
A Hierarchical Genotyping Framework Using DNA Melting Temperatures Applied to Adenovirus Species Typing


Abstract
:1. Introduction
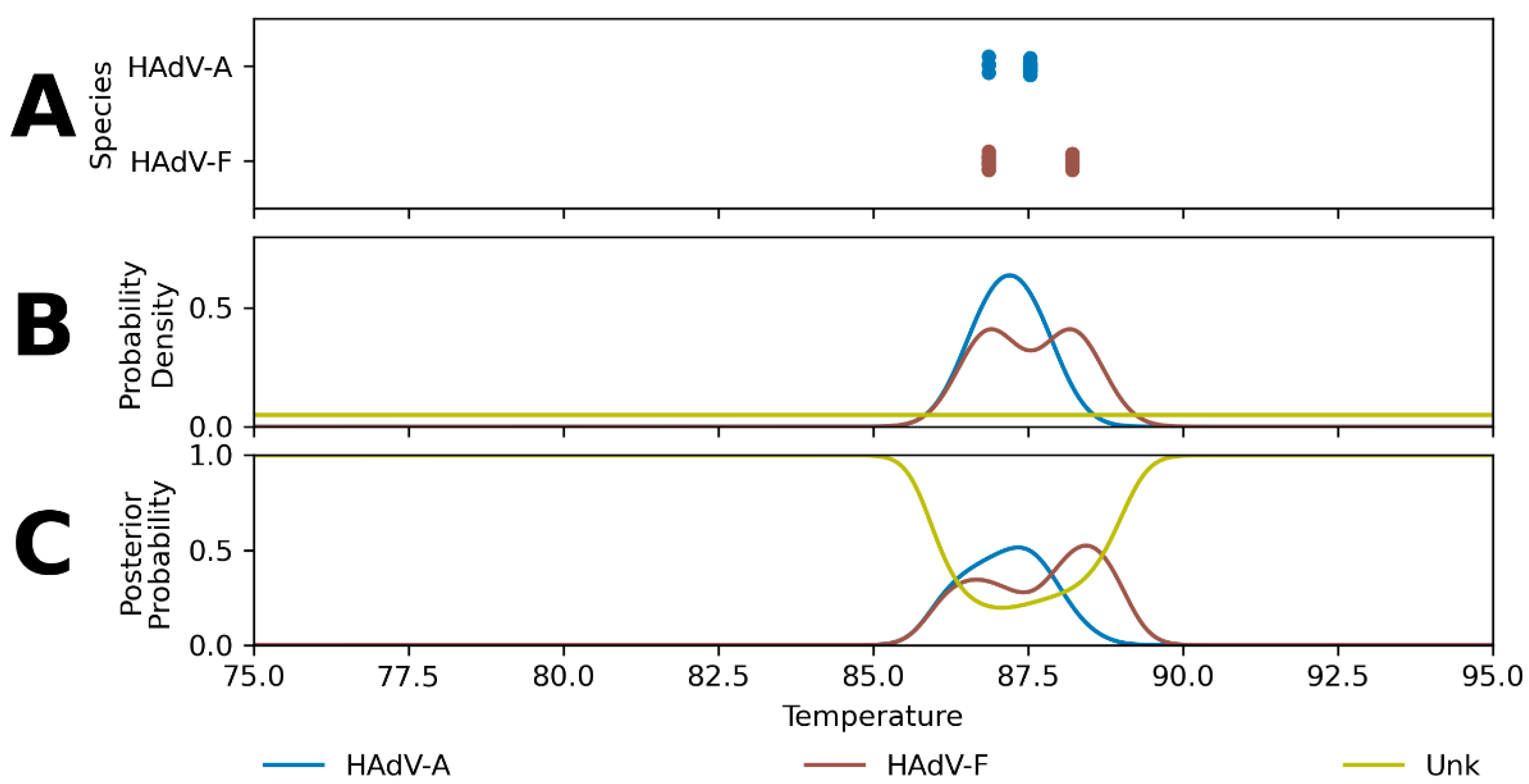
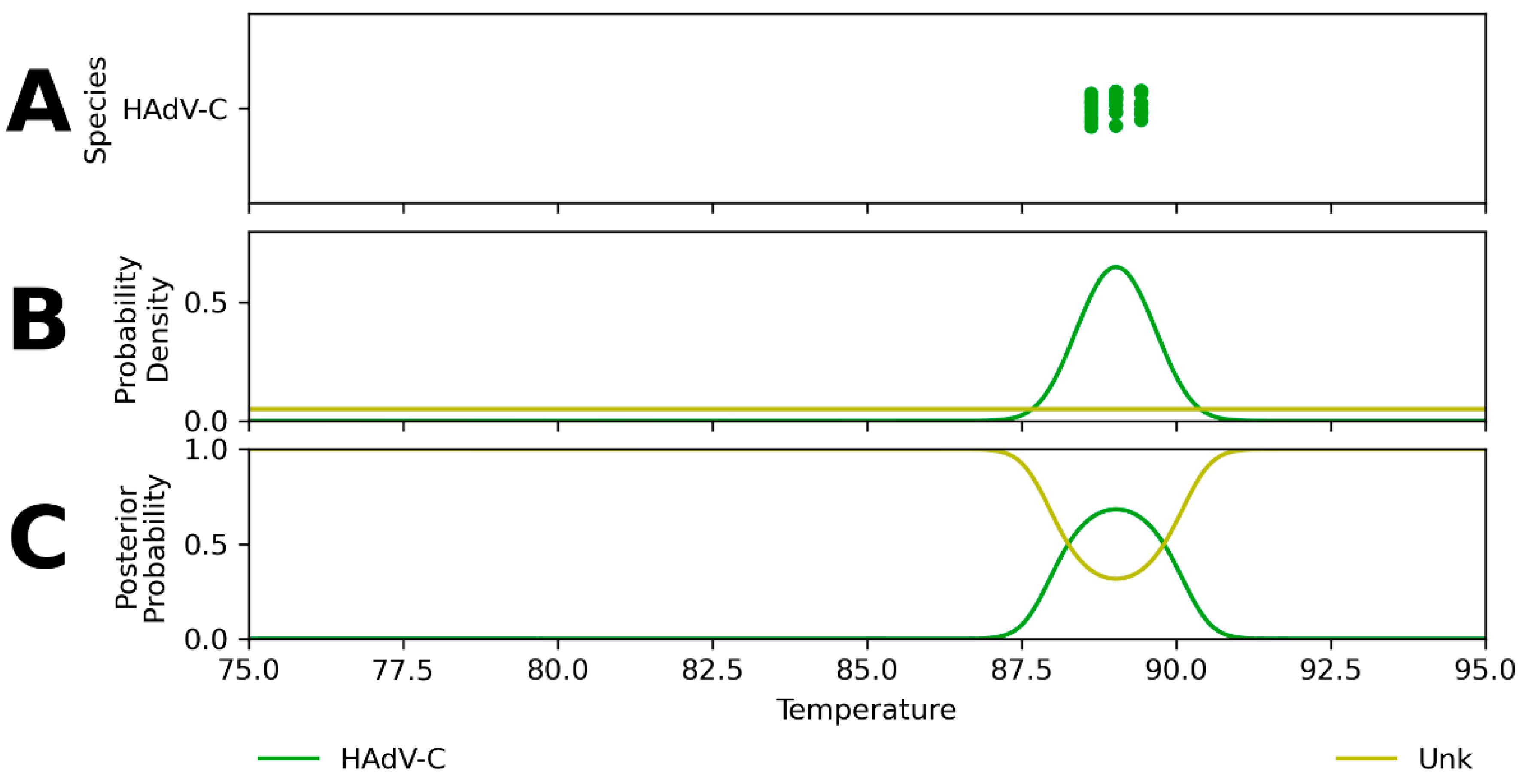
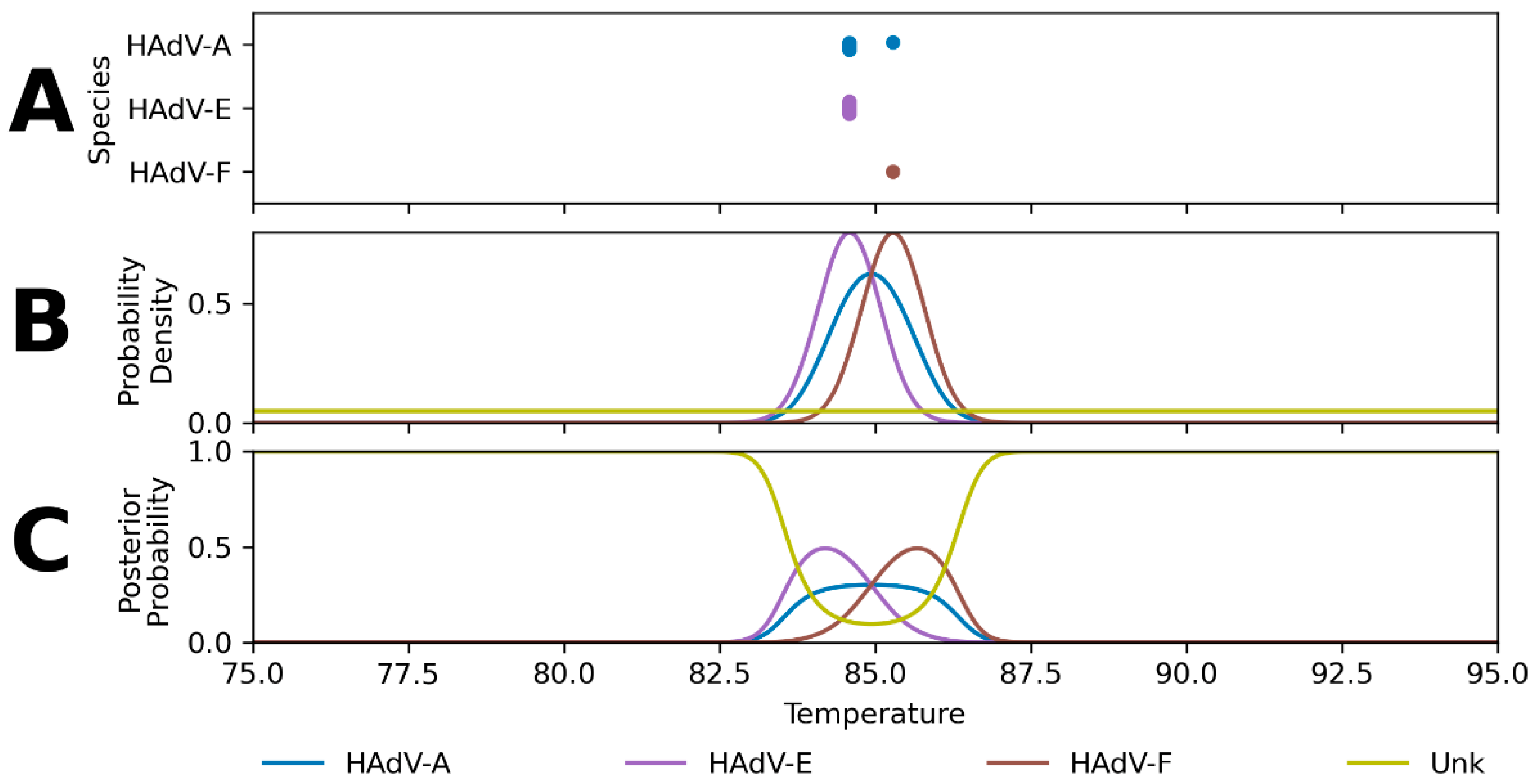
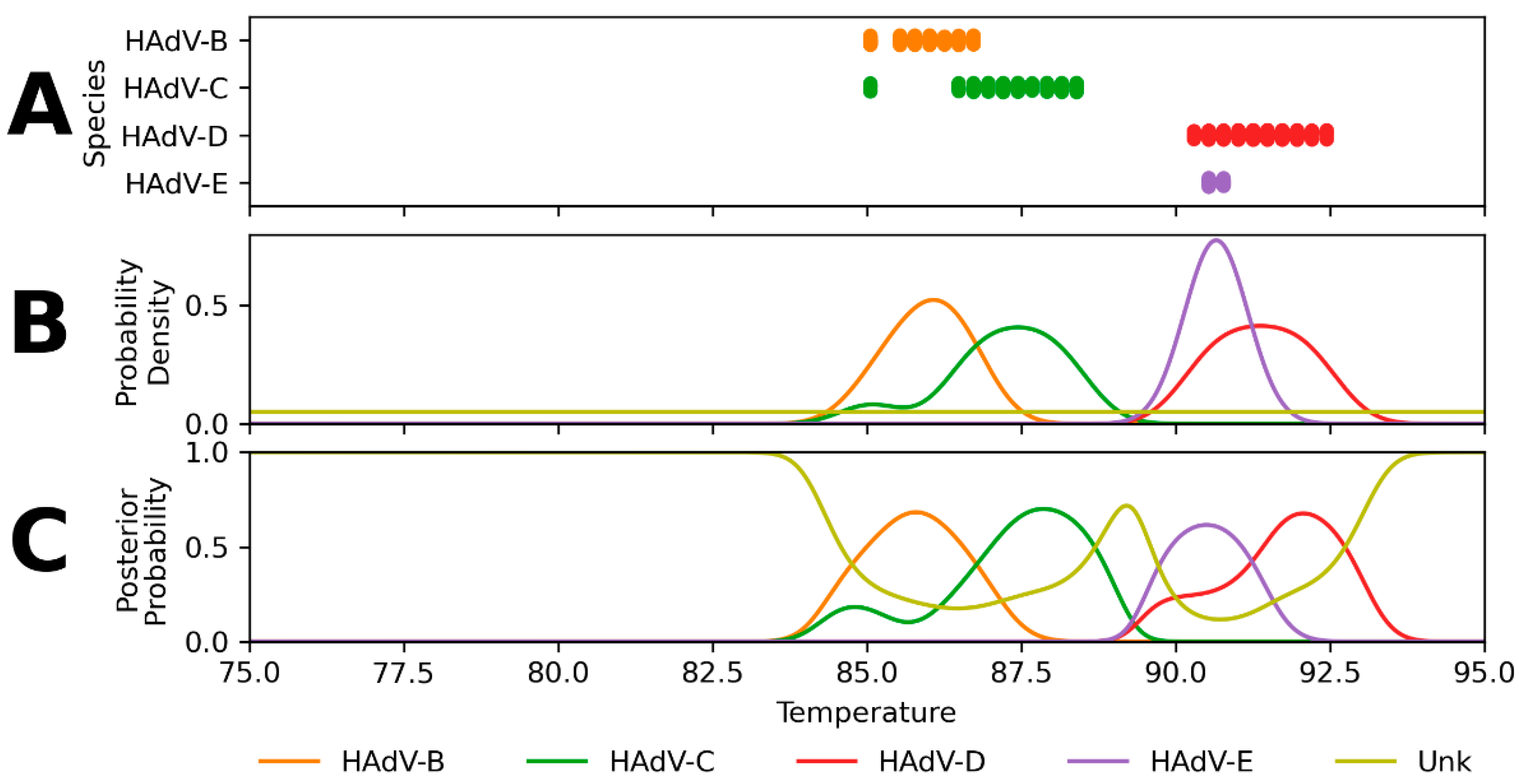
2. Results
2.1. Hierarchical Classification Model Performance
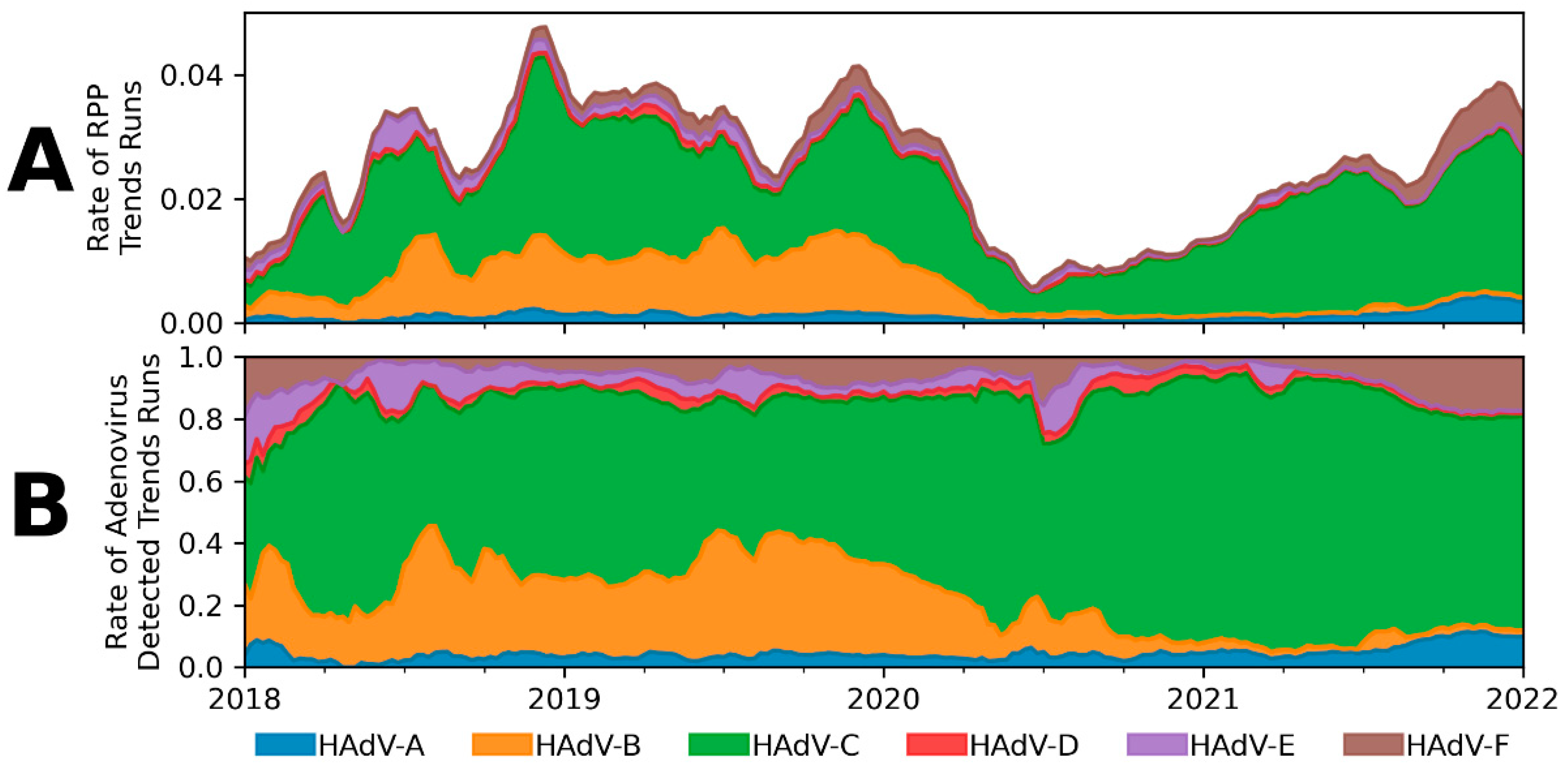
2.2. Application of Adenovirus Classification Model to Syndromic Trends
3. Discussion
3.1. Hierarchical Classification Model Performance
3.2. Generalization of the Genotyping Framework
3.3. Adenovirus Surveillance
4. Materials and Methods
4.1. Estimating Assay Reactivity
4.2. Predicting Distributions of Tm Values
4.3. Posterior Probabilities of a Genotype Group
4.4. Hierarchical Classification Model
4.5. Application to Syndromic Trends
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
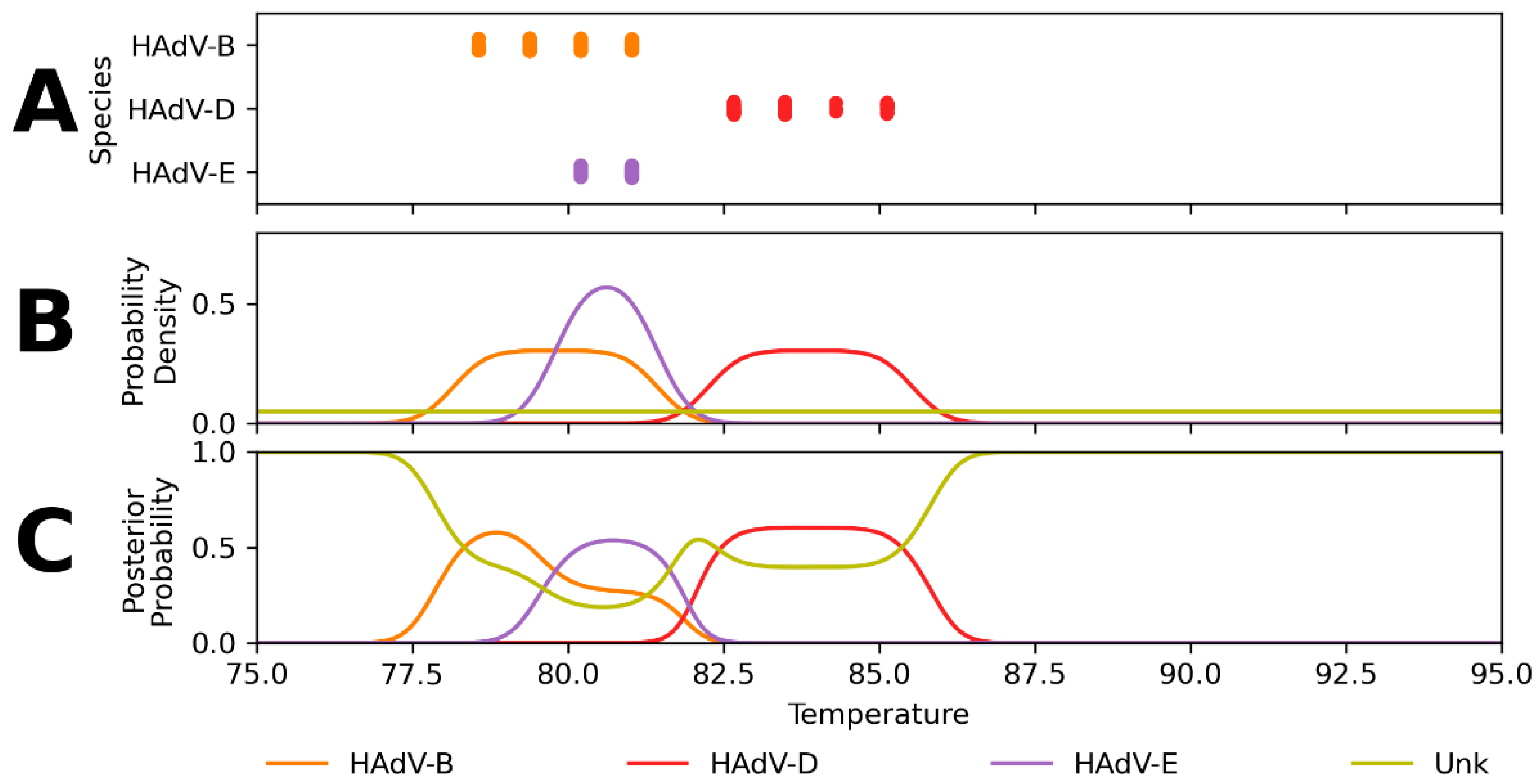
Appendix A. Expected Assay Reactivity and Estimated Tm Distributions
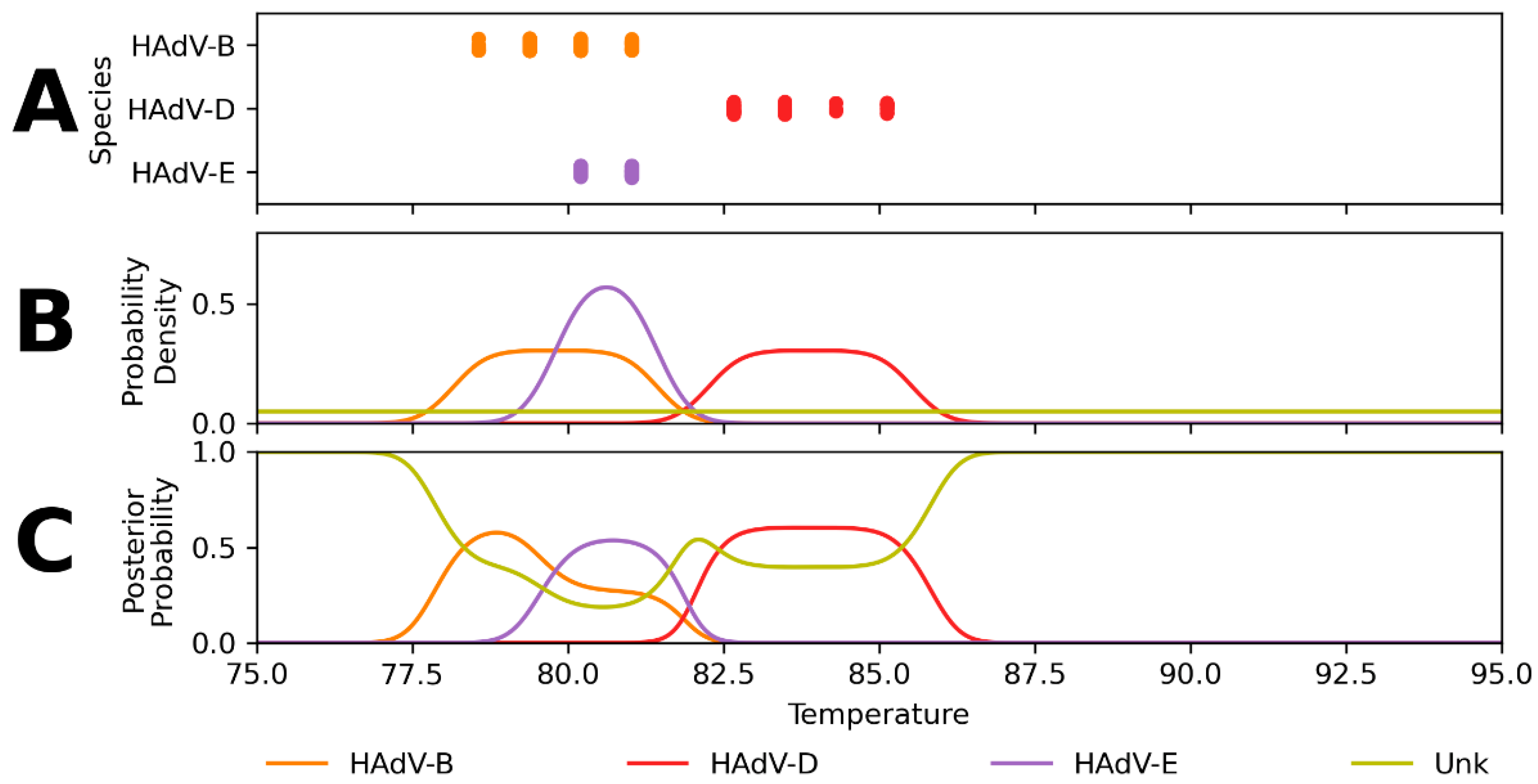

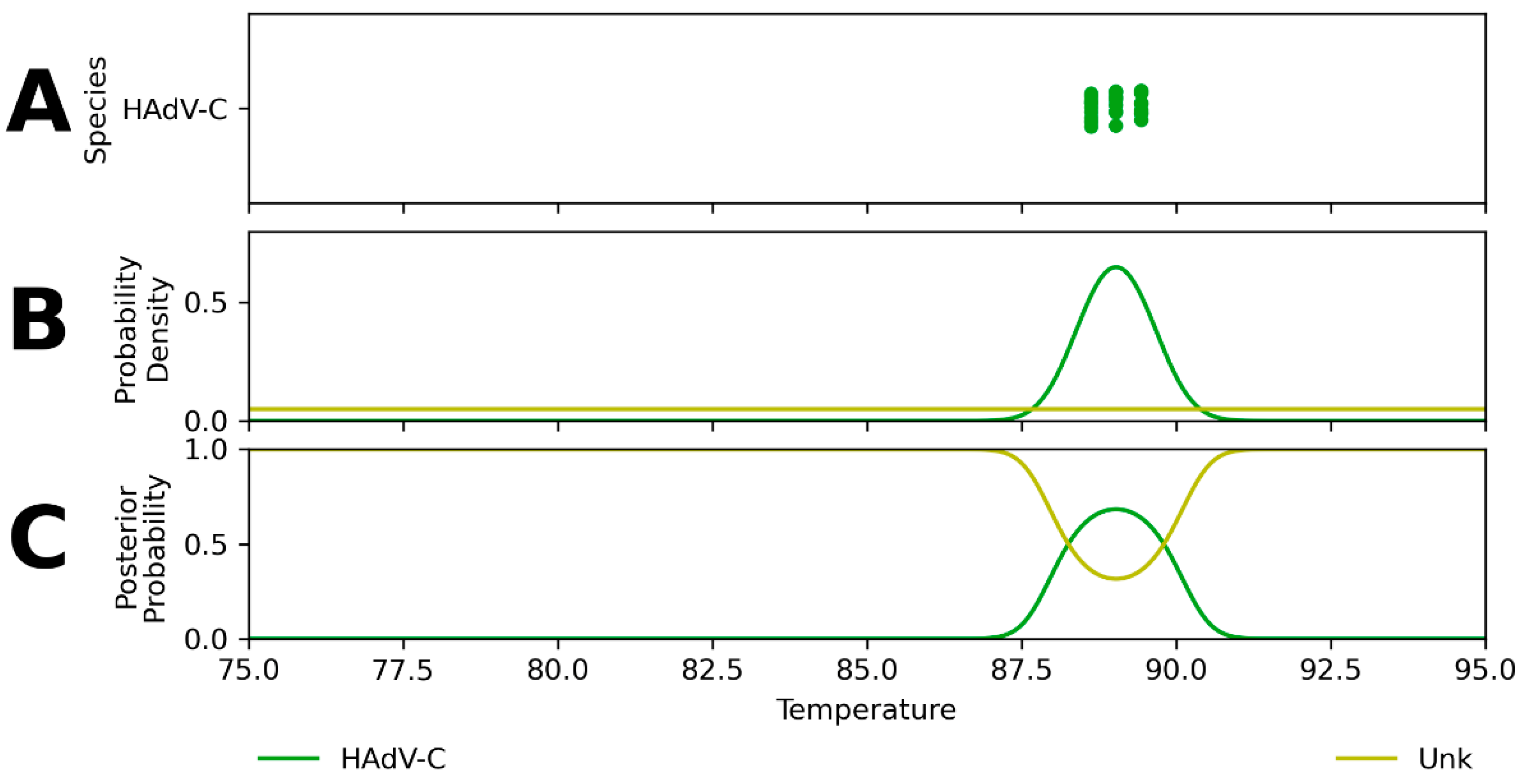

Appendix B. Performance of Assay Design-Informed Classification Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HAdV Species | Predicted Label | ||||||
