
Thymosin β4 Is an Endogenous Iron Chelator and Molecular Switcher of Ferroptosis

 ,
,  , , , and
, , , and
Abstract
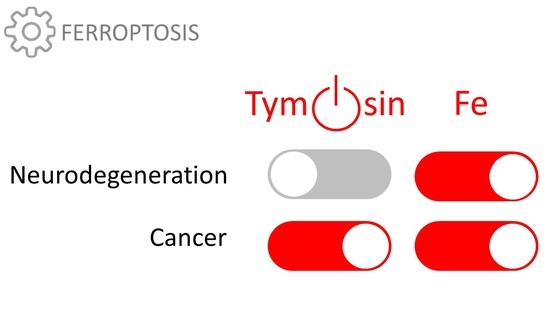
1. Introduction
2. Results
2.1. Thymosin β4 Metal Coordination
2.2. Thymosin β4 and Ferroptosis
3. Discussion
4. Conclusions
5. Experimental Procedures
5.1. Reagents
5.2. EPR Experimental Details
5.3. NMR
5.4. DFT
System Setup and Conformational Analysis
5.5. Molecular Dynamics Simulations
5.6. Cell Cultures
5.7. Cell Viability Measurements
5.8. Gene Expression
5.9. TEM Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in neurodegeneration–cause or consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, T.; Rama, R. Iron, oxidative stress and early neurological deterioration in ischemic stroke. Curr. Med. Chem. 2007, 14, 857–874. [Google Scholar] [CrossRef]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2015, 41, 274–286. [Google Scholar] [CrossRef]
- Cassimeris, L.; Safer, D.; Nachmias, V.T.; Zigmond, S.H. Thymosin beta 4 sequesters the majority of G-actin in resting human polymorphonuclear leukocytes. J. Cell Biol. 1992, 119, 1261–1270. [Google Scholar] [CrossRef]
- Philp, D.; Kleinman, H.K. Animal studies with thymosin β4, a multifunctional tissue repair and regeneration peptide. Ann. N. Y. Acad. Sci. 2010, 1194, 81–86. [Google Scholar] [CrossRef]
- Smart, N.; Rossdeutsch, A.; Riley, P.R. Thymosin β4 and angiogenesis: Modes of action and therapeutic potential. Angiogenesis 2007, 10, 229–241. [Google Scholar] [CrossRef]
- Faa, G.; Piras, M.; Mancuso, L.; Coni, P.; Pichiri, G.; Orrù, G.; Fanni, D.; Gerosa, C.; Cao, G.; Taibi, R.; et al. Thymosin beta-4 prenatal administration improves fetal development and halts side effects due to preterm delivery. Curr. Med. Chem. 2021, 25, 431–437. [Google Scholar]
- Goldstein, A.L. Thymosin β4: A New Molecular Target for Antitumor Strategies; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Pardon, M.-C. Anti-inflammatory potential of thymosin β4 in the central nervous system: Implications for progressive neu-rodegenerative diseases. Expert Opin. Biol. Ther. 2018, 18 (Suppl. 1), 165–169. [Google Scholar] [CrossRef]
- Erickson-Viitanen, S.; Ruggieri, S.; Natalini, P.; Horecker, B. Distribution of thymosin β4 in vertebrate classes. Arch. Biochem. Biophys. 1983, 221, 570–576. [Google Scholar] [CrossRef]
- Huff, T.; Rosorius, O.; Otto, A.M.; Müller, C.S.G.; Ballweber, E.; Hannappel, E.; Mannherz, H.G. Nuclear localisation of the G-actin sequestering peptide thymosin β4. J. Cell Sci. 2004, 117, 5333–5341. [Google Scholar] [CrossRef] [PubMed]
- Piludu, M.; Piras, M.; Pichiri, G.; Coni, P.; Orrù, G.; Cabras, T.; Messana, I.; Faa, G.; Castagnola, M. Thymosin Beta 4 May Translocate from the Cytoplasm in to the Nucleus in HepG2 Cells following Serum Starvation. An Ultrastructural Study. PLoS ONE 2015, 10, e0119642. [Google Scholar] [CrossRef]
- Zhang, Y.; Feurino, L.W.; Zhai, Q.; Wang, H.; Fisher, W.E.; Chen, C.; Yao, Q.; Li, M. Thymosin beta 4 is overexpressed in human pancreatic cancer cells and stimulates proinflammatory cytokine secretion and JNK activation. Cancer Biol. Ther. 2008, 7, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Park, M.J.; Lee, H.K.; Son, H.J.; Kim, C.N.; Kim, J.H.; Kang, D.W. Increased expression of thymosin β4 is in-dependently correlated with hypoxia inducible factor-1α (HIF-1α) and worse clinical outcome in human colorectal cancer. J. Pathol. Transl. Med. 2017, 51, 9–16. [Google Scholar] [CrossRef]
