
The Anticancer Activity Conferred by the Mud Crab Antimicrobial Peptide Scyreprocin through Apoptosis and Membrane Disruption

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
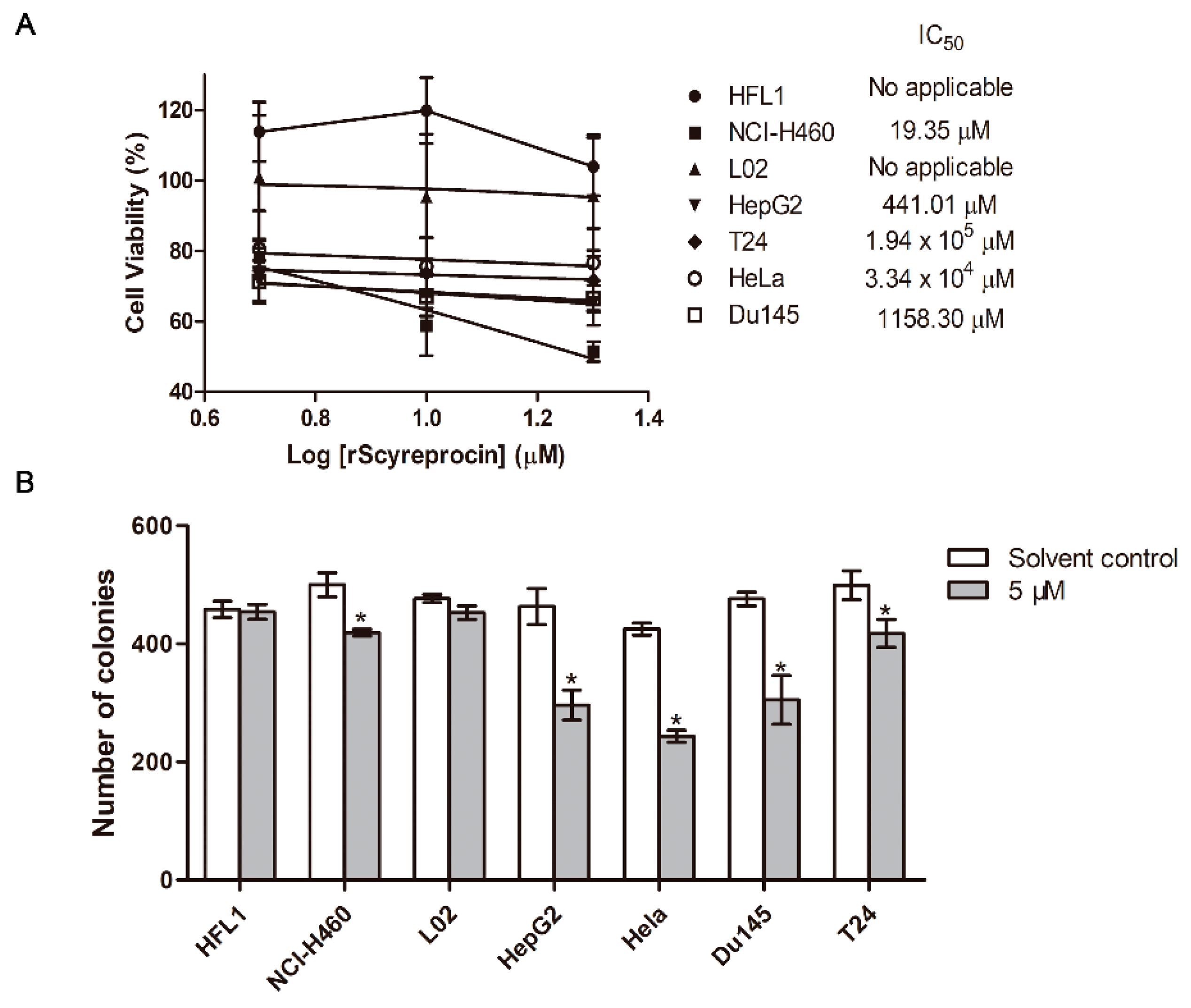
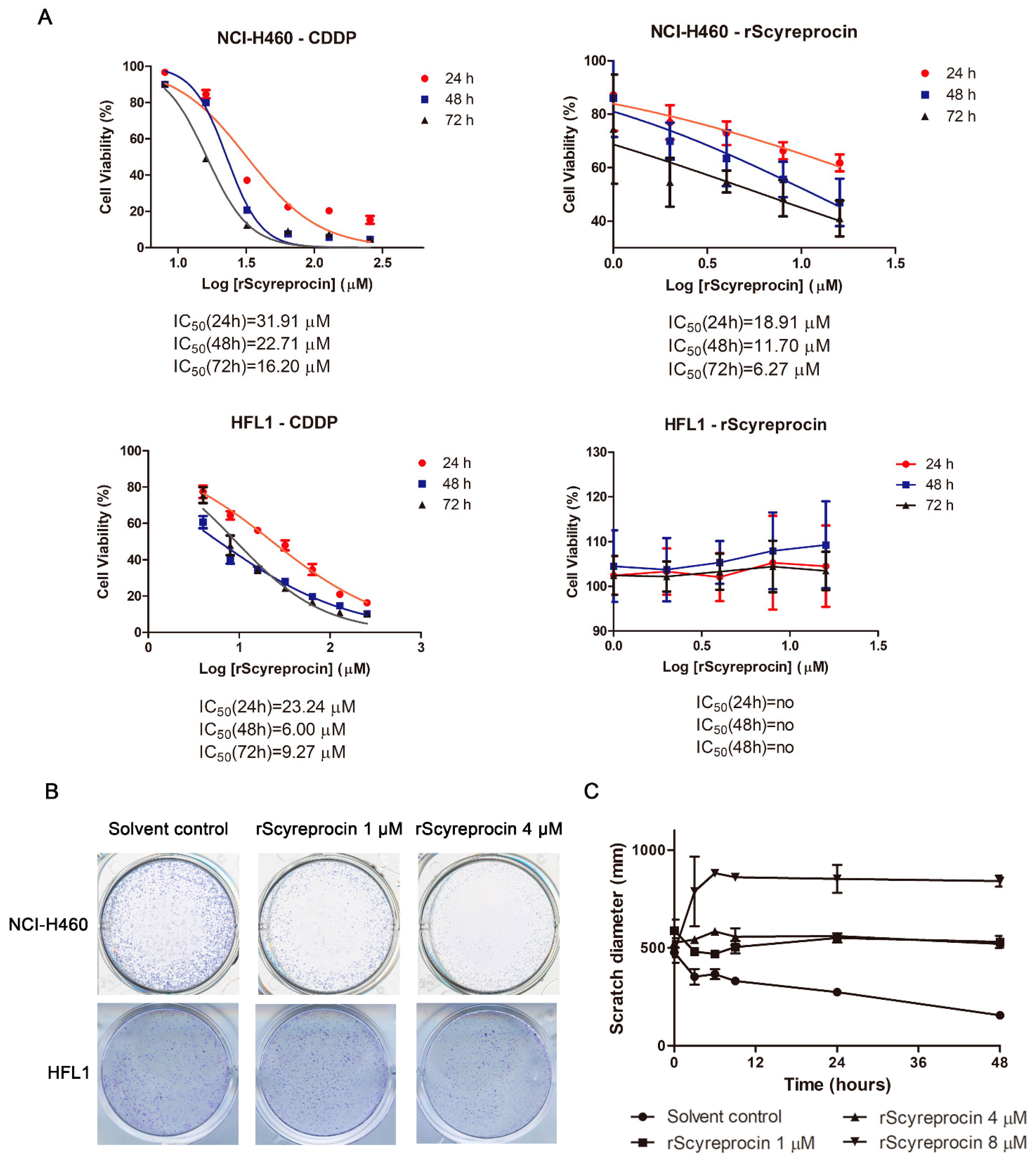
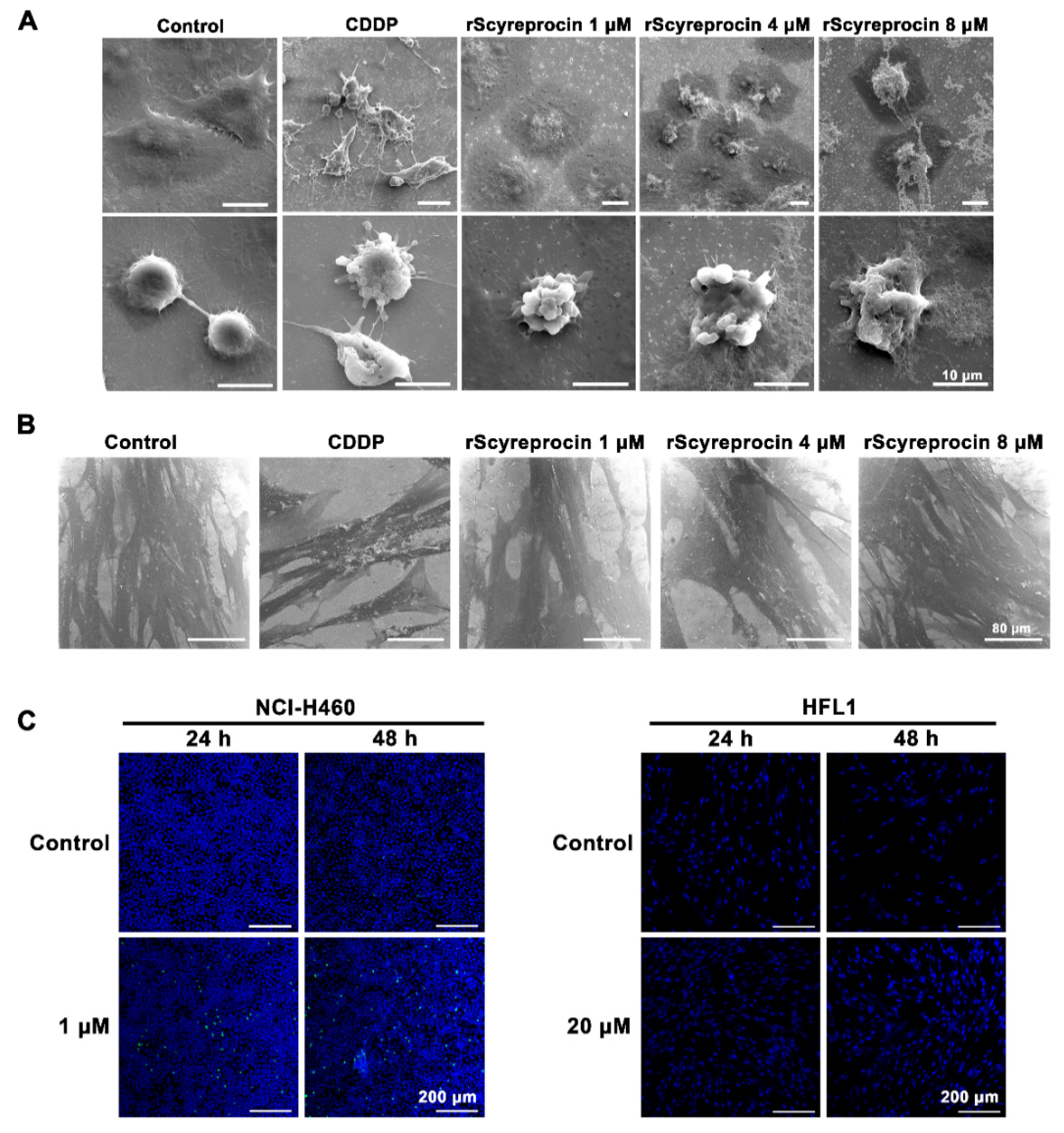
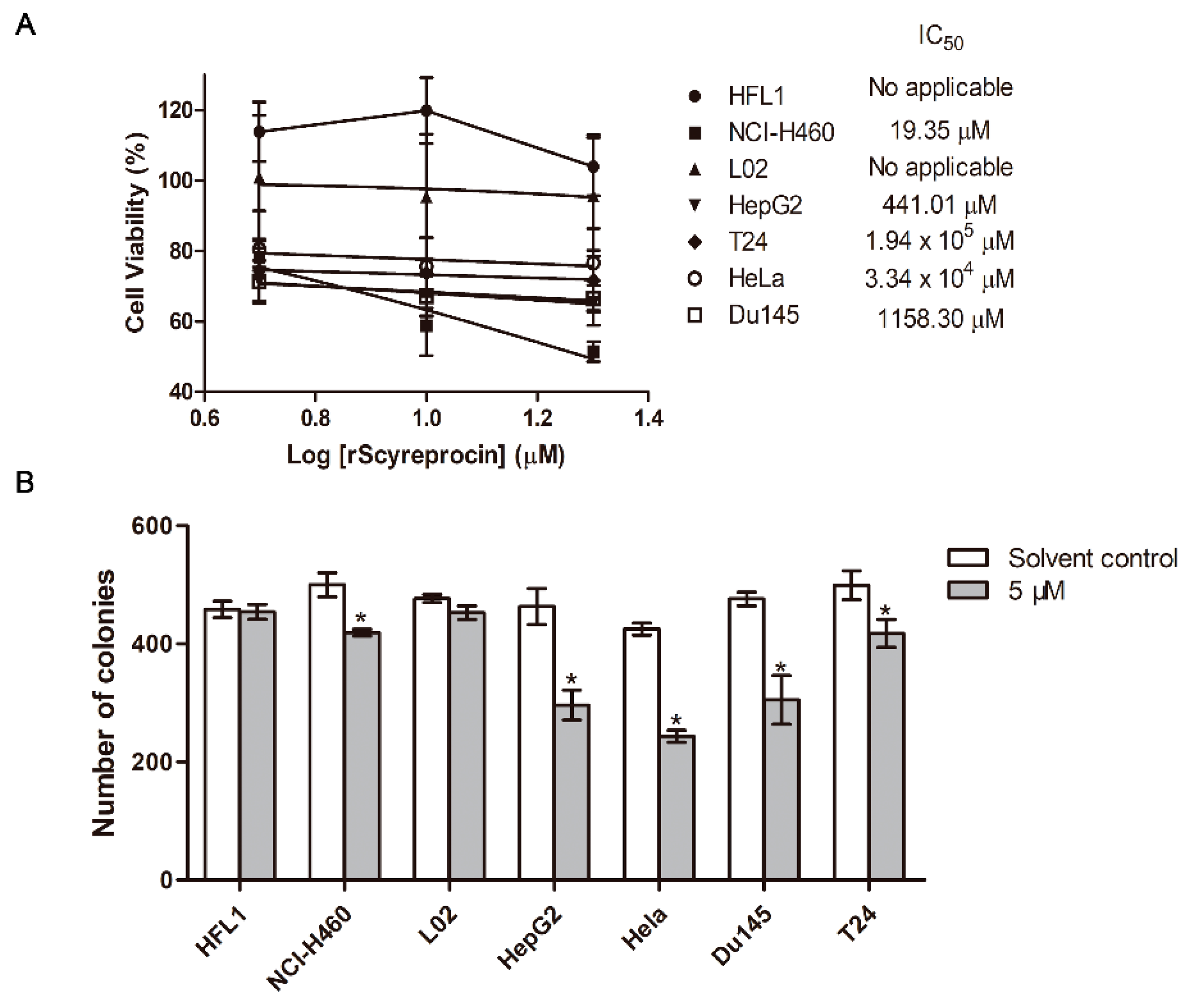
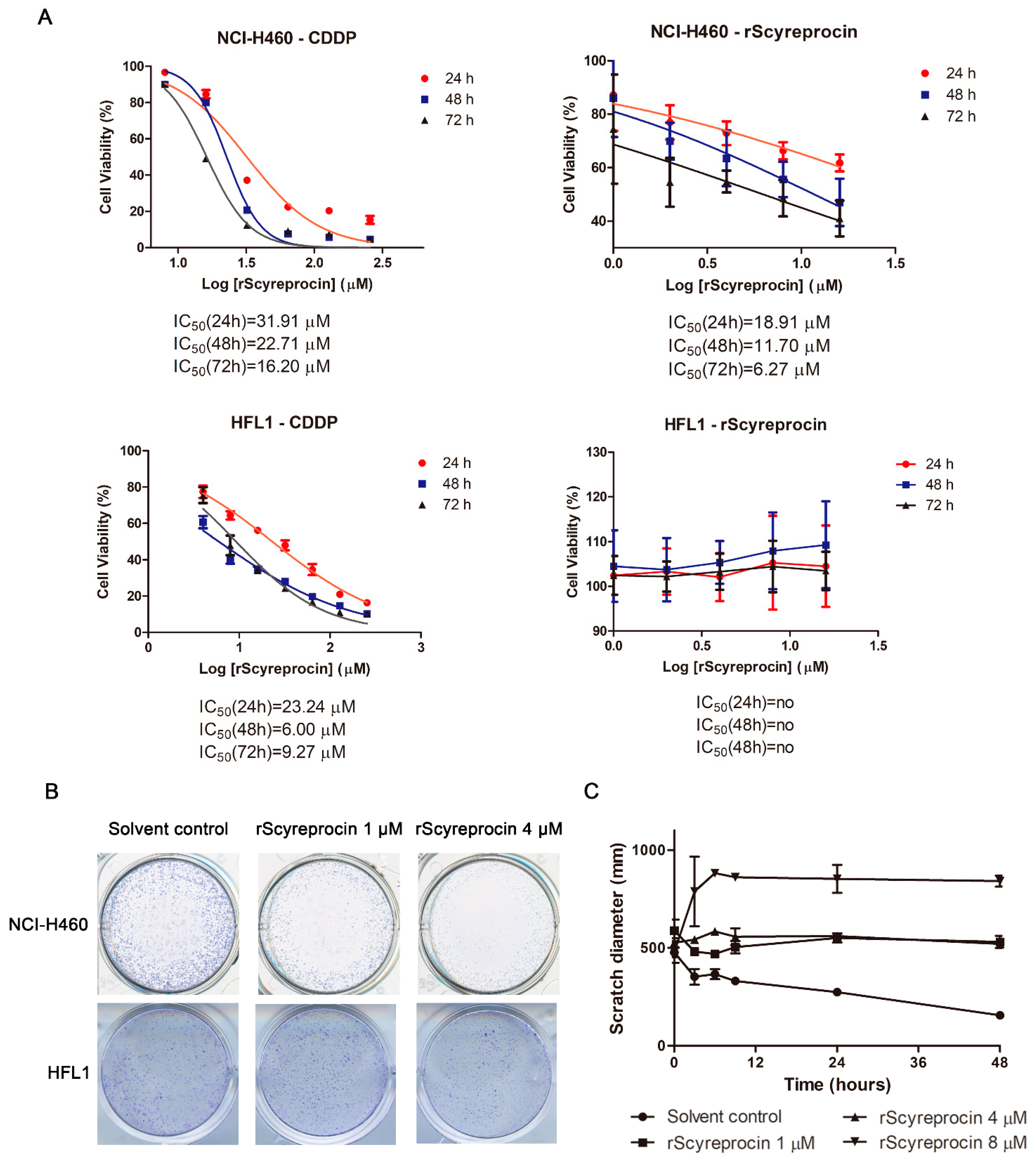
2.1. Recombinant scyreprocin (rScyreprocin) Inhibited Proliferation, Migration, and Colony Formation Ability of Cancer Cells but Not Non-Cancer Cells
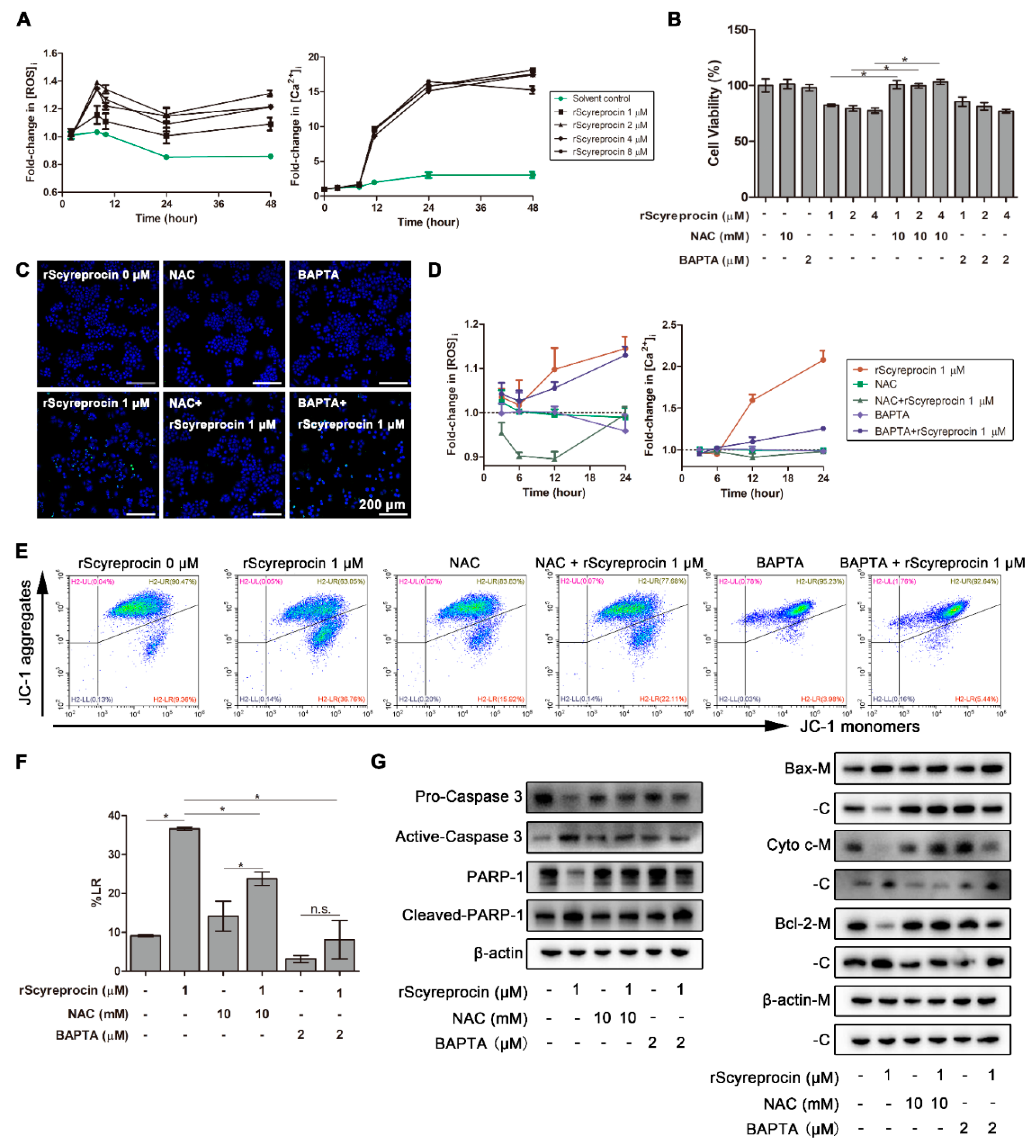
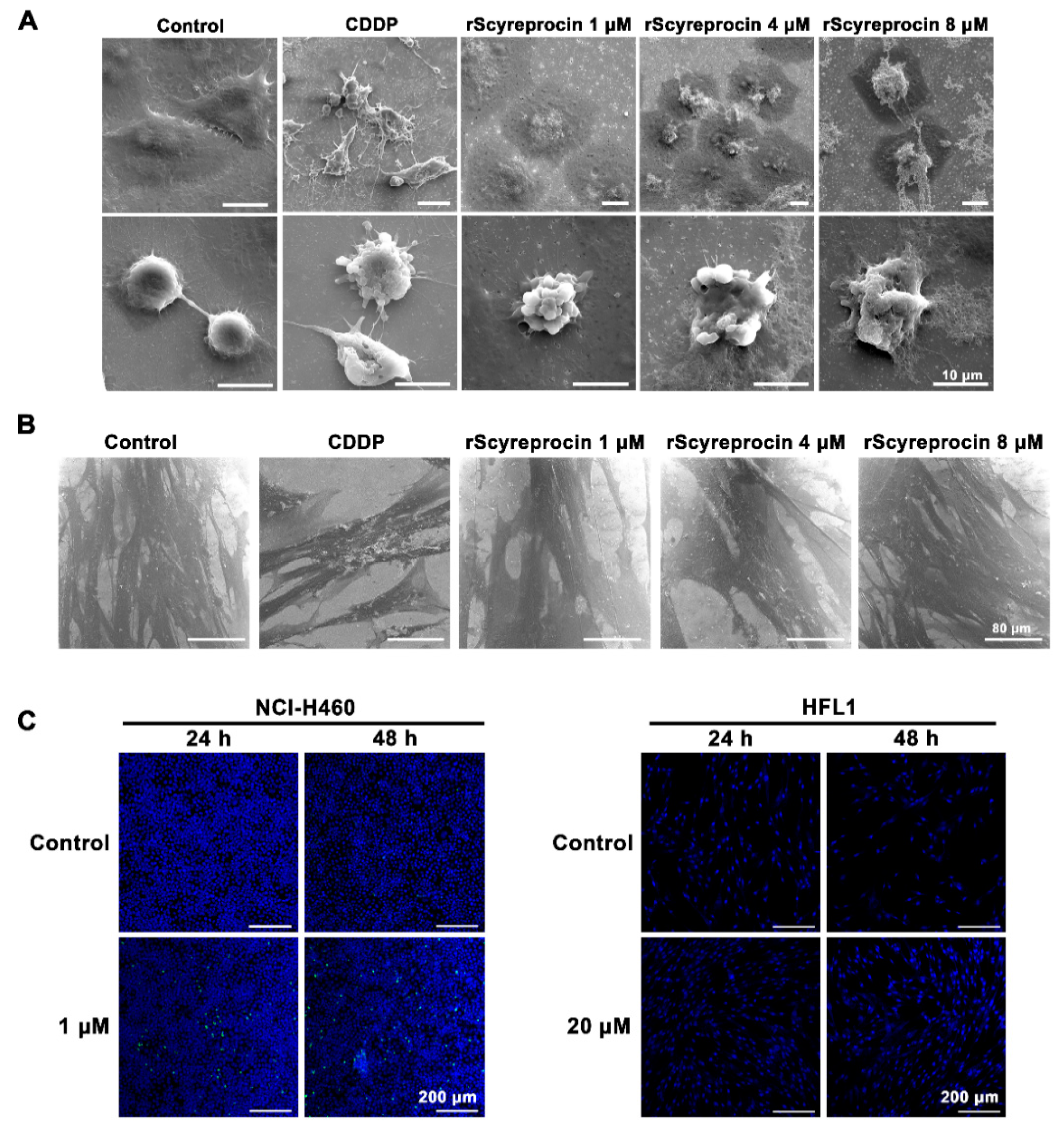
2.2. rScyreprocin Induced Apoptosis and Membrane Disruption of Cancer Cells
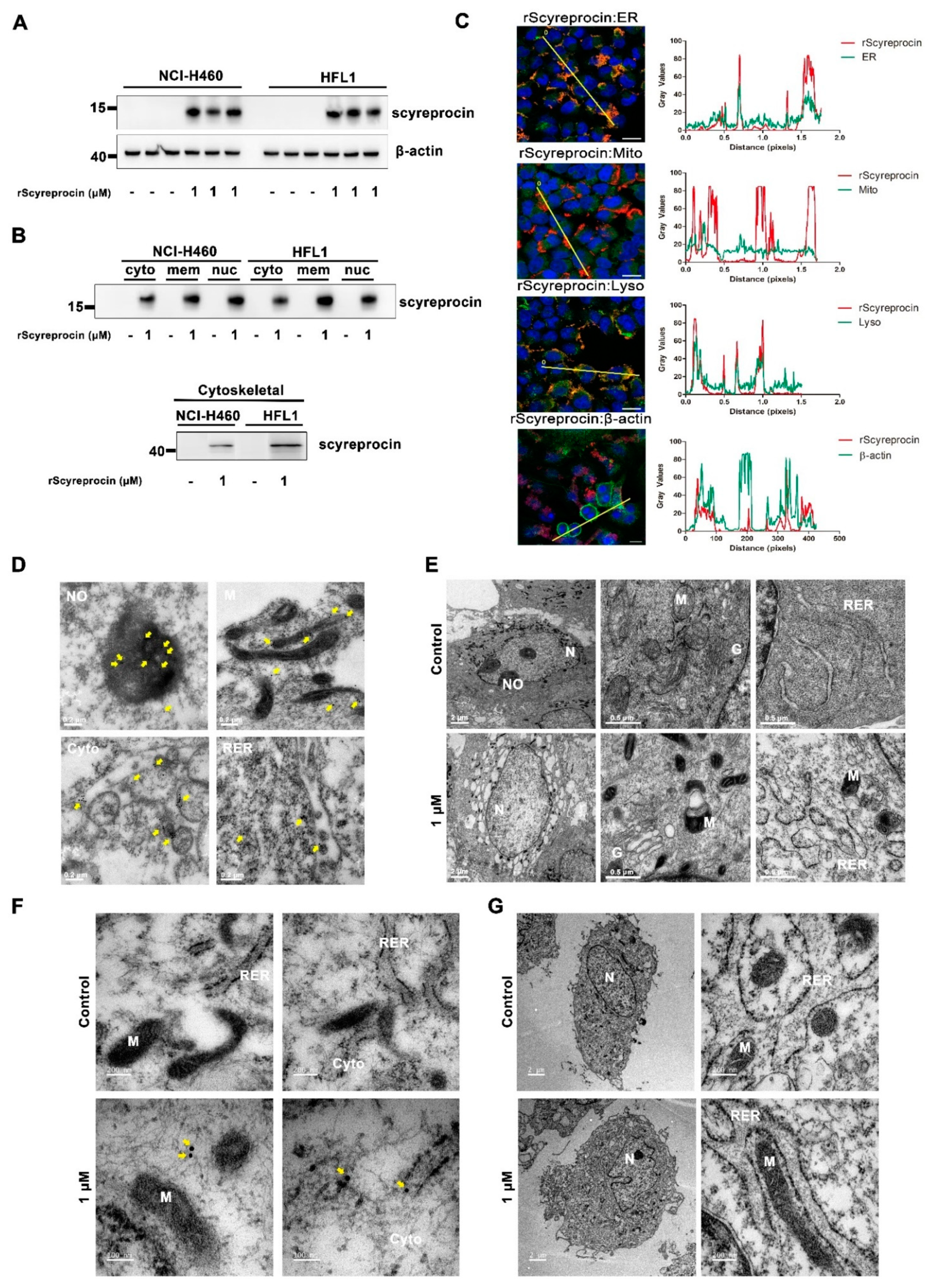
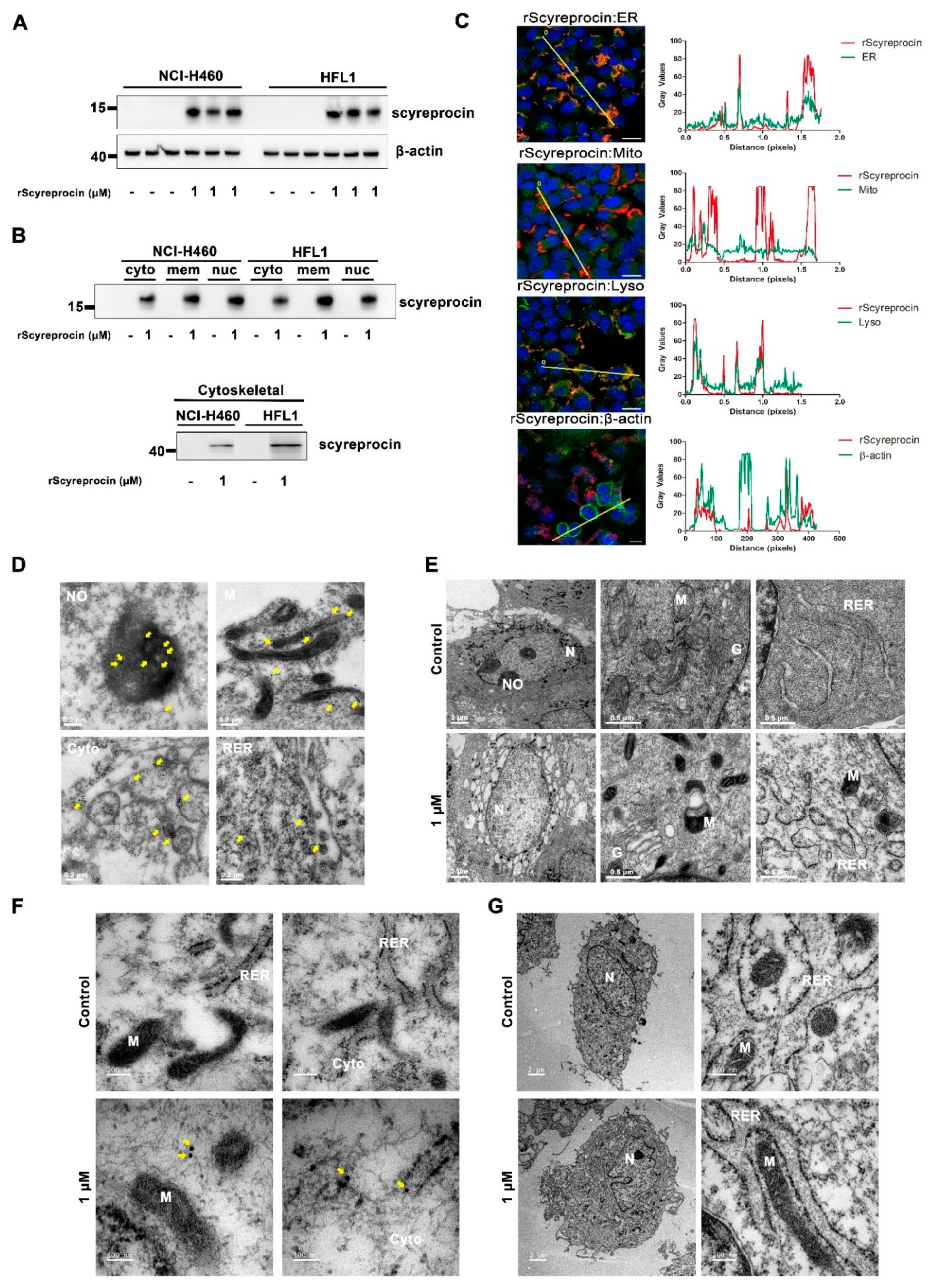
2.3. rScyreprocin Entered into Cells and Distributed in Organelles
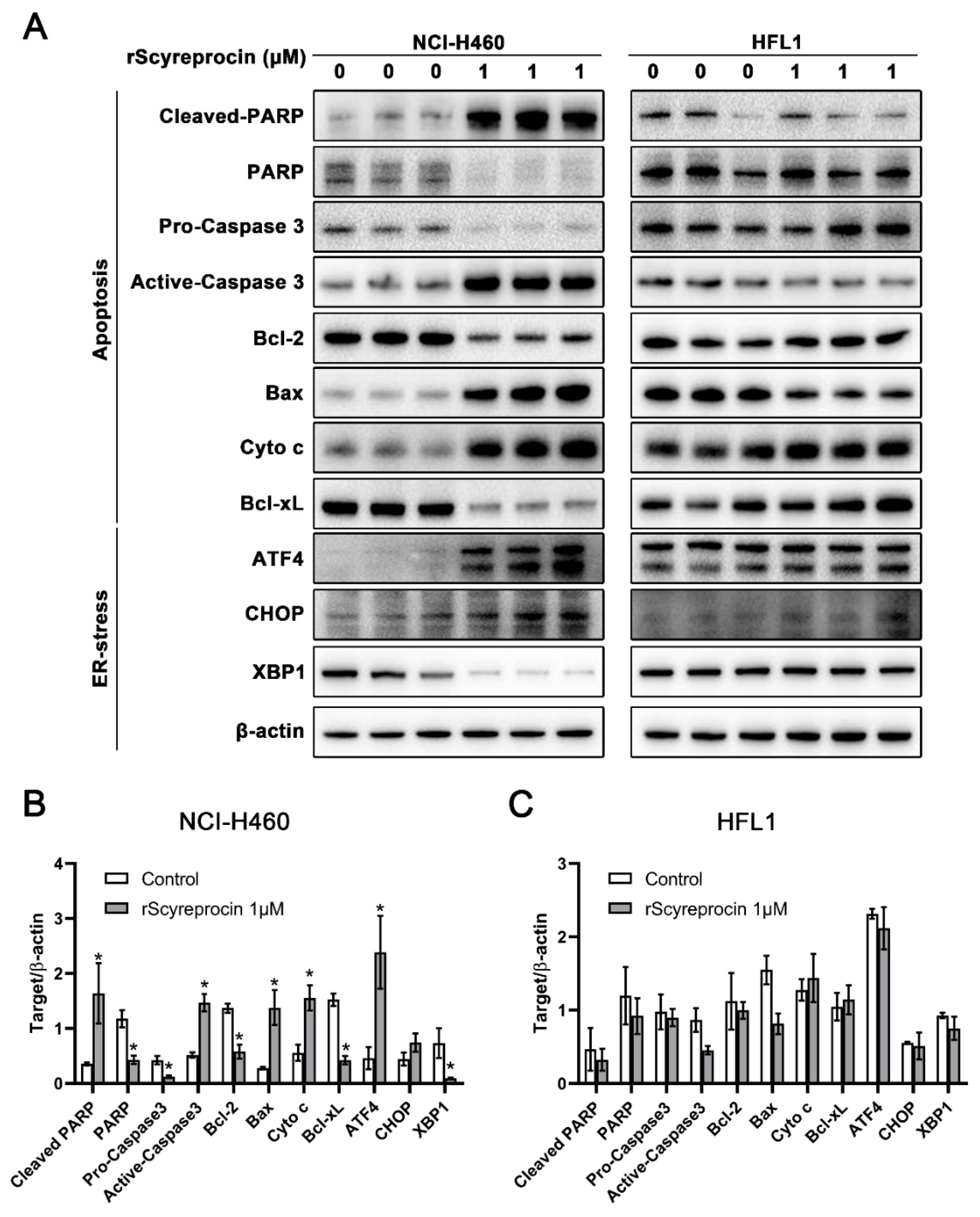
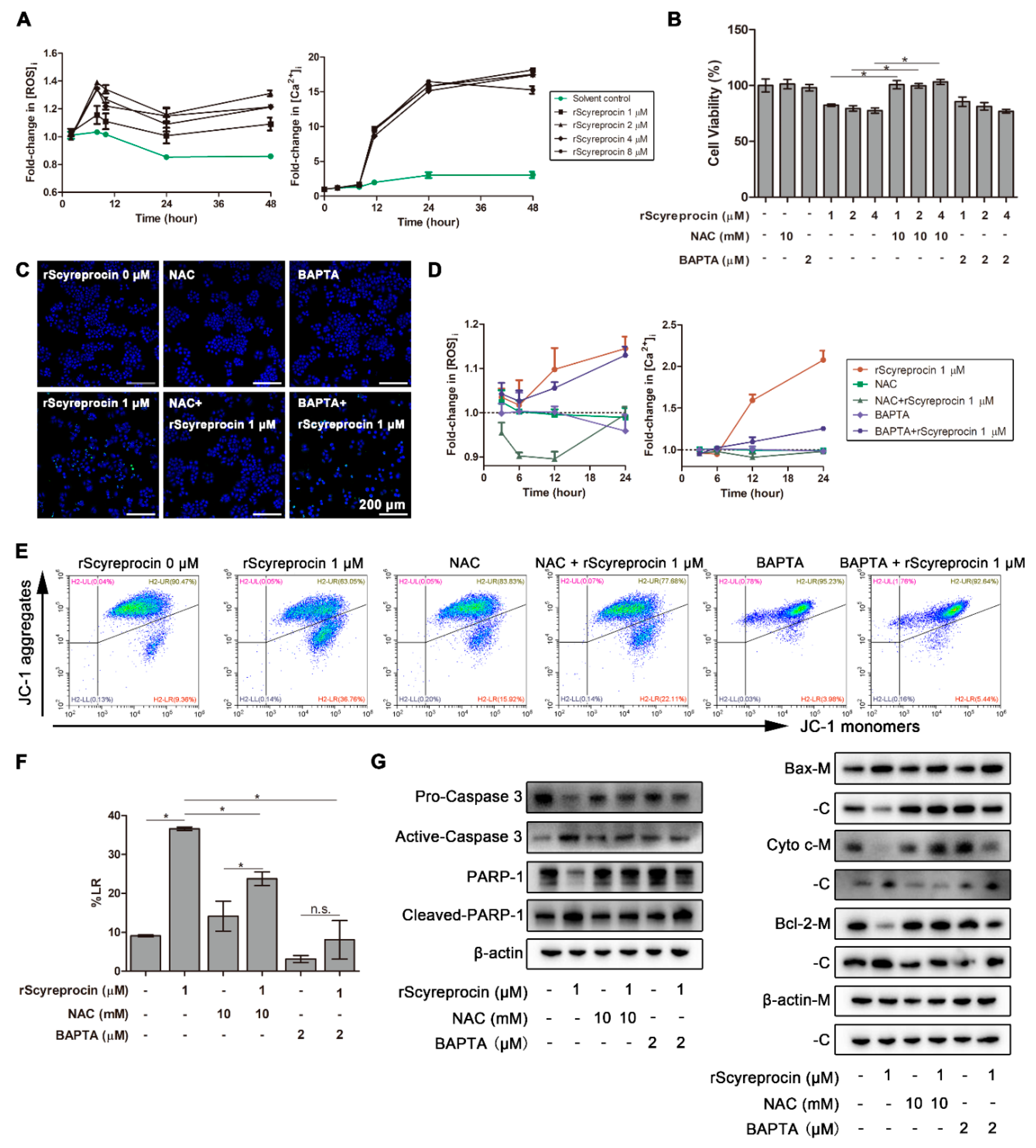
2.4. rScyreprocin Induced Endoplasmic Reticulum (ER) Stress and Apoptosis in H460 Cells
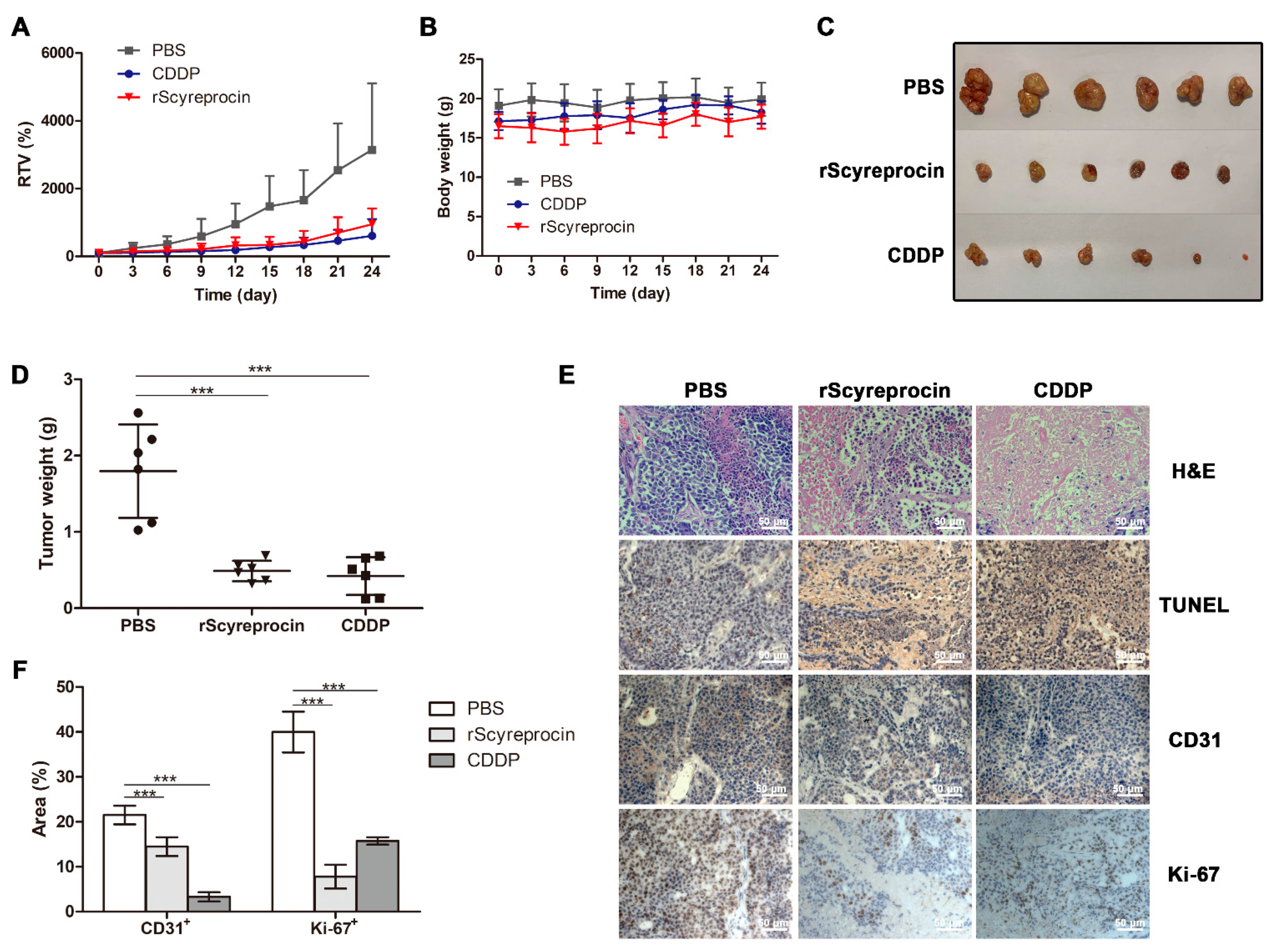
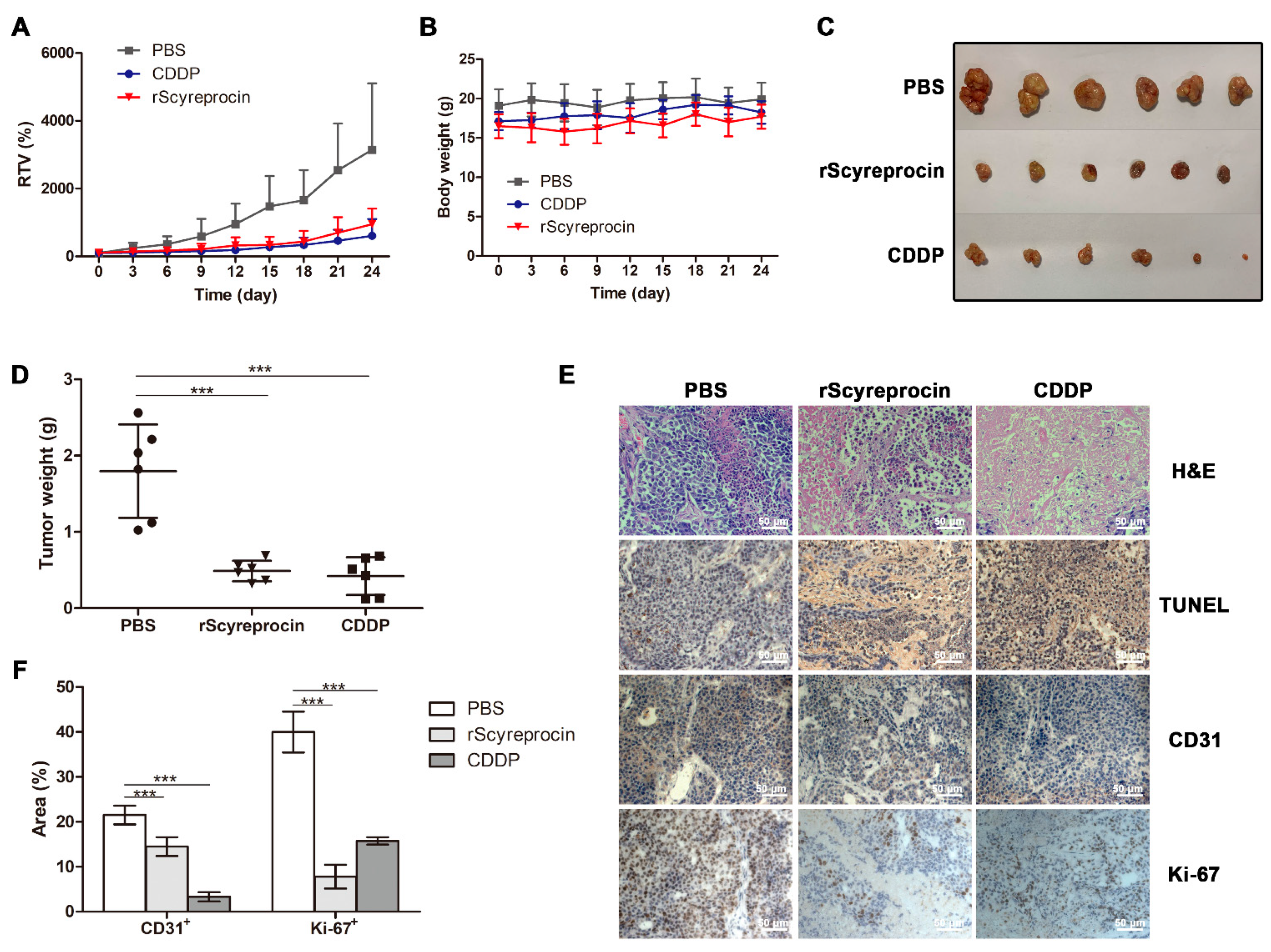
2.5. rScyreprocin Exerted Anti-Tumor Effect in Nude Mice Model
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Recombinant Protein and Antibody Preparation
4.3. Cell Lines and Cell Culture