|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | ||
| Known Label | A | 10 | 0 | 0 | 0 | 0 | 38 |
| B | 2 | 79 | 0 | 0 | 2 | 0 | |
| C | 0 | 0 | 57 | 0 | 0 | 0 | |
| D | 0 | 0 | 0 | 56 | 1 | 0 | |
| E | 0 | 0 | 0 | 17 | 27 | 1 | |
| F | 23 | 1 | 0 | 0 | 0 | 31 | |
| HAdV Species | Precision | Recall | F1-Score | Support |
|---|---|---|---|---|
| A | 0.26 | 0.21 | 0.23 | 48 |
| B | 0.99 | 0.95 | 0.97 | 83 |
| C | 1.00 | 1.00 | 1.00 | 57 |
| D | 0.77 | 0.93 | 0.84 | 60 |
| E | 0.90 | 0.61 | 0.73 | 44 |
| F | 0.45 | 0.56 | 0.50 | 55 |
| Macro average | 0.73 | 0.71 | 0.71 | |
| Weighted average | 0.75 | 0.75 | 0.75 | |
| Accuracy (±s.d.) | 0.75 (±0.43) | |||
References
- Alizon, S.; Luciani, F.; Regoes, R.R. Epidemiological and clinical consequences of within-host evolution. Trends Microbiol. 2011, 19, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Holm-Hansen, C.C.; Midgley, S.E.; Fischer, T.K. Global emergence of enterovirus D68: A systematic review. Lancet Infect. Dis. 2016, 16, e64–e75. [Google Scholar] [CrossRef]
- Oermann, C.M.; Schuster, J.E.; Conners, G.P.; Newland, J.G.; Selvarangan, R.; Jackson, M.A. Enterovirus D68: A focused review and clinical highlights from the 2014 U.S. outbreak. Ann. Am. Thorac. Soc. 2015, 12, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, L.; Dien