- Wirsching, H.-G.; Krishnan, S.; Florea, A.-M.; Frei, K.; Krayenbühl, N.; Hasenbach, K.; Reifenberger, G.; Weller, M.; Tabatabai, G. Thymosin beta 4 gene silencing decreases stemness and invasiveness in glioblastoma. Brain 2014, 137, 433–448. [Google Scholar] [CrossRef]
- Huang, D.; Wang, S.; Wang, A.; Chen, X.; Zhang, H. Thymosin beta 4 silencing suppresses proliferation and invasion of non-small cell lung cancer cells by repressing Notch1 activation. Acta Biochim. Biophys. Sin. 2016, 48, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Lachowicz, J.I.; Jaremko, M.; Jaremko, L.; Pichiri, G.; Coni, P.; Piludu, M. Metal coordination of thymosin β4: Chemistry and possible implications. Coord. Chem. Rev. 2019, 396, 117–123. [Google Scholar] [CrossRef]
- Al-Harthi, S.; Lachowicz, J.I.; Nowakowski, M.E.; Jaremko, M.; Jaremko, Ł. Towards the functional high-resolution coor-dination chemistry of blood plasma human serum albumin. J. Inorg. Biochem. 2019, 198, 110716. [Google Scholar] [CrossRef]
- Zarbock, J.; Oschkinat, H.; Hannappel, E.; Kalbacher, H.; Voelter, W.; Holak, T.A. Solution Conformation of Thymosin beta4: A Nuclear Magnetic Resonance and Simulated Annealing Study. Biochemistry 1990, 29, 7814–\7821. [Google Scholar] [CrossRef]
- Emwas, A.-H.; Szczepski, K.; Poulson, B.G.; Chandra, K.; McKay, R.T.; Dhahri, M.; AlAhmari, F.; Jaremko, L.; Lachowicz, J.I.; Jaremko, M. NMR as a “Gold Standard” Method in Drug Design and Discovery. Molecules 2020, 25, 4597. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Low, T.; Goldstein, A.L. Chemical characterization of thymosin beta 4. J. Biol. Chem. 1982, 257, 1000–1006. [Google Scholar] [CrossRef]
- Mujika, J.I.; Torre, G.D.; Formoso, E.; Grande-Aztatzi, R.; Grabowski, S.J.; Exley, C.; Lopez, X. Aluminum's preferential binding site in proteins: Sidechain of amino acids versus backbone interactions. J. Inorg. Biochem. 2018, 181, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. Metal Binding Affinity and Selectivity in Metalloproteins: Insights from Computational Studies. Annu. Rev. Biophys. 2008, 37, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. Clinical Relevance of Anemia and Transfusion Iron Overload in Myelodysplastic Syndromes. Hematology 2008, 2008, 166–175. [Google Scholar] [CrossRef]
- Shenoy, N.; Vallumsetla, N.; Rachmilewitz, E.; Verma, A.; Ginzburg, Y. Impact of iron overload and potential benefit from iron chelation in low-risk myelodysplastic syndrome. Blood 2014, 124, 873–881. [Google Scholar] [CrossRef]
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.P.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophago-cytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood Am. J. Hematol. 2018, 131, 2581–2593. [Google Scholar]
- Imoto, S.; Kono, M.; Suzuki, T.; Shibuya, Y.; Sawamura, T.; Mizokoshi, Y.; Sawada, H.; Ohbuchi, A.; Saigo, K. Haemin-induced cell death in human monocytic cells is consistent with ferroptosis. Transfus. Apher. Sci. 2018, 57, 524–531. [Google Scholar] [CrossRef]