4.4. Colony Formation
4.5. Cell Viability Assay
4.6. Electron Microscopy Observation
4.7. Scratch-Wound Assay
4.8. Cell Apoptosis Detection
4.9. Cell Immunofluorescent Labeling Assay
4.10. Analysis of Mitochondria Membrane Potential (MMP)
4.11. Measurement of Intracellular Reactive Oxygen Species (ROS) and Intracellular Ca2+ Concentration
4.12. Western Blotting Assay
4.13. Extraction of Proteins from Different Cell Compartments
4.14. Assessment of In Vivo Anticancer Activity of Rscyreprocin
4.15. H&E, TUNEL, and Immunohistochemical Staining
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer, I. Cancer Today; World Health Organization: Lyon, France, 2020. [Google Scholar]
- Siegel, R.L.; Miller, D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Dy, G.K.; Adjei, A.A. Small Cell Lung Cancer. Mayo Clin. Proc. 2008, 83, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, W.; Anderson, G.; Bartolucci, A.; Bell, D.; Toni, V. Chemotherapy in non-small cell lung cancer: A meta-analysis using updated data on individual patients from 52 randomised clinical trials. BMJ Clin. Res. 1995, 311, 899–909. [Google Scholar] [CrossRef]
- Howington, J.A.; Blum, M.G.; Chang, A.C.; Balekian, A.A.; Murthy, S.C. Treatment of Stage I and II Non-small Cell Lung Cancer: Diagnosis and Management of Lung Cancer, 3rd ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2013, 143, e278S–e313S. [Google Scholar] [CrossRef]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucl. Acid Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Pinedo, H.M. Gastrointestinal toxicity of cis-diamminedichloroplatinum (II). Neth. J. Med. 1982, 25, 270–274. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Kroemer, G. Molecular mechanism of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- D’Addario, G.; Pintillie, M.; Leighl, N.B.; Feld, R.; Cerny, T.; Shepherd, F.A. Platinum-based versus non-platinum-based chemotherapy in advanced non-small cell lung cancer (NSCLC): A meta-analysis of the published literature. J. Clin. Oncol. 2005, 23, 2926–2936. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.W.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochi. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Patrzykat, A.; Gallant, J.W.; Seo, J.-K.; Pytyck, J.; Douglas, S.E. Novel antimicrobial peptides derived from flatfish genes. Antimicrob. Agents Chemother. 2003, 47, 2464–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varkey, J.; Nagaraj, R. Antibacterial activity of human neutrophil defensin HNP-1 analogs without cysteines. Antimicrob. Agents Chemother. 