
Novel All-Nitrogen Molecular Crystals Composed of Tetragonal N4 Molecules



Abstract
:1. Introduction
2. Computational Details
2.1. Method
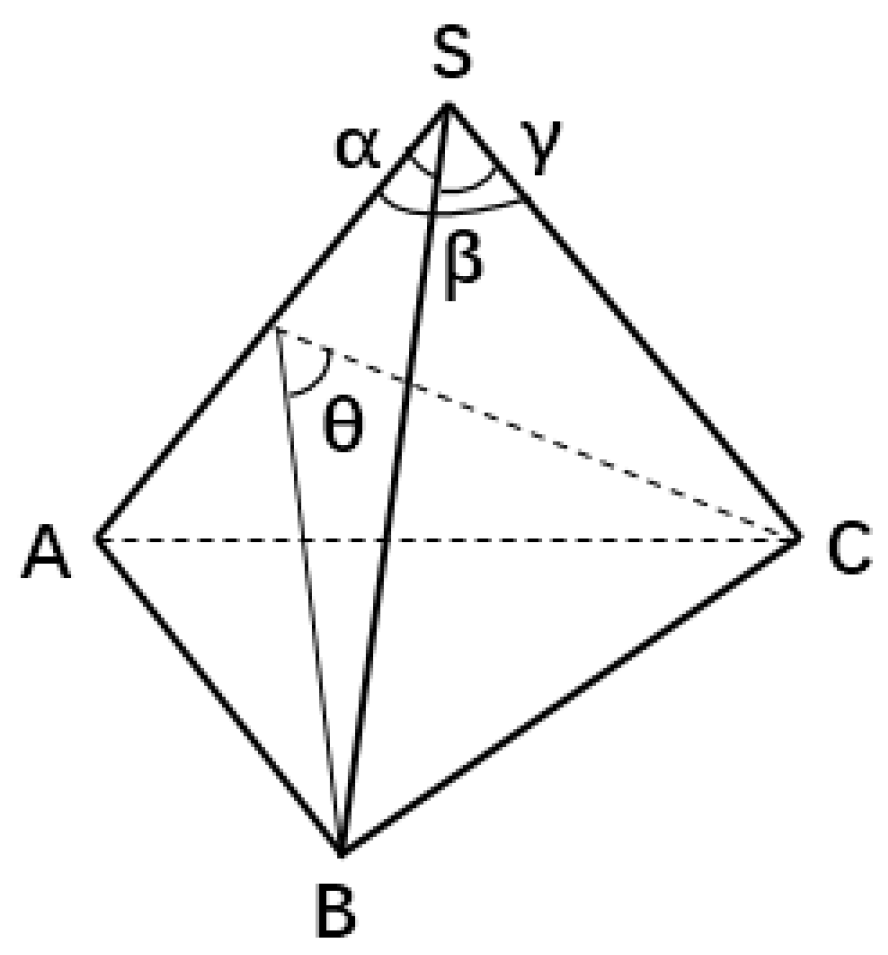
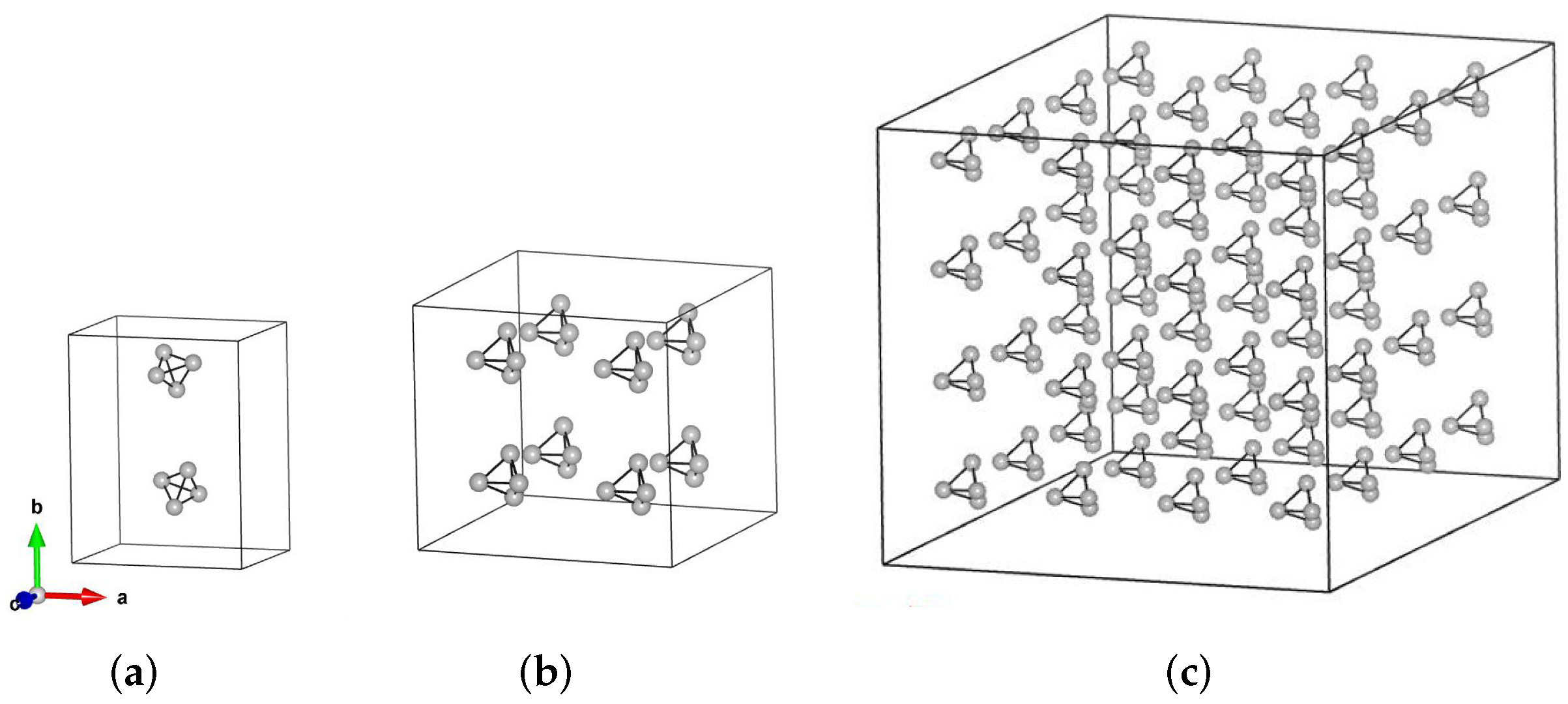
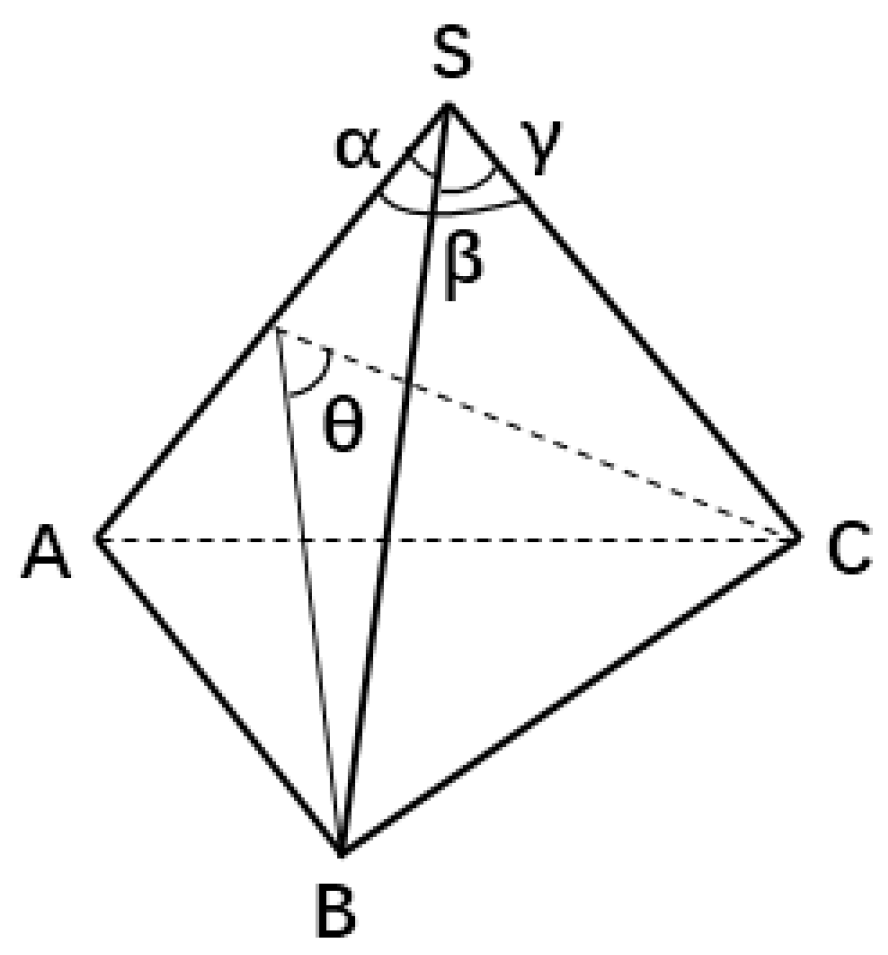
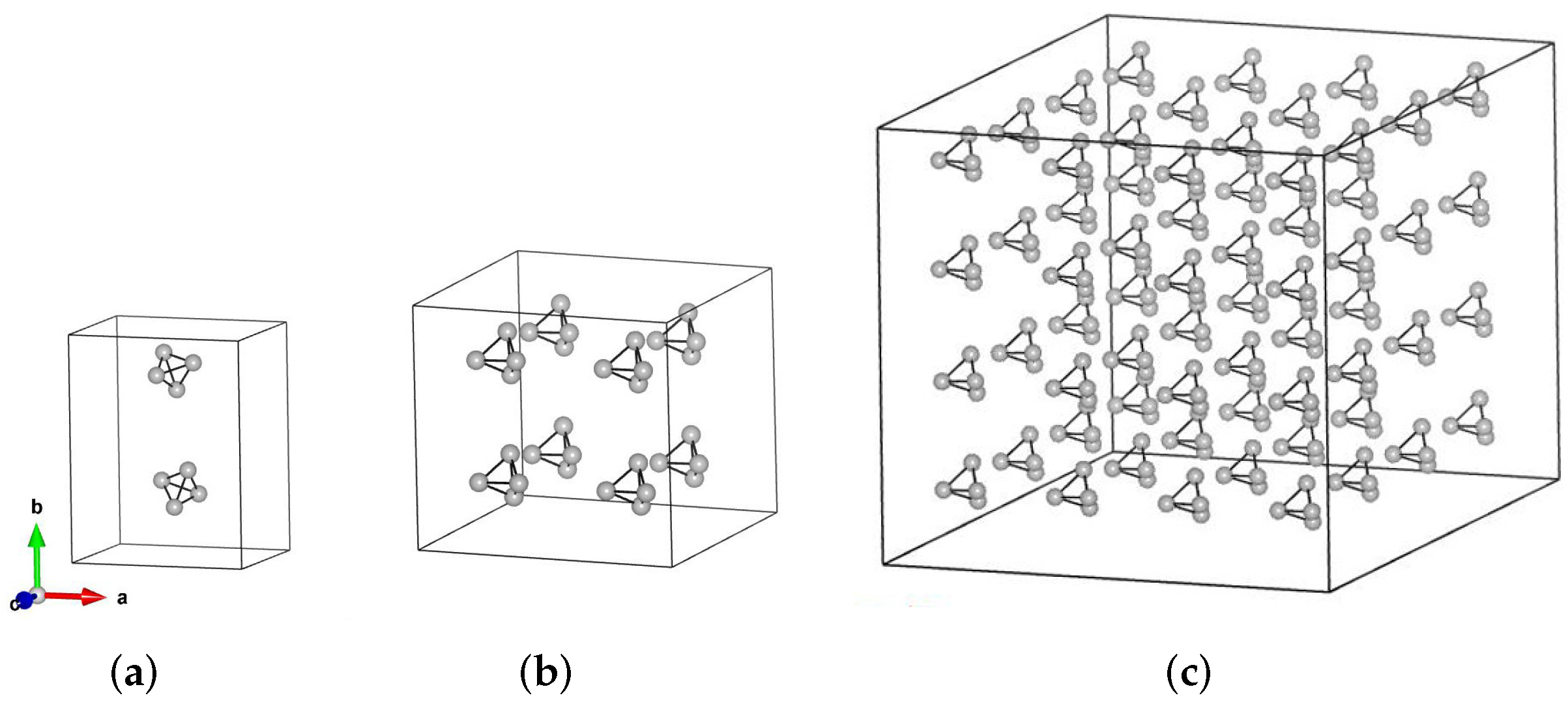
2.2. Characterization of Atomic Structure
3. Results and Discussion
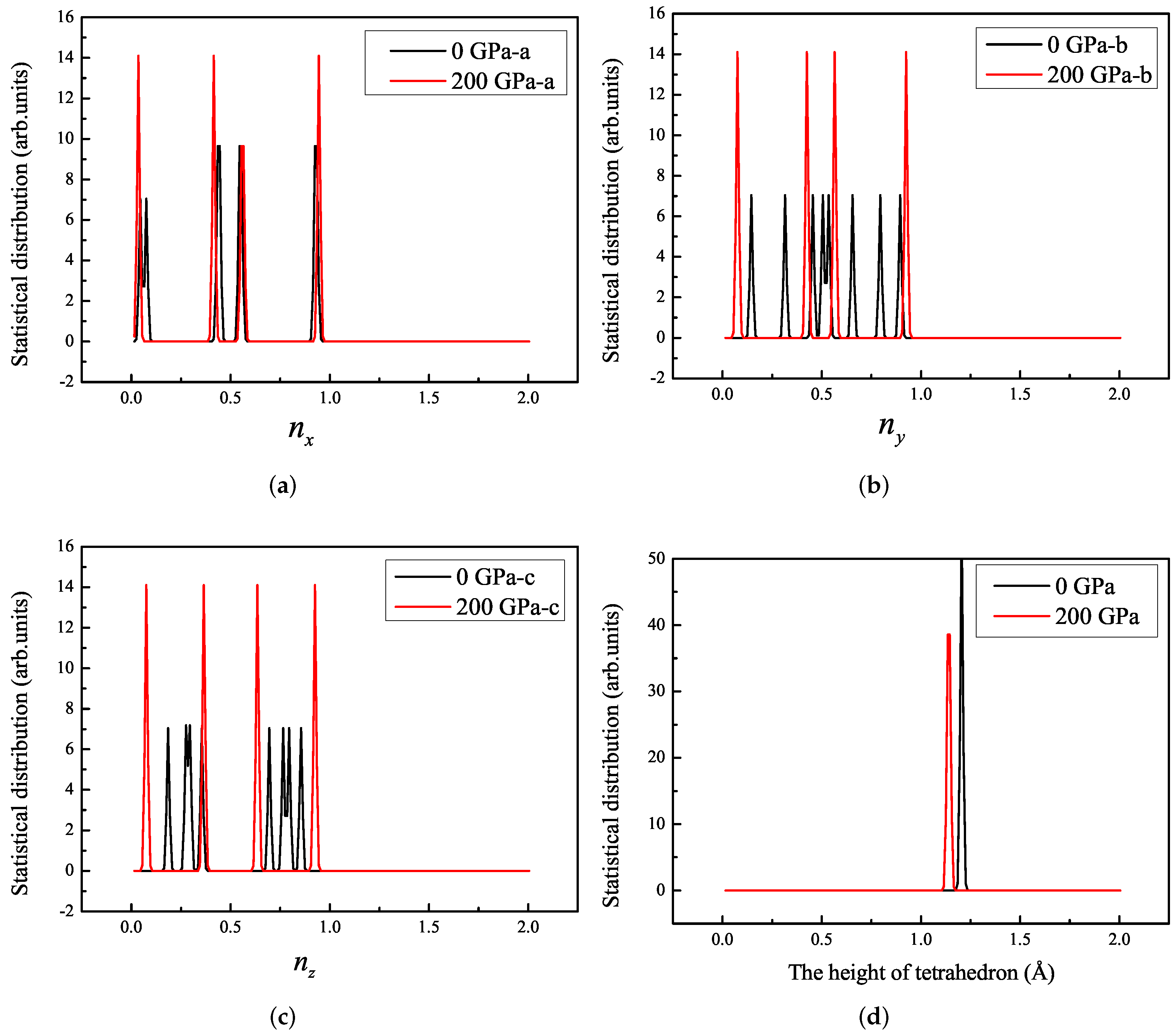
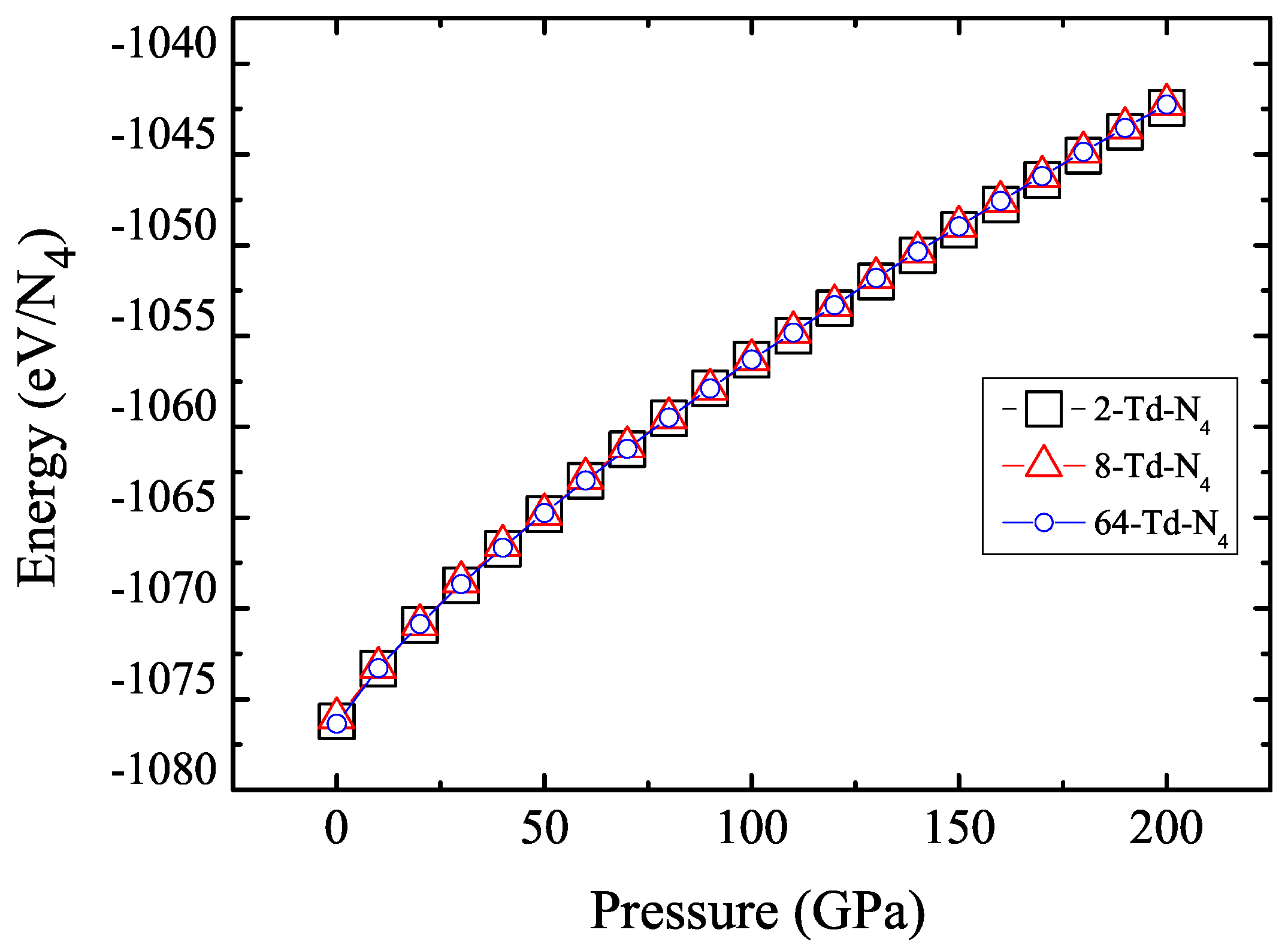
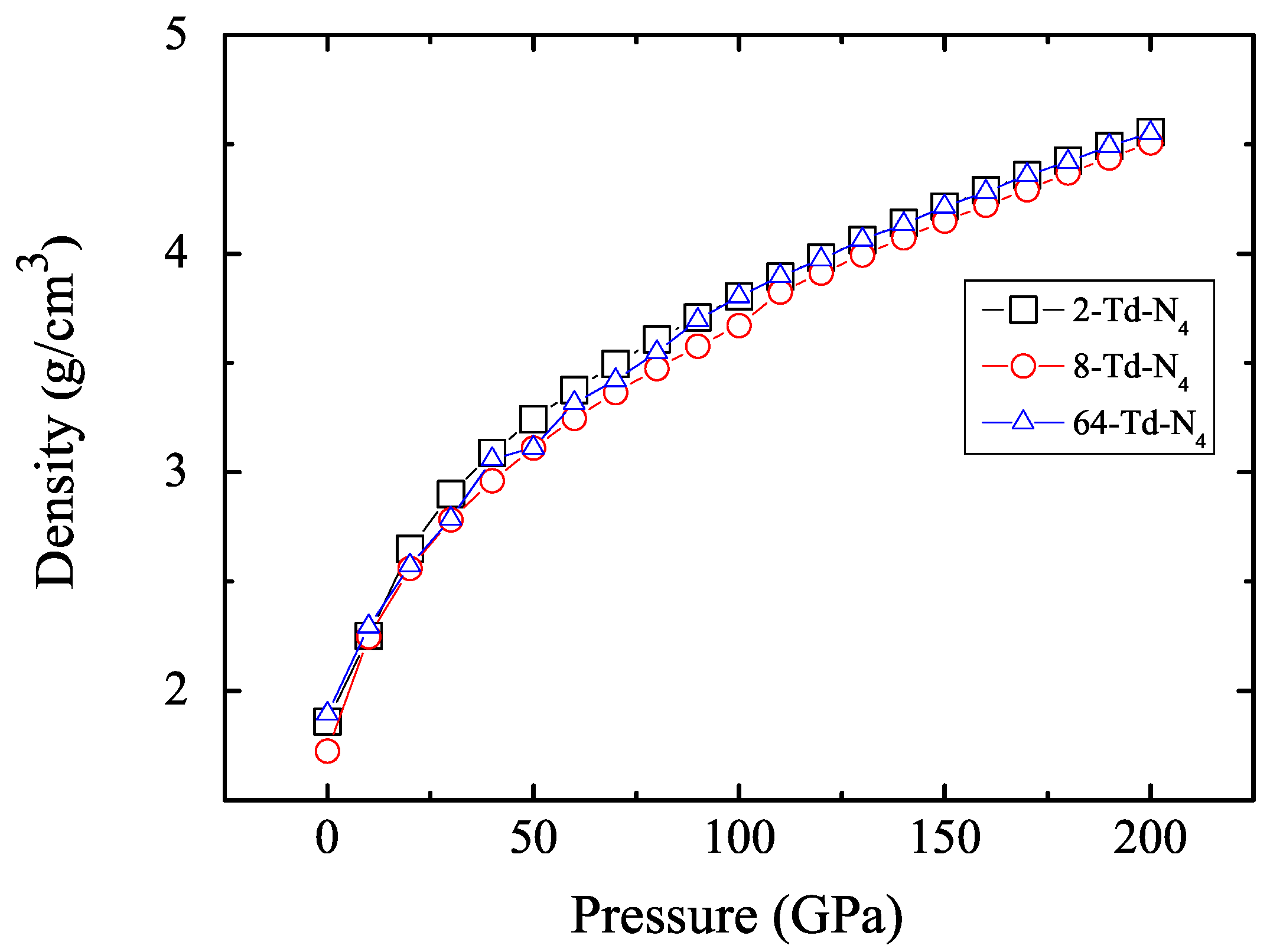
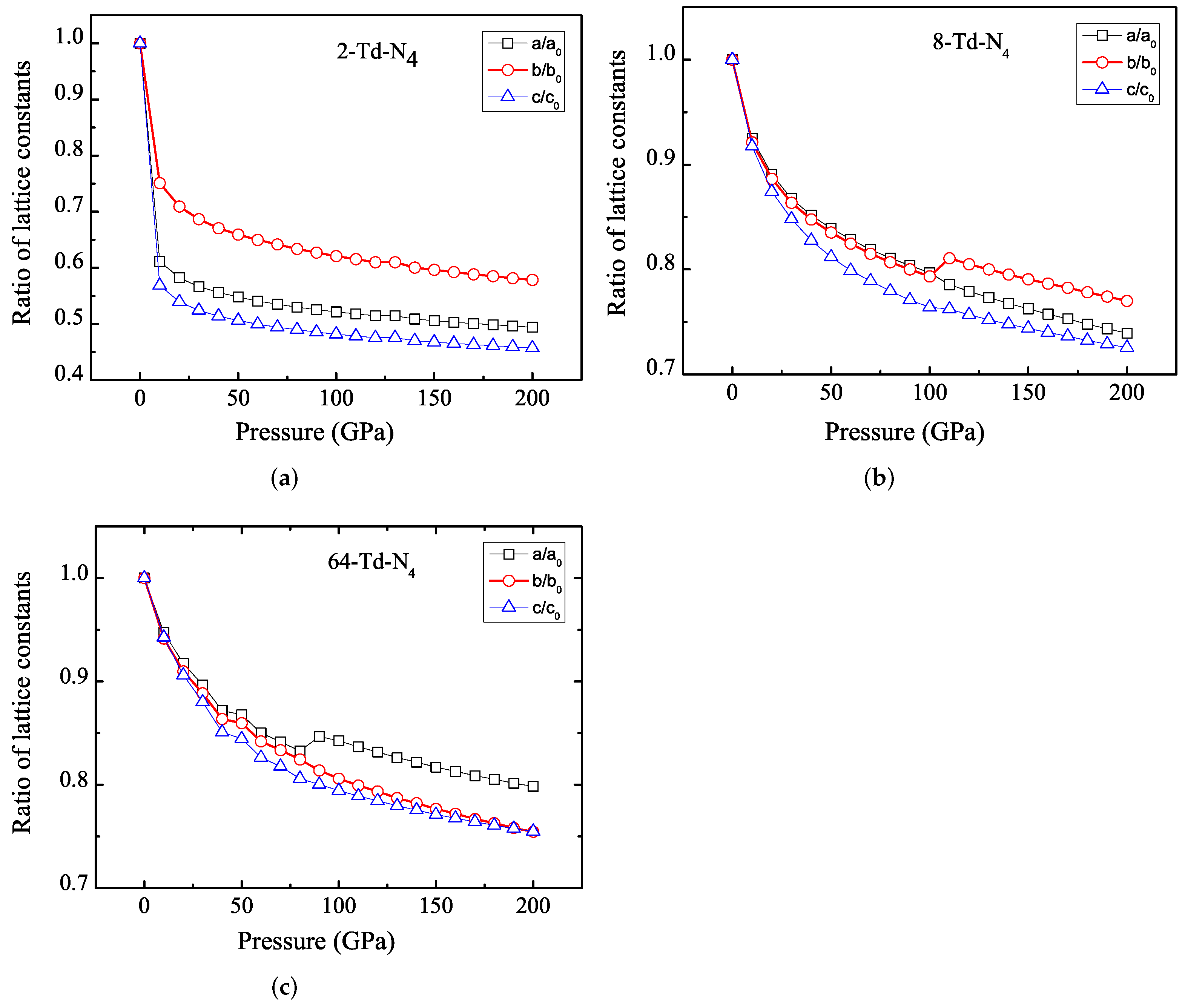
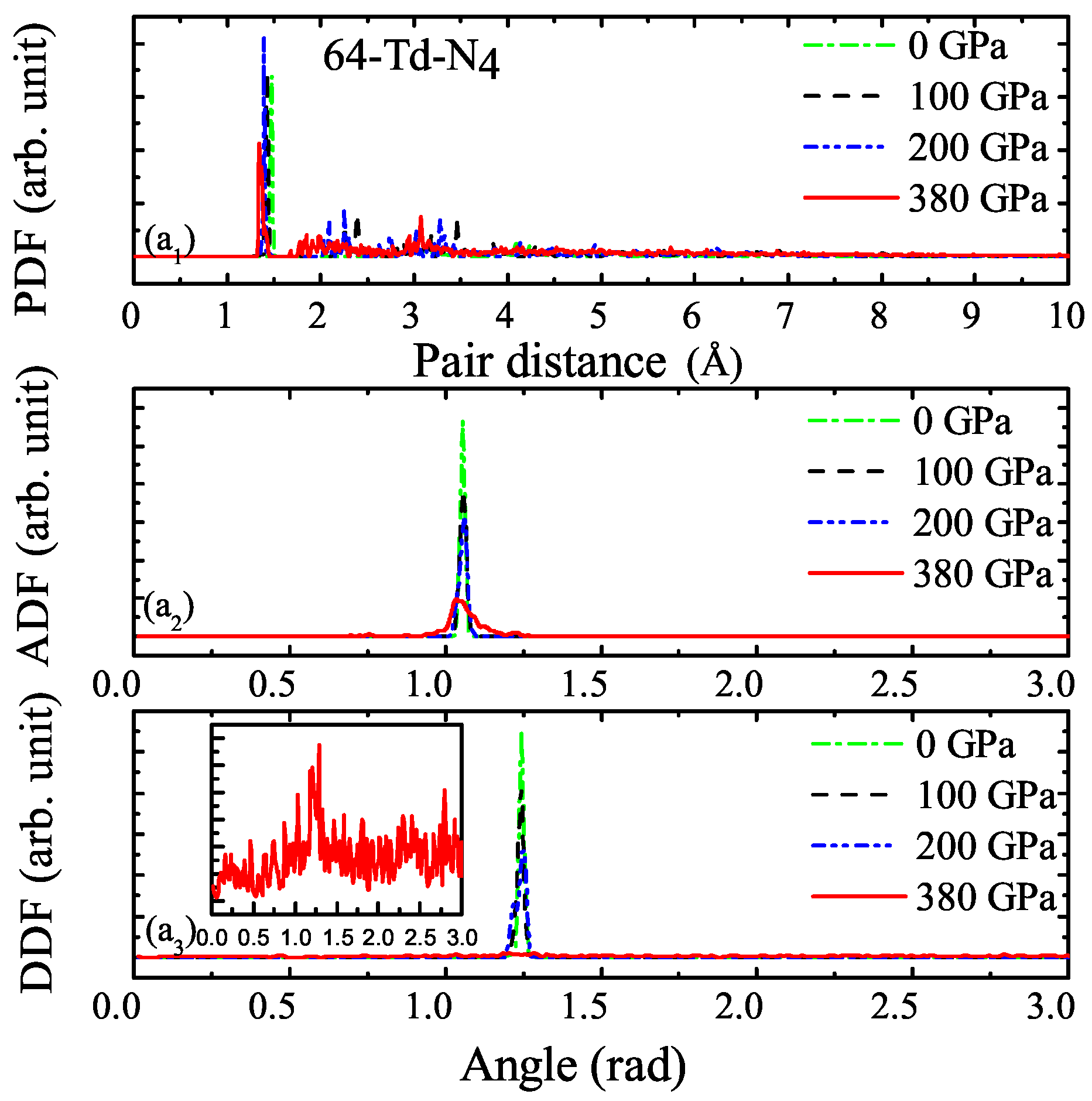
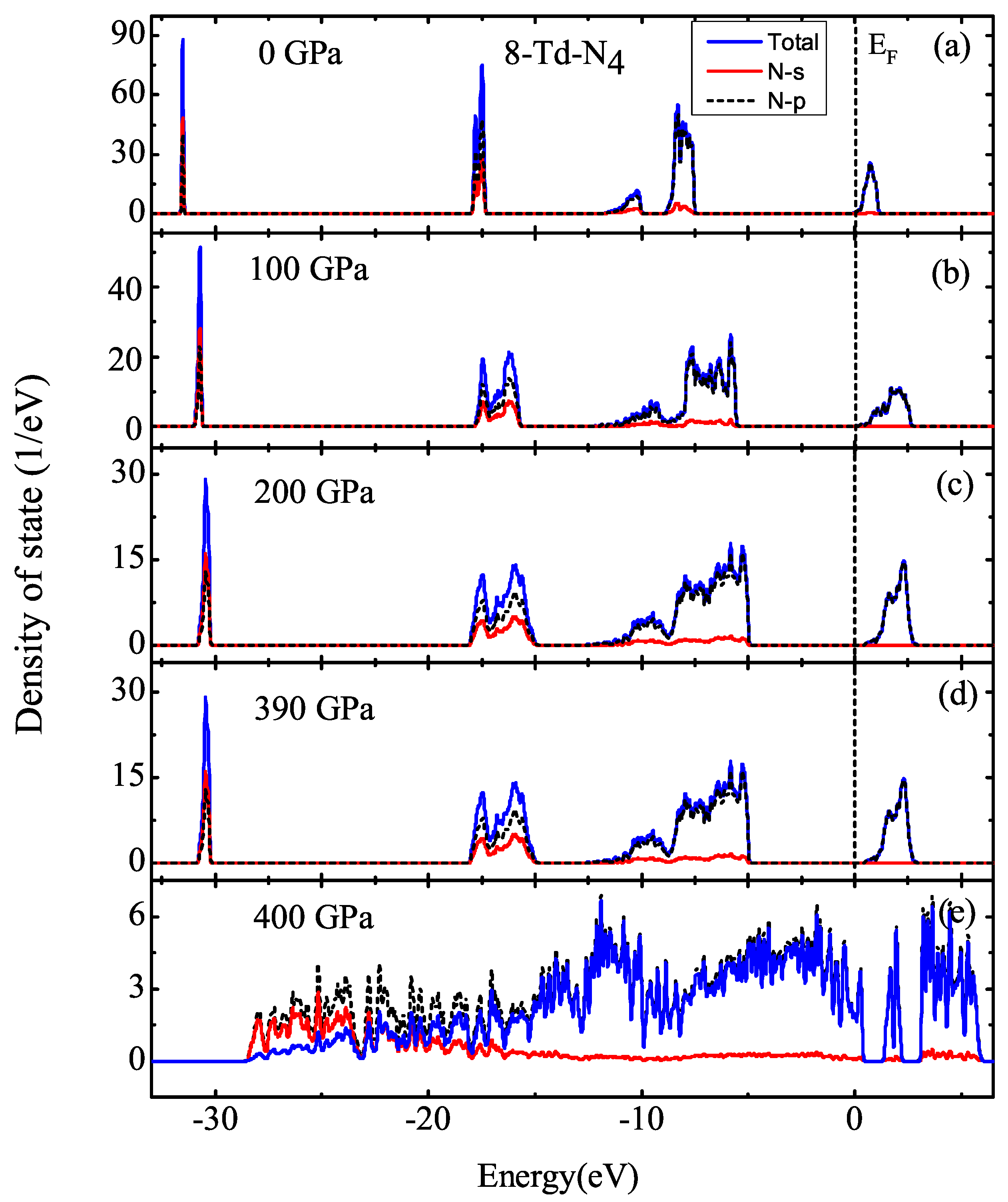
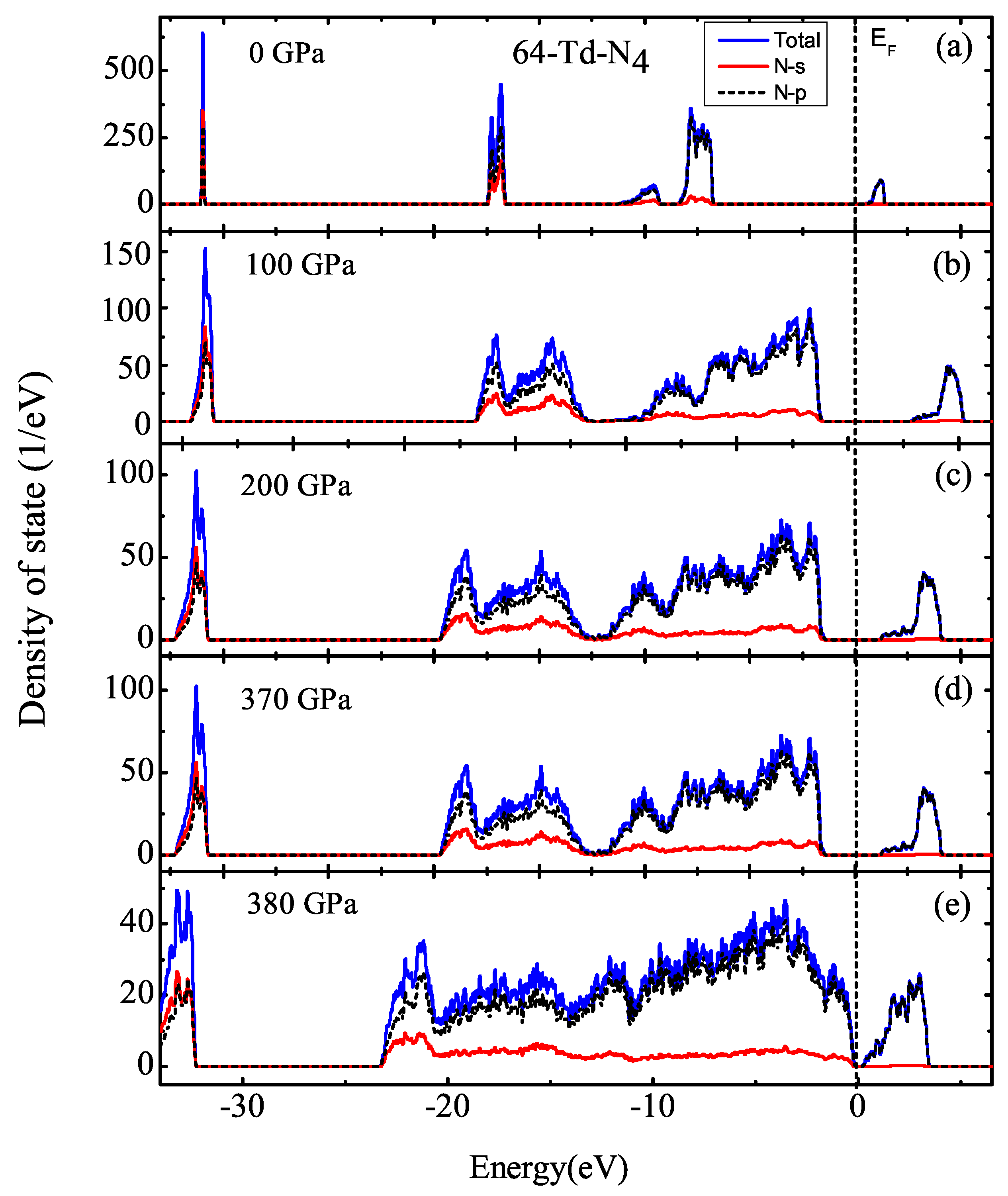
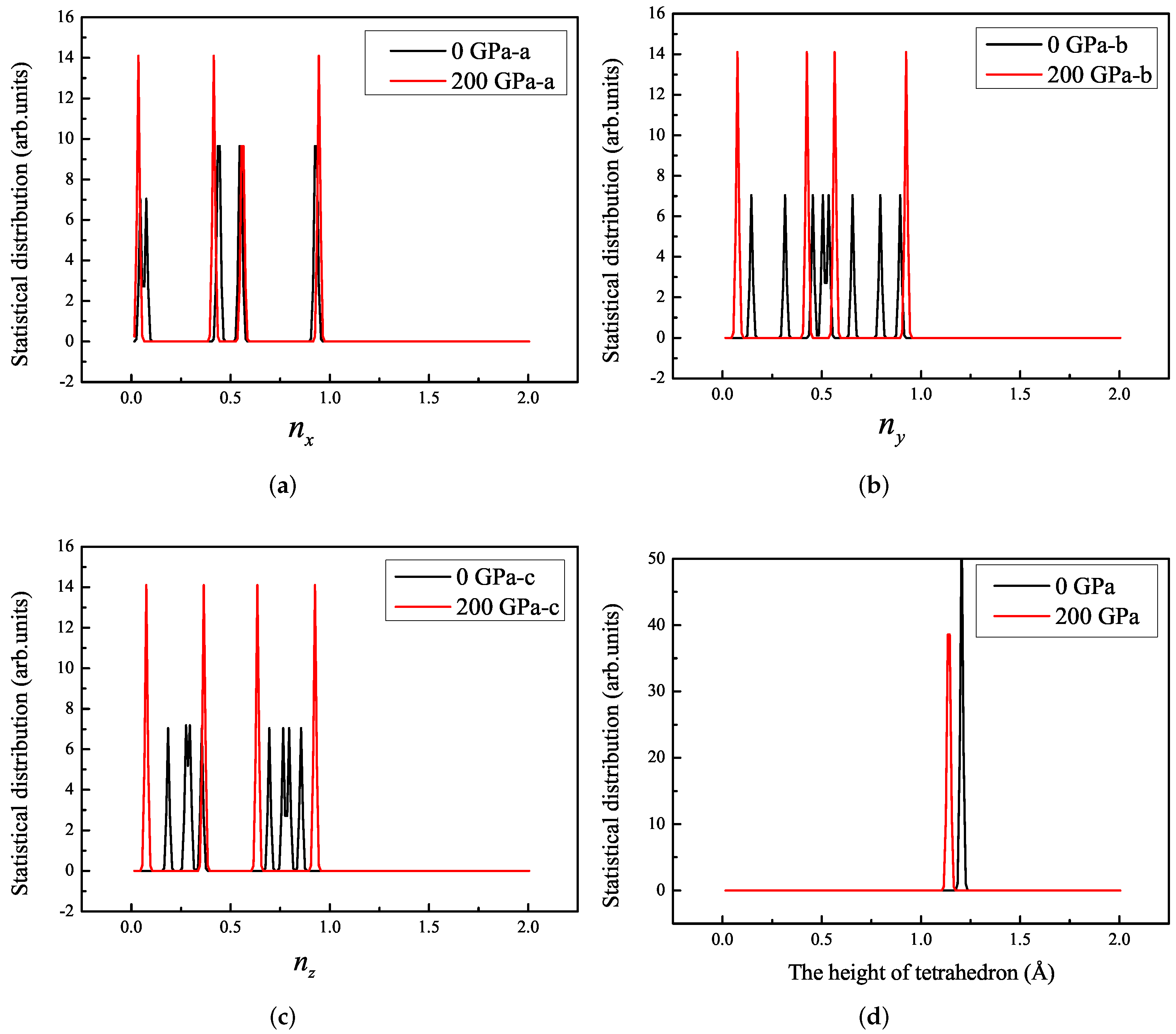
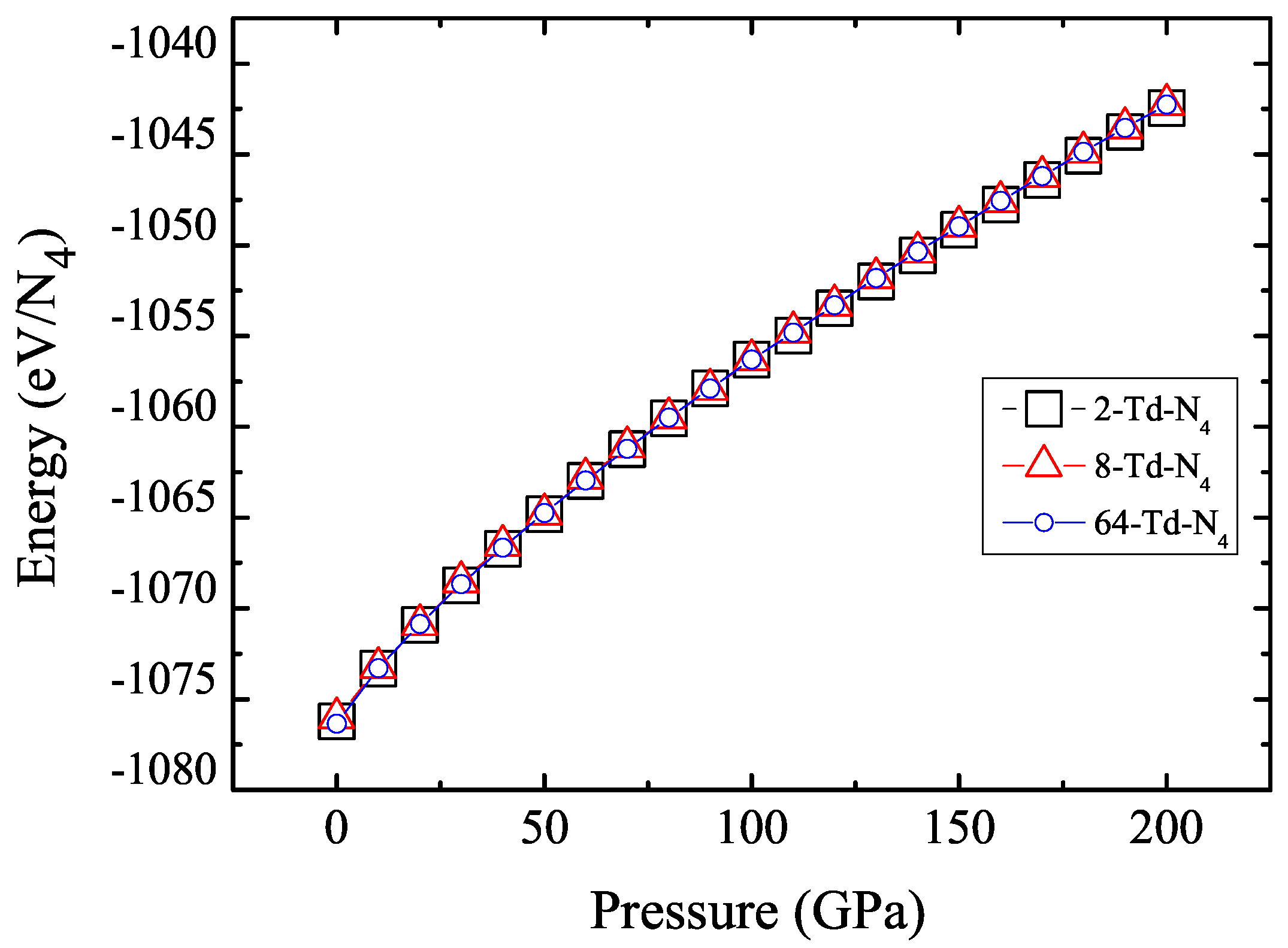
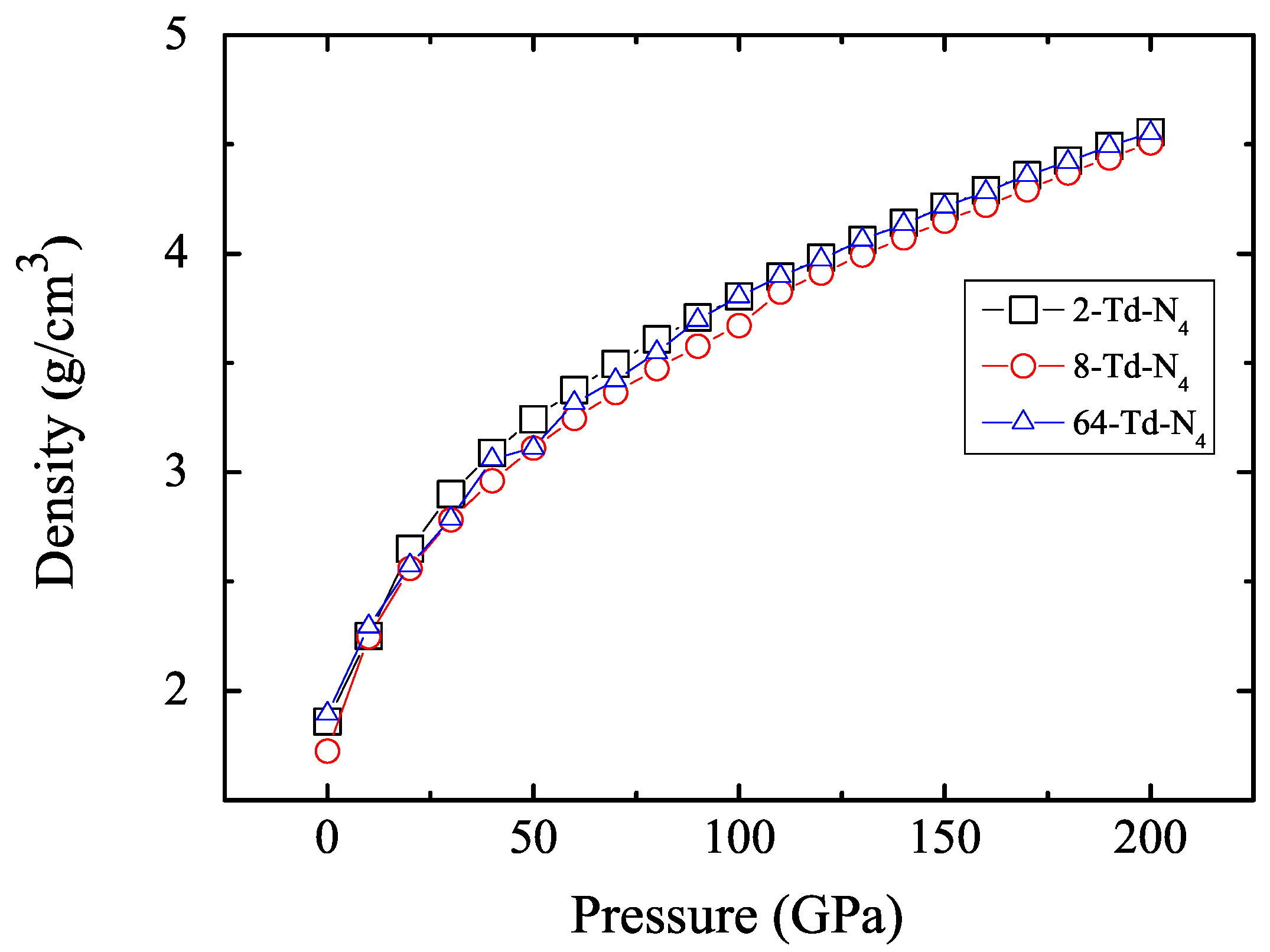
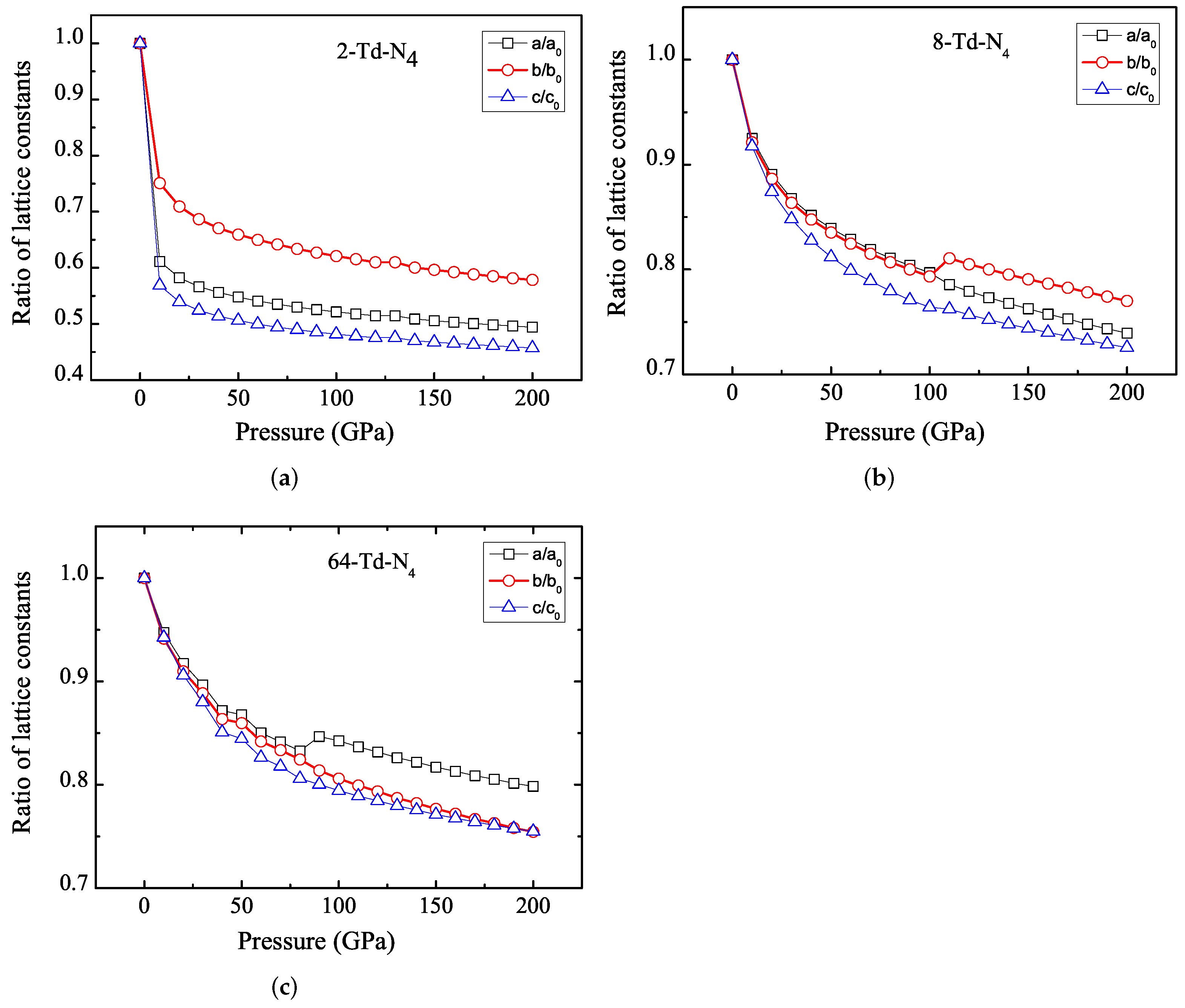
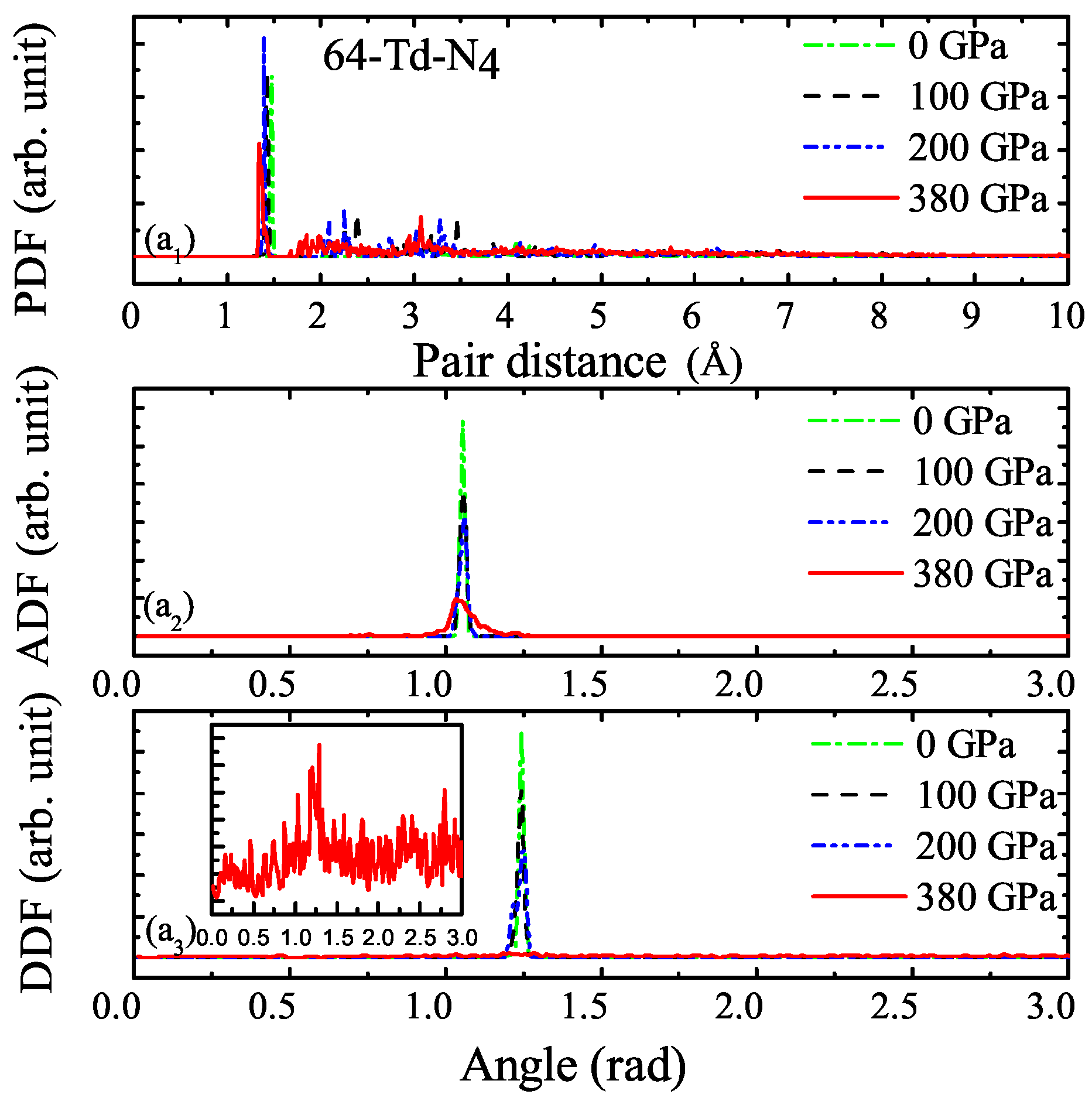
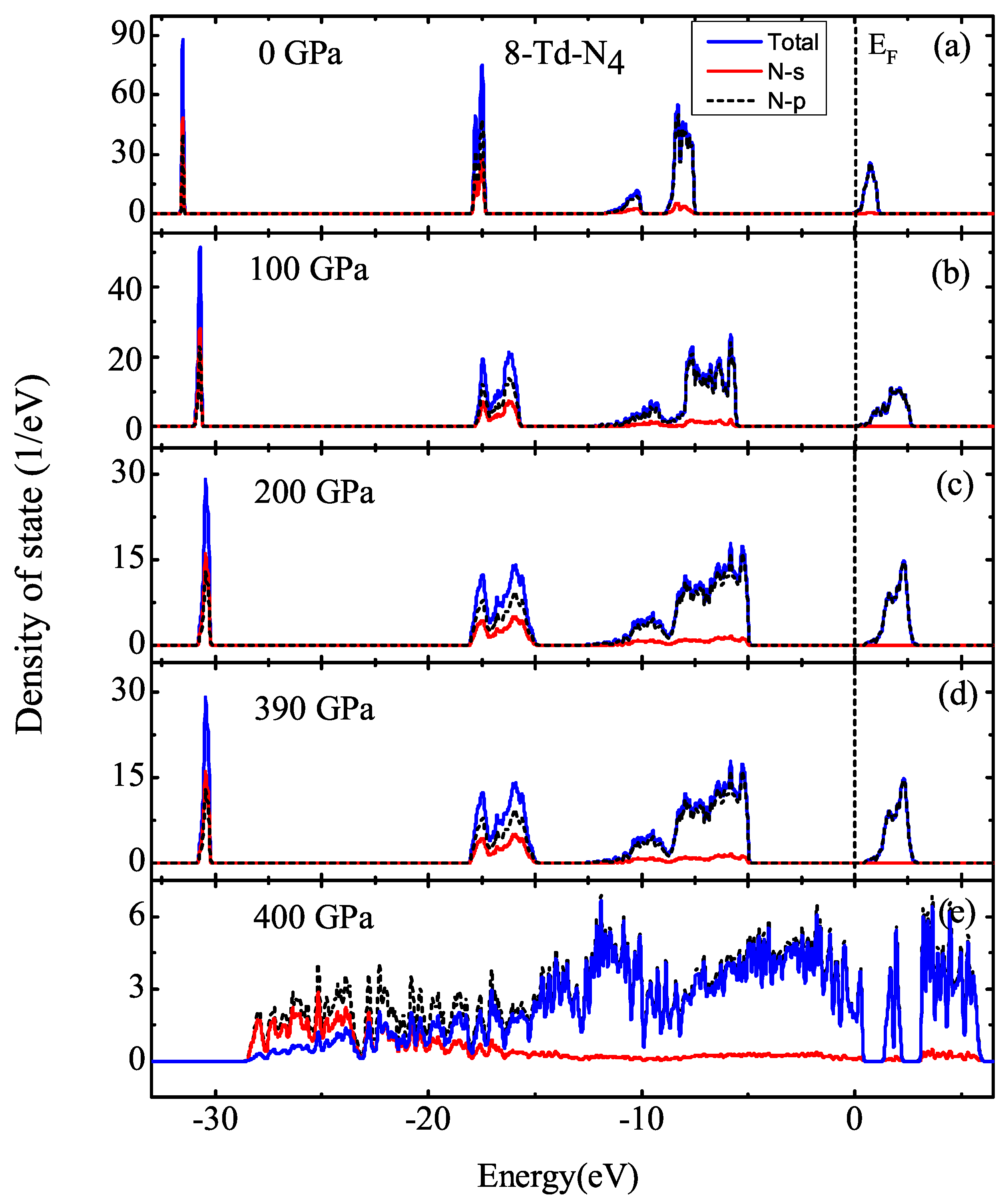
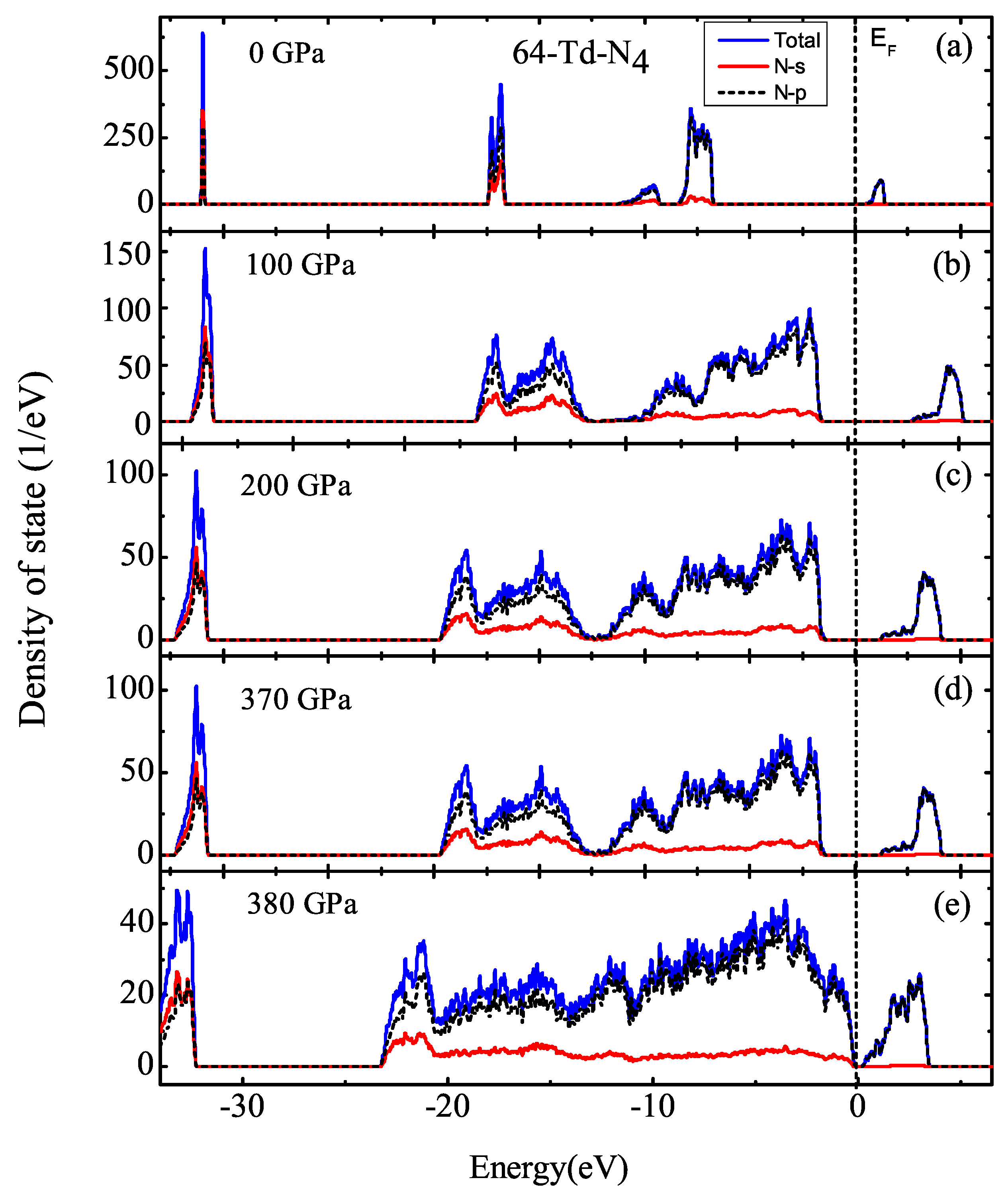
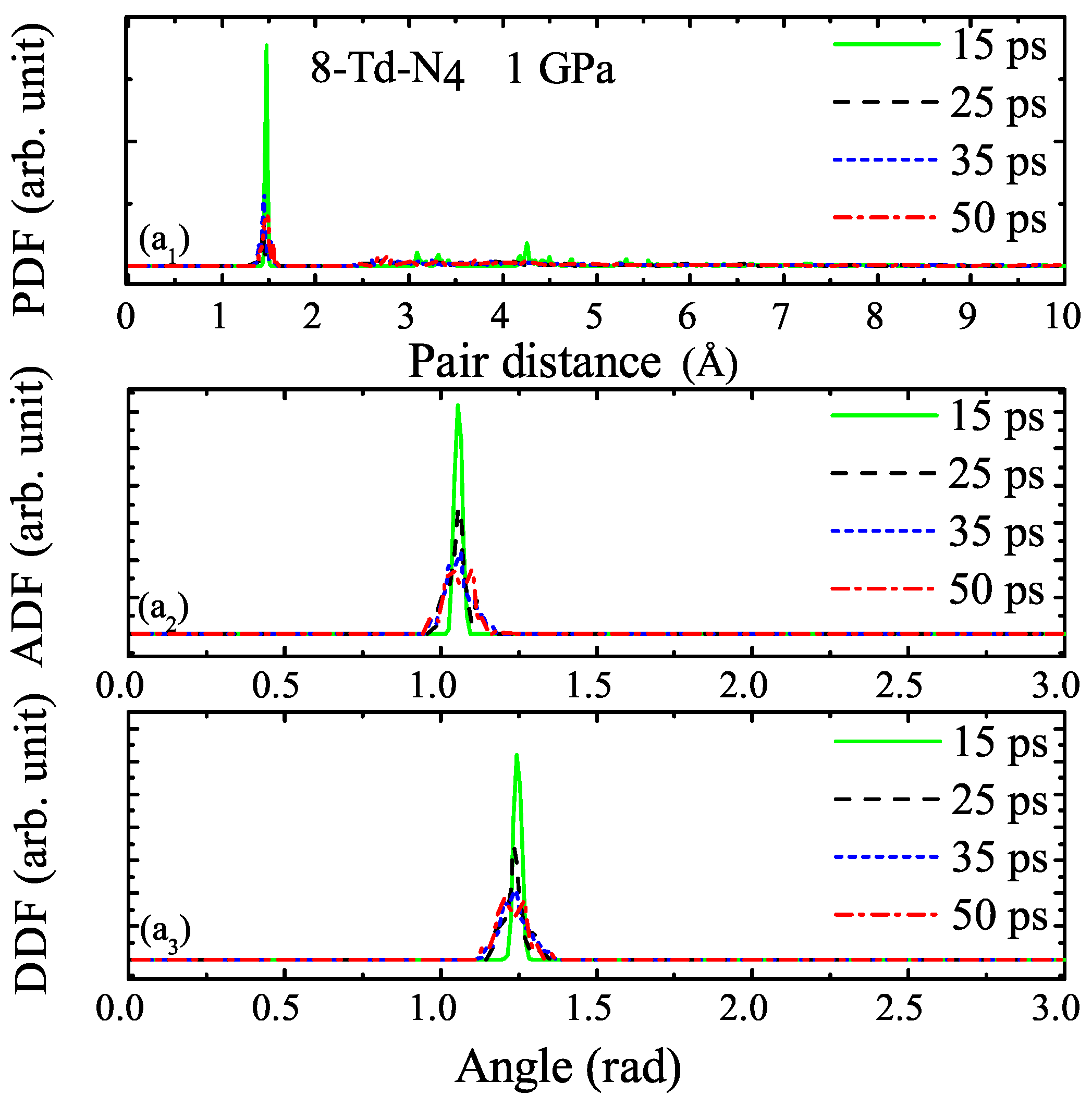
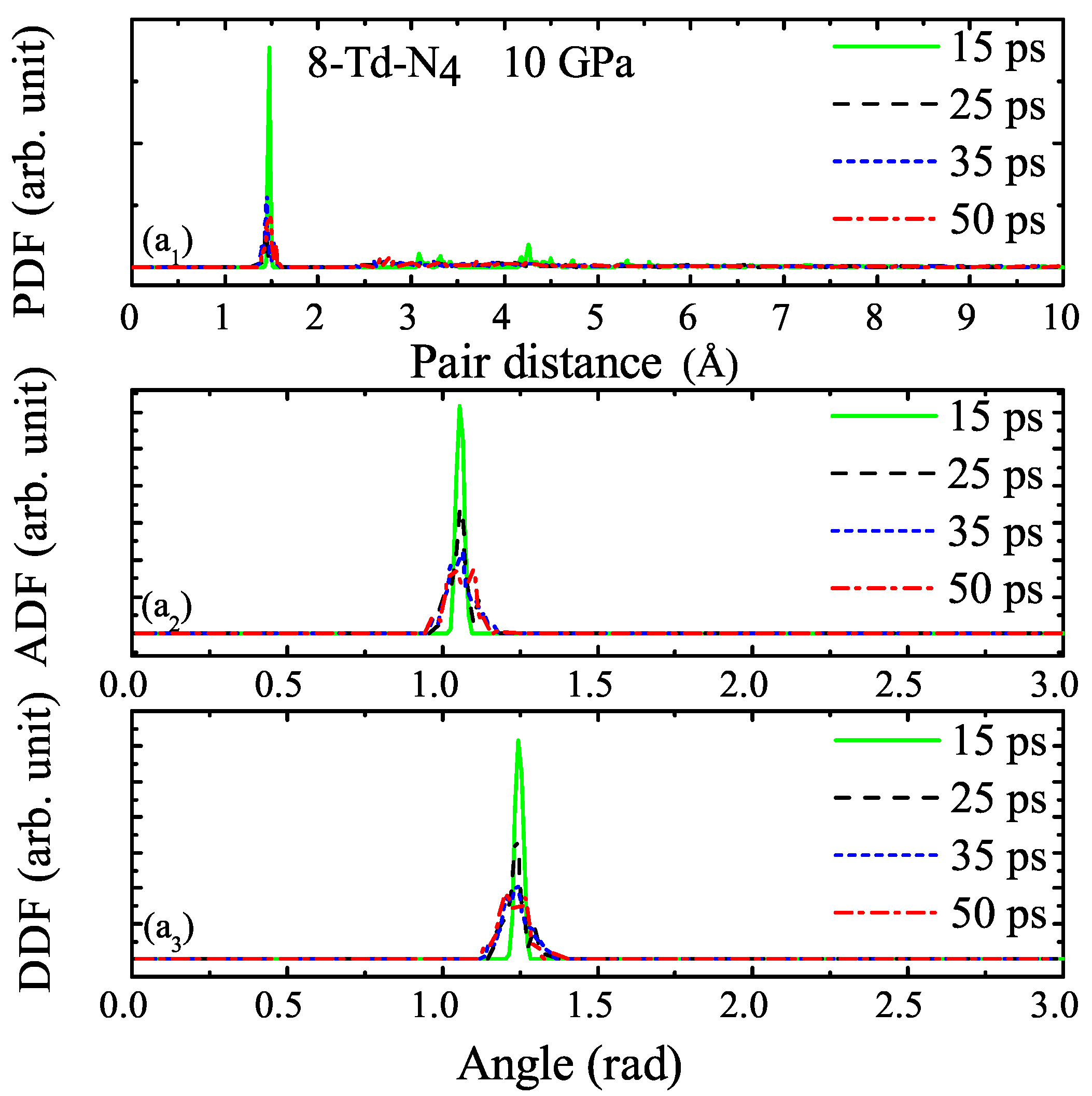
3.1. Properties under Hydrostatic Pressures
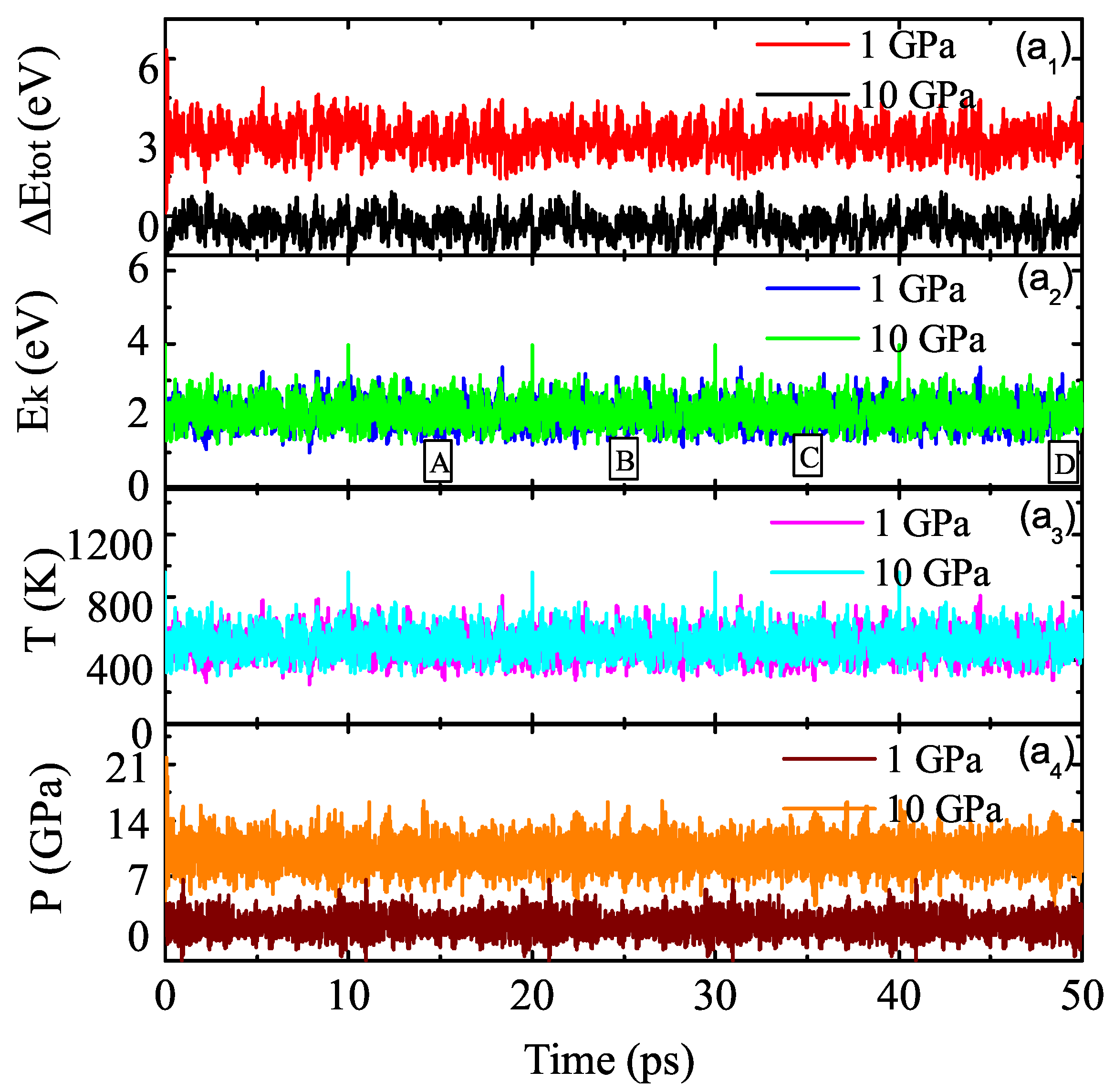
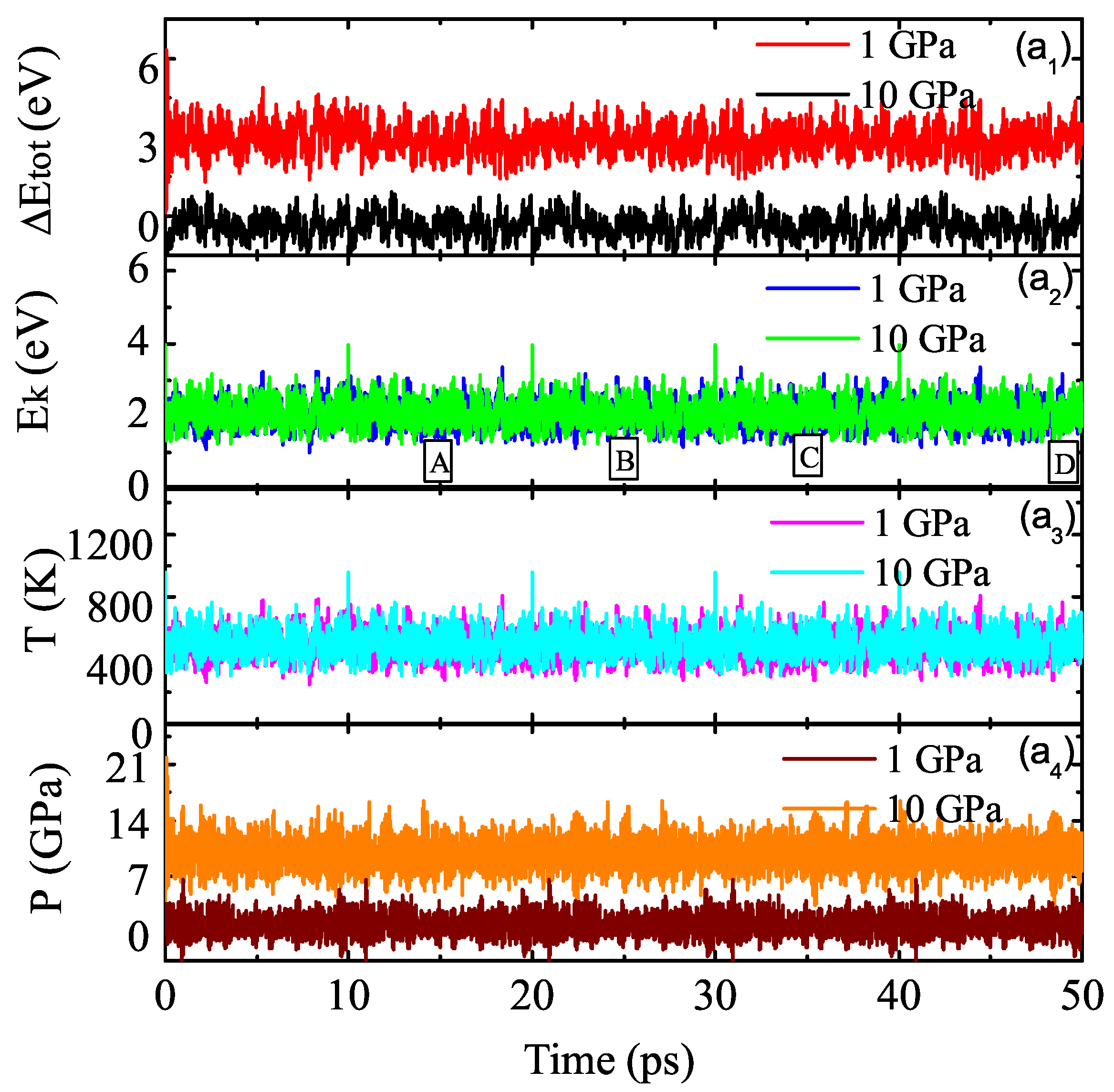
3.2. Properties of Kinetics and Thermodynamics
3.3. Prediction of Detonation Performance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Curtius, T. Ueber stickstoffwasserstoffsaure (azoimid) N3H. Berichte Dtsch. Chem. Ges. 1890, 23, 3023–3033. [Google Scholar] [CrossRef] [Green Version]