Bard, J.; Galvin, B.; Nawrocki, J.; Niesters, H.G.M.; Stellrecht, K.A.; St George, K.; Daly, J.A.; Blaschke, A.J.; Robinson, C.; et al. Enterovirus D68 outbreak detection through a syndromic disease epidemiology network. J. Clin. Virol. 2020, 124, 104262. [Google Scholar] [CrossRef]
- Rossen, J.W.A.; Friedrich, A.W.; Moran-Gilad, J. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect. 2018, 24, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Poritz, M.A.; Blaschke, A.J.; Byington, C.L.; Meyers, L.; Nilsson, K.; Jones, D.E.; Thatcher, S.A.; Robbins, T.; Lingenfelter, B.; Amiott, E.; et al. FilmArray, an automated nested multiplex PCR system for multi-pathogen detection: Development and application to respiratory tract infection. PLoS ONE 2011, 6, e26047. [Google Scholar] [CrossRef]
- Ririe, K.M.; Rasmussen, R.P.; Wittwer, C.T. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal. Biochem. 1997, 245, 154–160. [Google Scholar] [CrossRef]
- Wittwer, C.T.; Herrmann, M.G.; Gundry, C.N.; Elenitoba-Johnson, K.S.J. Real-time multiplex PCR assays. Methods 2001, 25, 430–442. [Google Scholar] [CrossRef]
- Howley, P.M.; Israel, M.A.; Law, M.F.; Martin, M.A. A rapid method for detecting and mapping homology between heterologous DNAs. Evaluation of polyomavirus genomes. J. Biol. Chem. 1979, 254, 4876–4883. [Google Scholar] [CrossRef]