2005, 49, 4561–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.-A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739–15751. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Seger, D.; Makovitzki, A.; Kalchenko, V.; Eshhar, Z.; Degani, H.; Shai, Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006, 66, 5371–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Assodeh, A.; Klonisch, T.; Halayko, A.J.; Kadkhoda, K.; Kroczak, T.J.; Gibson, S.B.; Booy, E.P.; Naderi-Manesh, H.; Los, M. Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J. Cell Mol. Med. 2010, 12, 1005–1022. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Kim, Y.J.; Kim, H.; Kim, S.C.; Cho, J.H. Buforin IIb induces endoplasmic reticulum stress-mediated apoptosis in HeLa cells. Peptides 2015, 69, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, F.; Chen, H.-Y.; Peng, H.; Hao, H.; Wang, K.-J. A novel antimicrobial peptide scyreprocin from mud crab Scylla paramamosain showing potent antifungal and anti-biofilm activity. Front. Microbiol. 2020, 11, 1589. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, F.; Qiao, K.; Zhang, H.; Chen, H.Y.; Wang, K.J. Two Male-Specific Antimicrobial Peptides SCY2 and Scyreprocin as Crucial Molecules Participated in the Sperm Acrosome Reaction of Mud Crab Scylla paramamosain. Int. J. Mol. Sci. 2022, 23, 3373. [Google Scholar] [CrossRef]