- Perera, S.; Bartlett, R.J. Coupled-cluster calculations of Raman intensities and their application to N4 and N5−. Chem. Phys. Lett. 1999, 314, 381–387. [Google Scholar] [CrossRef]
- Samartzis, P.C.; Wodtke, A.M. All-nitrogen chemistry: How far are we from N60? Int. Rev. Phys. Chem. 2006, 25, 527–552. [Google Scholar] [CrossRef]
- Thrush, B.A. The detection of free radicals in the high intensity photolysis of hydrogen azide. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1956, 235, 143–147. [Google Scholar] [CrossRef]
- Bittererova, M.; Ostmark, H.; Brinck, T. A theoretical study of the azide (N3) doublet states, a new route to tetraazatetrahedrane (N4): (N) + (N3) → (N4). J. Chem. Phys. 2002, 116, 9740–9748. [Google Scholar] [CrossRef]
- Prasad, R. Theoretical study of fine and hyperfine interactions in , , and . J. Chem. Phys. 2003, 119, 9549–9558. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W.; Sheehy, J.A.; Boatz, J.A. : A novel homoleptic polynitrogen ion as a high energy density material. Angew. Chem. Int. Ed. 1999, 38, 2004–2009. [Google Scholar] [CrossRef]
- Christe, K.O.; Dixon, D.A.; McLemore, D.; Wilson, W.W.; Sheehy, J.A.; Boatz, J.A. On a quantitative scale for Lewis acidity and recent progress in polynitrogen chemistry. J. Fluor. Chem. 2000, 101, 151–153. [Google Scholar] [CrossRef]
- Christe, K.O.; Zhang, X.; Sheehy, J.A.; Bau, R. Crystal structure of , normal coordinate analyses of , , , SF4, SeF4, and TeF4, and simple method for calculating the effects of fluorine bridging on the structure and vibrational spectra of ions in a strongly interacting ionic solid. Cheniinform 2001, 32, 6338. [Google Scholar]
- Vij, A.; Pavlovich, J.G.; Wilson, W.W.; Vij, V.; Christe, K.O. Experimental detection of the pentaazacyclopentadienide (pentazolate) anion, cyclo-. Angew. Chem. Int. Ed. 2002, 41, 3051–3054. [Google Scholar] [CrossRef]
- Vogler, A.; Wright, R.E.; Kunkely, H. Photochemical reductive cis-elimination in cis-diazidobis (triphenylphosphane) platinum (ii) evidence of the formation of bis (triphenylphosphane) platinum(O) and hexaazabenzene. Angew. Chem. Int. Ed. Engl. 1980, 19, 717–718. [Google Scholar] [CrossRef]
- Trinquier, G.; Malrieu, J.P.; Daudey, J.P. Ab initio study of the regular polyhedral molecules N4, P4, As4, N8, P8 and As8. Chem. Phys. Lett. 1981, 80, 552–557. [Google Scholar] [CrossRef]
- Lauderdale, W.J.; Stanton, J.F.; Bartlett, R.J. Stability and energetics of metastable molecules: Tetraazatetrahedrane (N4), hexaazabenzene (N6), and octaazacubane (N8). J. Phys. Chem. 1992, 96, 1173–1178. [Google Scholar] [CrossRef]
