Kuśtrowski, P., Datka, J., Rutkowska-Zbik, D., & Góra-Marek, K. (2022). Modulation of ODH Propane Selectivity by Zeolite Support Desilication: Vanadium Species Anchored to Al-Rich Shell as Crucial Active Sites. International Journal of Molecular Sciences, 23(10), 5584. https://doi.org/10.3390/ijms23105584