- SantaLucia, J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhnoo, I.; Wadell, G.; Svensson, L.; Johansson, M.E. Importance of enteric adenoviruses 40 and 41 in acute gastroenteritis in infants and young children. J. Clin. Microbiol. 1984, 20, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Wen, L.; Wu, Z.; Liu, N.; Yang, C.; Liu, W.; Ba, Z.; Wang, J.; Yi, S.; Li, H.; et al. Human adenovirus type 7 infection associated with severe and fatal acute lower respiratory illness and nosocomial transmission. J. Clin. Microbiol. 2015, 53, 746–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Wan, C.; Ke, C.; Seto, J.; Dehghan, S.; Zou, L.; Zhou, J.; Cheng, Z.; Jing, S.; Zeng, Z.; et al. Re-emergent human adenovirus genome type 7d caused an acute respiratory disease outbreak in Southern China after a twenty-one year absence. Sci. Rep. 2014, 4, 7365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, C.C.; O’Brien, S.J.; Tompkins, D.S.; Bolton, F.J.; Berry, L.; Dodds, J.; Choudhury, D.; Halstead, F.; Iturriza-Gómara, M.; Mather, K.; et al. Changes in causes of acute gastroenteritis in the United Kingdom over 15 years: Microbiologic findings from 2 prospective, population-based studies of infectious intestinal disease. Clin. Infect. Dis. 2012, 54, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.M.; Lee, J.S.; Lee, J.Y.; Singh, G.; Dyer, D.W.; Seto, D.; Chodosh, J.; Rajaiya, J. Adenoviromics: Mining the human adenovirus species D genome. Front. Microbiol. 2018, 9, 2178. [Google Scholar] [CrossRef]
- Lynch, J.P.; Kajon, A.E. Adenovirus: Epidemiology, global spread of novel serotypes, and advances in treatment and prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [Green Version]
- Kandel, R.; Srinivasan, A.; D’Agata, E.M.C.; Lu, X.; Erdman, D.; Jhung, M. Outbreak of adenovirus type 4 infection in a long-term care facility for the elderly. Infect. Control. Hosp. Epidemiol. 2010, 31, 755–757. [Google Scholar] [CrossRef]
- Gray, G.C.; Callahan, J.D.; Hawksworth, A.W.; Fisher, C.A.; Gaydos, J.C. Respiratory diseases among U.S. Military personnel: Countering emerging threats. Emerg. Infect. Dis. 1999, 5, 379–387. [Google Scholar] [CrossRef]
- Binder, A.M.; Biggs, H.M.; Haynes, A.K.; Chommanard, C.; Lu, X.; Erdman, D.D.; Watson, J.T.; Gerber, S.I. Morbidity and mortality weekly report human adenovirus surveillance—United States, 2003–2016. Morb. Mortal. Wkly. Rep. 2017, 66, 1039–1042. [Google Scholar] [CrossRef] [Green Version]