- Engelke, R.; Stine, J.R. Is N8 cubane stable? J. Phys. Chem. 1990, 94, 5689–5694. [Google Scholar] [CrossRef]
- Leininger, M.L.; Sherrill, C.D.; Schaefer, H.F., III. N8: A structure analogous to pentalene, and other high-energy density minima. J. Phys. Chem. 1995, 99, 2324–2328. [Google Scholar] [CrossRef]
- Gagliardi, L.; Evangelisti, S.; Roos, B.O.; Widmark, P.-O. A theoretical study of ten N8 isomers. J. Mol. Struct. THEOCHEM 1998, 428, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.W.; Gordon, M.S.; Boatz, J.A. Cubic fuels? Int. J. Quantum Chem. 2000, 76, 434–446. [Google Scholar] [CrossRef]
- Zheng, J.P.; Waluk, J.; Spanget-Larsen, J.; Blake, D.M.; Radziszewski, J.G. Tetrazete (N4). Can it be prepared and observed? Chem. Phys. Lett. 2000, 328, 227–233. [Google Scholar] [CrossRef]
- de Castro, S.C.; Schaefer, H.F.; Pitzer, R.M. Electronic structure of the molecular ion. J. Chem. Phys. 1981, 74, 550–558. [Google Scholar] [CrossRef]
- Rennie, E.E.; Mayer, P.M. Confirmation of the “long-lived” tetra-nitrogen (N4) molecule using neutralization-reionization mass spectrometry and ab initio calculations. J. Chem. Phys. 2004, 120, 10561–10578. [Google Scholar] [CrossRef]