- Soares, M.P.; Hamza, I. Macrophages and Iron Metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Grohm, J.; Plesnila, N.; Culmsee, C. Bid mediates fission, membrane permeabilization and perinuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain Behav. Immun. 2010, 24, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2019, 99, 151058. [Google Scholar] [CrossRef]
- Gerdes, H.-H.; Carvalho, R.N. Intercellular transfer mediated by tunneling nanotubes. Curr. Opin. Cell Biol. 2008, 20, 470–475. [Google Scholar] [CrossRef]
- Davis, D.M.; Sowinski, S. Membrane nanotubes: Dynamic long-distance connections between animal cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 431–436. [Google Scholar] [CrossRef]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C.-M. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J. Cell Sci. 2005, 118, 3695–3703. [Google Scholar] [CrossRef]
- Luchetti, F.; Canonico, B.; Arcangeletti, M.; Guescini, M.; Cesarini, E.; Stocchi, V.; Degli Esposti, M.; Papa, S. Fas Signalling Promotes Intercellular Communication in T Cells. PLoS ONE 2012, 7, e35766. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2019, 28, 101328. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Hou, Q.; Hsu, Y.-T. Bax translocates from cytosol to mitochondria in cardiac cells during apoptosis: Development of a GFP-Bax-stable H9c2 cell line for apoptosis analysis. Am. J. Physiol. Circ. Physiol. 2005, 289, H477–H487. [Google Scholar] [CrossRef] [PubMed]
- Jungas, T.; Motta, I.; Duffieux, F.; Fanen, P.; Stoven, V.; Ojcius, D.M. Glutathione Levels and BAX Activation during Apoptosis Due to Oxidative Stress in Cells Expressing Wild-type and Mutant Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 2002, 277, 27912–27918. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, R.L.; Nakayama, M.; Kawaguchi, K.; Jin, K.; Stetler, R.A.; Simon, R.P.; Graham, S.H. Expression of the Apoptosis-Effector Gene, Bax, Is Up-Regulated in Vulnerable Hippocampal CA1 Neurons Following Global Ischemia. J. Neurochem. 2002, 67, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Mai, J.K.; Krajewska, M.; Sikorska, M.; Mossakowski, M.J.; Reed, J.C. Upregulation of bax protein levels in neurons following cerebral ischemia. J. Neurosci. 1995, 15, 6364–6376. [Google Scholar] [CrossRef]
- Wei, C.; Kumar, S.; Kim, I.-K.; Gupta, S. Thymosin beta 4 protects cardiomyocytes from oxidative stress by targeting ant-ioxidative enzymes and anti-apoptotic genes. PLoS ONE 2012, 7, e42586. [Google Scholar] [CrossRef]
- Kumar, S.; Gupta, S. Thymosin beta 4 prevents oxidative stress by targeting antioxidant and anti-apoptotic genes in cardiac fibroblasts. PLoS ONE 2011, 6, e26912. [Google Scholar] [CrossRef]
- Guo, S.; Wharton, W.; Moseley, P.; Shi, H. Heat shock protein 70 regulates cellular redox status by modulating glutathi-one-related enzyme activities. Cell Stress Chaperones 2007, 12, 245. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Sun, X.Y.; Cao, J.; Mivechi, N.F.; Giffard, R.G. Over-expression of HSP-70 protects astrocytes from combined oxygen-glucose deprivation. NeuroReport 1996, 7, 429–432. [Google Scholar] [CrossRef]