- Ghebremedhin, B. Human adenovirus: Viral pathogen with increasing importance. Eur. J. Microbiol. Immunol. 2014, 4, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BioFire Diagnostics LLC. BioFire® Respiratory Panel 2.1 (RP2.1) De Novo Instructions for Use. 2021, p. 1. Available online: https://docs.biofiredx.com/wp-content/uploads/BFR0001-3590-01-RP2.1-De-Novo-CLSI-Procedure.pdf (accessed on 9 February 2022).
- Meyers, L.; Ginocchio, C.C.; Faucett, A.N.; Nolte, F.S.; Gesteland, P.H.; Leber, A.; Janowiak, D.; Donovan, V.; Dien Bard, J.; Spitzer, S.; et al. Automated real-time collection of pathogen-specific diagnostic data: Syndromic infectious disease epidemiology. JMIR Public Health Surveill. 2018, 4, e59. [Google Scholar] [CrossRef] [PubMed]
- Hossin, M.; Sulaiman, M.N. A review on evaluation metrics for data classification evaluations. Int. J. Data Min. Knowl. Manag. Process 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Xu, W.; McDonough, M.C.; Erdman, D.D. Species-specific identification of human adenoviruses by a multiplex PCR assay. J. Clin. Microbiol. 2000, 38, 4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Fujisawa, T.; Suga, S.; Taniguchi, K.; Nagao, M.; Ito, M.; Ochiai, H.; Konagaya, M.; Hanaoka, N.; Fujimoto, T. Species differences in circulation and inflammatory responses in children with common respiratory adenovirus infections. J. Med. Virol. 2018, 90, 873–880. [Google Scholar] [CrossRef]
- Probst, V.; Datyner, E.K.; Haddadin, Z.; Rankin, D.A.; Hamdan, L.; Rahman, H.K.; Spieker, A.; Stewart, L.S.; Guevara, C.; Yepsen, E.; et al. Human adenovirus species in children with acute respiratory illnesses. J. Clin. Virol. 2021, 134, 104716. [Google Scholar] [CrossRef]
- Dou, Y.; Li, Y.; Ma, C.; Zhu, H.; Du, J.; Liu, H.; Liu, Q.; Chen, R.; Tan, Y. Rapid diagnosis of human adenovirus B, C and E in the respiratory tract using multiplex quantitative polymerase chain reaction. Mol. Med. Rep. 2018, 18, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- Kendall Scott, M.; Chommanard, C.; Lu, X.; Appelgate, D.; Grenz, L.; Schneider, E.; Gerber, S.I.; Erdman, D.D.; Thomas, A. Human adenovirus associated with severe respiratory infection, Oregon, USA, 2013–2014. Emerg. Infect. Dis. 2016, 22, 1044–1051. [Google Scholar] [CrossRef] [Green Version]
- National Adenovirus Type Reporting System Publications | CDC. Available online: https://www.cdc.gov/adenovirus/reporting-surveillance/natrs/index.html (accessed on 9 February 2022).