- Hirshberg, B.; Gerber, R.B.; Krylov, A.I. Calculations predict a stable molecular crystal of N8. Nat. Chem. 2013, 6, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Greschner, M.J.; Zhang, M.; Majumdar, A.; Liu, H.; Peng, F.; Tse, J.S.; Yao, Y. A New Allotrope of Nitrogen as High-Energy Density Material. J. Phys. Chem. A 2016, 120, 2920–2925. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, L.; Yao, M.; Miao, M.; Liu, B. Novel all-nitrogen molecular crystals of aromatic nl0. Adv. Sci. 2020, 7, 1902320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.J.; Rice, J.E. Theoretical characterization of tetrahedral N4. J. Chem. Phys. 1991, 94, 1215–1221. [Google Scholar] [CrossRef]
- Yarkony, D.R. Theoretical studies of spin-forbidden radiationless decay in polyatomic systems: Insights from recently developed computational methods. J. Am. Chem. Soc. 1992, 114, 5406–5411. [Google Scholar] [CrossRef]
- Dunn, K.M.; Morokuma, K. Transition state for the dissociation of tetrahedral N4. J. Chem. Phys. 1995, 102, 4904–4908. [Google Scholar] [CrossRef]
- Jia, W.; Fu, J.; Cao, Z.; Wang, L.; Chi, X.; Gao, W.; Wang, L.-W. Fast plane wave density functional theory molecular dynamics calculations on multi-GPU machines. J. Comput. Phys. 2013, 251, 102–115. [Google Scholar] [CrossRef]