- Stewart, B.W.; Wild, C.P. International agency for research on cancer. In World Cancer Report; World Health Organization: Lyon, France, 2014. [Google Scholar]
- World-Health-Organization. Antimicrobial Resistance: Global Report on Surveillance; World-Health-Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Hanaoka, Y.; Yamaguchi, Y.; Yamamoto, H.; Ishii, M.; Nagase, T.; Kurihara, H.; Akishita, M.; Ouchi, Y. In vitro and in vivo anticancer activity of human β-defensin-3 and its mouse homolog. Anticancer. Res. 2016, 36, 5999–6004. [Google Scholar] [CrossRef] [Green Version]
- Arias, M.; Haney, E.F.; Hilchie, A.L.; Corcoran, J.A.; Hyndman, M.E.; Hancock, R.E.W.; Vogel, H.J. Selective anticancer activity of synthetic peptides derived from the host defence peptide tritrpticin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tu, J.; Zhou, C.; Li, J.; Zhao, L. The effect of Lfcin-B on non-small cell lung cancer H460 cells is mediated by inhibiting VEGF expression and inducing apoptosis. Arch. Pharm. Res. 2014, 38, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X.; Xu, X.M.; Hong, S.G.; Chen, J.G.; Zhang, L.R. RGD-Tachyplesin inhibits tumor growth. Cancer Res. 2001, 61, 2434–2438. [Google Scholar] [CrossRef] [PubMed]