- Park, S.W.; Pons-Salort, M.; Messacar, K.; Cook, C.; Meyers, L.; Farrar, J.; Grenfell, B.T. Epidemiological dynamics of enterovirus D68 in the United States and implications for acute flaccid myelitis. Sci. Transl. Med. 2021, 13, abd2400. [Google Scholar] [CrossRef]
- Nawrocki, J.; Olin, K.; Holdrege, M.C.; Hartsell, J.; Meyers, L.; Cox, C.; Powell, M.; Cook, C.V.; Jones, J.; Robbins, T.; et al. The effects of social distancing policies on non-SARS-CoV-2 respiratory pathogens. Open Forum Infect. Dis. 2021, 8, ofab133. [Google Scholar] [CrossRef]
- NCBI. BLASTN (RRID:SCR_001598). Published 2022. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&BLAST_PROGRAMS=megaBlast&PAGE_TYPE=BlastSearch (accessed on 9 February 2022).
- Robbins, T.; Wilson, T.; Cox, C.; Jones, J.; Galvin, B.; Kiser, A. System and Method for Identifying Analytes in Assay Using Normalized Tm Values. World Intellectual Property Organization—International Bureau WO 2021113554. 10 June 2021. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021113554 (accessed on 9 February 2022).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]

| HAdV Species | Predicted Label | ||||||
|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | ||
| Known Label | A | 46 | 0 | 0 | 0 | 0 | 2 |
| B | 0 | 81 | 0 | 0 | 2 | 0 | |
| C | 0 | 0 | 57 | 0 | 0 | 0 | |
| D | 0 | 0 | 0 | 53 | 4 | 3 | |
| E | 0 | 0 | 0 | 0 | 43 | 1 | |
| F | 6 | 1 | 0 | 0 | 0 | 48 | |
| HAdV Species | Precision | Recall | F1-Score | Support |
|---|---|---|---|---|
| A | 0.88 | 0.96 | 0.92 | 48 |
| B | 0.99 | 0.98 | 0.98 | 83 |
| C | 1.00 | 1.00 | 1.00 | 57 |
| D | 1.00 | 0.88 | 0.94 | 60 |
| E | 0.88 | 0.98 | 0.92 | 44 |
| F | 0.89 | 0.87 | 0.88 | 55 |
| Macro average | 0.94 | 0.94 | 0.94 | |
| Weighted average | 0.95 | 0.95 | 0.95 | |
| Accuracy (±s.d.) | 0.95 (±0.04) | |||
| RPP HAdV Assay | HAdV Reactive Species |
|---|---|
| RPP HAdV-1 | B, D, E |
| RPP HAdV-2 | A, F |
| RPP HAdV-3 | C |
| RPP HAdV-4 | A, E, F |
| RPP HAdV-5 | B, C, D, E |
| HAdV Species | Limit of Detection Study | Inclusivity Study | Cross-Reactivity Study | Species Run Count | |||
|---|---|---|---|---|---|---|---|
| Isolate Serotype | Run Count | Isolate Serotype | Run Count | Isolate Serotype | Run Count | ||
| A | A18 | 39 | A12 | 3 | A31 | 3 | 48 |
| A31 | 3 | ||||||
| B | B7a | 40 | B3 | 3 | B3 | 3 | 83 |
| B7d/d2 | 3 | ||||||
| B7h | 3 | 7a | 3 | ||||
| B11 | 4 | ||||||
| B14 | 3 | B14 | 3 | ||||
| B16 | 3 | ||||||
| B21 | 3 | B21 | 3 | ||||
| B34 | 3 | ||||||
| B35 | 3 | ||||||
| B50 | 3 | ||||||
| C | C2 | 39 | C1 | 3 | C2 | 3 | 57 |
| C5 | 3 | C5 | 3 | ||||
| C6 | 3 | C6 | 3 | ||||
| D | D37 | 40 | D8 | 4 | D8 | 3 | 60 |
| D20 | 3 | ||||||
| D20 | 3 | D26 | 3 | ||||
| D37 | 4 | ||||||
| E | E4a | 38 | E4 | 3 | E4 | 3 | 44 |
| F | F41 | 40 | F40 | 6 | F40 | 3 | 55 |
| F41 | 3 | F41 | 3 | ||||
| Total Run Count | 347 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galvin, B.; Jones, J.; Powell, M.; Olin, K.; Jones, M.; Robbins, T. A Hierarchical Genotyping Framework Using DNA Melting Temperatures Applied to Adenovirus Species Typing. Int. J. Mol. Sci. 2022, 23, 5441. https://doi.org/10.3390/ijms23105441
Galvin B, Jones J, Powell M, Olin K, Jones M, Robbins T. A Hierarchical Genotyping Framework Using DNA Melting Temperatures Applied to Adenovirus Species Typing. International Journal of Molecular Sciences. 2022; 23(10):5441. https://doi.org/10.3390/ijms23105441
Chicago/Turabian StyleGalvin, Ben, Jay Jones, Michaela Powell, Katherine Olin, Matthew Jones, and Thomas Robbins. 2022. "A Hierarchical Genotyping Framework Using DNA Melting Temperatures Applied to Adenovirus Species Typing" International Journal of Molecular Sciences 23, no. 10: 5441. https://doi.org/10.3390/ijms23105441
APA StyleGalvin, B., Jones, J., Powell, M., Olin, K., Jones, M., & Robbins, T. (2022). A Hierarchical Genotyping Framework Using DNA Melting Temperatures Applied to Adenovirus Species Typing. International Journal of Molecular Sciences, 23(10), 5441. https://doi.org/10.3390/ijms23105441