- Jia, W.; Cao, Z.; Wang, L.; Fu, J.; Chi, X.; Gao, W.; Wang, L.-W. The analysis of a plane wave pseudopotential density functional theory code on a GPU machine. Comput. Phys. Commun. 2013, 184, 9–18. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Emzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Quigley, D.; Probert, M. Constant pressure Langevin dynamics: Theory and application. Comput. Phys. Commun. 2005, 169, 322–325. [Google Scholar] [CrossRef]
- Bittererova, M.; Brinck, T.; Ostmark, H. Theoretical study of the singlet electronically excited states of N4. Chem. Phys. Lett. 2001, 340, 597–603. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Jacobs, S.J. Chemistry of detonations. I. a simple method for calculating detonation properties of C–H–N–O 356 explosives. J. Chem. Phys. 1968, 48, 23–35. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Ablard, J.E. Chemistry of detonations. II. buffered equilibria. J. Chem. Phys. 1968, 48, 36–42. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Dickinson, C. Chemistry of detonations. III. evaluation of the simplified calculational method for chapmanjouguet detonation pressures on the basis of available experimental information. J. Chem. Phys. 1968, 48, 43–50. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Hurwitz, H. Chemistry of detonations. IV. evaluation of a simple predictional method for detonation velocities of C-H-N-O explosives. J. Chem. Phys. 1968, 48, 3685–3692. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Lai, W.P.; Yu, T.; Ge, Z.X.; Luo, Y.J.; Xu, T.; Yin, S.W. Theoretical investigations on fundamental performances of all-nitrogen materials: 11. enthalpy of formation prediction. Chin. J. Energ. Mater. 2017, 25, 552–556. [Google Scholar]
- Bondarchuk, S.V. Bipentazole (N10): A Low-Energy Molecular Nitrogen Allotrope with High Intrinsic Stability. J. Phys. Chem. Lett. 2020, 11, 5544–5548. [Google Scholar] [CrossRef]
- Li, Y.C.; Pang, S.P. Progress of all-nitrogen ultrahigh-energetic materials. Chin. J. Explos. Propellants 2012, 35, 1–8. [Google Scholar]
- Lu, Y.H.; He, J.X.; Lei, Q.; Ren, X.T.; Guo, Y.Y.; Ding, N.; Cao, Y.Y. Research progress in all-nitrogen compounds. Chern. Propellants Polym. Mater. 2013, 9. [Google Scholar]
- Liu, Y.Z.; Lai, W.P.; Yu, T.; Ge, Z.X.; Xu, T.; Luo, Y.J.; Yin, S.W. Theoretical investigations on fundamental performances of all-nitrogen materials: 1. crystal density rediction. Chin. J. Energetic Mater. 2017, 25, 100–105. (In Chinese) [Google Scholar]
- Chen, S.-L.; Yang, Z.-R.; Wang, B.-J.; Shang, Y.; Sun, L.-Y.; He, C.-T.; Zhou, H.-L.; Zhang, W.-X.; Chen, X.-M. Molecular perovskite high-energetic materials. Sci. China Mater. 2018, 61, 1123–1128. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure (GPa) | Density (g/cm) | Energy (eV) |