- Neale, C.; Hsu, J.C.Y.; Yip, C.M.; Pomès, R. Indolicidin binding induces thinning of a lipid bilayer. Biophys. J. 2014, 106, L29–L31. [Google Scholar] [CrossRef] [Green Version]
- Fressinaud, C.; Eyer, J. Neurofilaments and NFL-TBS.40-63 peptide penetrate oligodendrocytes through clathrin-dependent endocytosis to promote their growth and survival in vitro. Neuroscience 2015, 298, 42–51. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.H.; Liebes, L.; Zou, Y.; Perez-Soler, R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J. Biol. Chem. 2003, 278, 33714–33723. [Google Scholar] [CrossRef] [Green Version]
- Harr, M.W.; Distelhorst, C.W. Apoptosis and autophagy: Decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2010, 2, a005579. [Google Scholar] [CrossRef]
- Meldolesi, J.; Pozzan, T. The endoplasmic reticulum Ca2+ store: A view from the lumen. Trends Biochem. Sci. 1998, 23, 10–14. [Google Scholar] [CrossRef]
- Gordeeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 2003, 68, 1077–1080. [Google Scholar] [CrossRef]
- Bonilla, M.; Nastase, K.; Cunningham, K. Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J. 2002, 21, 2343–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, L.; Hajnóczky, G. Mitochondria and endoplasmic reticulum: The lethal interorganelle cross-talk. J. Bioenerg. Biomembr. 