|---|---|---|
| 30 | 2.79 | −8549.92 |
| 50 | 3.11 | −8518.47 |
| 80 | 3.48 | −8476.25 |
| 100 | 3.67 | −8450.27 |
| Explosive | ||||
|---|---|---|---|---|
| TNT | 1.65 | −14.17 | 20.7/19.4 | 7.01/6.92 |
| RDX | 1.80 | 16.79 | 34.4/34.9 | 8.80/8.82 |
| HMX | 1.91 | 17.87 | 38.5/39.1 | 9.15/9.15 |
| CL-20 | 2.04 | 95.04 | 44.3/45.9 | 9.64/9.60 |
| N | 2.09 | 180.8 | 74.2 | 12.27 |
| N | 2.20 | 180.8 | 81.0 | 12.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, S.; Wang, F. Novel All-Nitrogen Molecular Crystals Composed of Tetragonal N4 Molecules. Int. J. Mol. Sci. 2022, 23, 5503. https://doi.org/10.3390/ijms23105503
Pang S, Wang F. Novel All-Nitrogen Molecular Crystals Composed of Tetragonal N4 Molecules. International Journal of Molecular Sciences. 2022; 23(10):5503. https://doi.org/10.3390/ijms23105503
Chicago/Turabian StylePang, Suna, and Feng Wang. 2022. "Novel All-Nitrogen Molecular Crystals Composed of Tetragonal N4 Molecules" International Journal of Molecular Sciences 23, no. 10: 5503. https://doi.org/10.3390/ijms23105503
APA StylePang, S., & Wang, F. (2022). Novel All-Nitrogen Molecular Crystals Composed of Tetragonal N4 Molecules. International Journal of Molecular Sciences, 23(10), 5503. https://doi.org/10.3390/ijms23105503