2005, 37, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Drago, I.; Pizzo, P.; Pozzan, T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ. 2007, 14, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, G.J.; Árpád, B.; Ildikó, M.; Gábor, P.; Ferhan, A.; Iván, K.; Roberta, F.B.; Róbert, A.l.; László, H.; Puskás, L.G. Achiral mannich-base curcumin analogs induce unfolded protein response and mitochondrial membrane depolarization in PANC-1 cells. Int. J. Mol. Sci. 2017, 18, 2105. [Google Scholar] [CrossRef]
- Wu, J.M.; Jan, P.S.; Yu, H.C.; Haung, H.Y.; Fang, H.J.; Chang, Y.I.; Cheng, J.W.; Chen, H.M. Structure and function of a custom anticancer peptide, CB1a. Peptides 2009, 30, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Pittala, S.; Krelin, Y.; Shoshan-Barmatz, V. Targeting liver cancer and associated pathologies in mice with a mitochondrial VDAC1-based peptide. Neoplasia 2018, 20, 594–609. [Google Scholar] [CrossRef]
- Lin, W.; Yang, J.; He, X.; Mo, G.; Jing, H.; Yan, X.; Lin, D.; Ren, L. Structure and function of a potent lipopolysaccharide-binding antimicrobial and anti-inflammatory peptide. J. Med. Chem. 2013, 9, 3546–3556. [Google Scholar] [CrossRef]
- Chen, H.M.; Chan, S.C.; Lee, J.C.; Chang, C.C.; Jack, R.W. Transmission electron microscopic observations of membrane effects of antibiotic Cecropin B on Escherichia coli. Microsc. Res. Technol. 2003, 62, 423–430. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Chen, H.-Y.; Hao, H.; Wang, K.-J. The Anticancer Activity Conferred by the Mud Crab Antimicrobial Peptide Scyreprocin through Apoptosis and Membrane Disruption. Int. J. Mol. Sci. 2022, 23, 5500. https://doi.org/10.3390/ijms23105500
Yang Y, Chen H-Y, Hao H, Wang K-J. The Anticancer Activity Conferred by the Mud Crab Antimicrobial Peptide Scyreprocin through Apoptosis and Membrane Disruption. International Journal of Molecular Sciences. 2022; 23(10):5500. https://doi.org/10.3390/ijms23105500
Chicago/Turabian StyleYang, Ying, Hui-Yun Chen, Hua Hao, and Ke-Jian Wang. 2022. "The Anticancer Activity Conferred by the Mud Crab Antimicrobial Peptide Scyreprocin through Apoptosis and Membrane Disruption" International Journal of Molecular Sciences 23, no. 10: 5500. https://doi.org/10.3390/ijms23105500
APA StyleYang, Y., Chen, H.-Y., Hao, H., & Wang, K.-J. (2022). The Anticancer Activity Conferred by the Mud Crab Antimicrobial Peptide Scyreprocin through Apoptosis and Membrane Disruption. International Journal of Molecular Sciences, 23(10), 5500. https://doi.org/10.3390/ijms23105500