- Plumier, J.-C.L.; Krueger, A.M.; Currie, R.W.; Kontoyiannis, D.; Kollias, G.; Pagoulatos, G.N. Transgenic mice expressing the human inducible Hsp70 have hippocampal neurons resistant to ischemic injury. Cell Stress Chaperon 1997, 2, 162–167. [Google Scholar] [CrossRef]
- Aufricht, C.; Lu, E.; Thulin, G.; Kashgarian, M.; Siegel, N.J.; Van Why, S.K. ATP releases HSP-72 from protein aggregates after renal ischemia. Am. J. Physiol. Content 1998, 274, F268–F274. [Google Scholar] [CrossRef] [PubMed]
- Bidmon, B.; Endemann, M.; Müller, T.; Arbeiter, K.; Herkner, K.; Aufricht, C. Heat shock protein-70 repairs proximal tubule structure after renal ischemia. Kidney Int. 2000, 58, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Copani, A.; Testa, D.; Ravagna, A.; Spadaro, F.; Tendi, E.; Nicoletti, V.; Stella, A.G. Nitric oxide synthase induction in astroglial cell cultures: Effect on heat shock protein 70 synthesis and oxidant/antioxidant balance. J. Neurosci. Res. 2000, 60, 613–622. [Google Scholar] [CrossRef]
- McLaughlin, B.; Hartnett, K.A.; Erhardt, J.A.; Legos, J.J.; White, R.F.; Barone, F.C.; Aizenman, E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc. Natl. Acad. Sci. USA 2003, 100, 715–720. [Google Scholar] [CrossRef]
- Yan, L.-J.; Christians, E.S.; Liu, L.; Xiao, X.; Sohal, R.S.; Benjamin, I.J. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002, 21, 5164–5172. [Google Scholar] [CrossRef]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal. 2007, 9, 2099–2118. [Google Scholar] [CrossRef]
- Hsu, Y.-Y.; Chen, C.-S.; Wu, S.-N.; Jong, Y.-J.; Lo, Y.-C. Berberine activates Nrf2 nuclear translocation and protects against oxidative damage via a phosphatidylinositol 3-kinase/Akt-dependent mechanism in NSC34 motor neuron-like cells. Eur. J. Pharm. Sci. 2012, 46, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2015, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Gasdaska, P.Y.; Gasdaska, J.R.; Cochran, S.; Powis, G. Cloning and sequencing of a human thioredoxin reductase. FEBS Lett. 1995, 373, 5–9. [Google Scholar] [CrossRef]
- Cai, L.L.; Ruberto, R.A.; Ryan, M.J.; Eaton, J.K.; Schreiber, S.L.; Viswanathan, V.S. Modulation of ferroptosis sensitivity by TXNRD1 in pancreatic cancer cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Yang, X.; Wu, Q.; An, P.; Jin, X.; Liu, W.; Huang, X.; Li, Y.; Yan, S.; et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Target. Ther. 2020, 5, 138. [Google Scholar] [CrossRef] [PubMed]
- Low, T.L.; Mercer, R.C. Isolation and structural studies of porcine, ovine and murine thymosin β4 by high-performance liquid chromatography. J. Chromatogr. A 1984, 301, 221–239. [Google Scholar] [CrossRef]
- Snyder, W.; Cook, M.; Nasset, E.; Karhausen, L.; Howells, G.P.; Tipton, I. Report of the task group on reference man; Pergamon Oxford: Oxford, UK, 1975; Volume 23. [Google Scholar]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- Cheah, J.H.; Kim, S.F.; Hester, L.D.; Clancy, K.W.; Patterson, S.E., III; Papadopoulos, V.; Snyder, S.H. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 2006, 51, 431–440. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhi-bition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Forciniti, S.; Greco, L.; Grizzi, F.; Malesci, A.; Laghi, L. Iron metabolism in cancer progression. Int. J. Mol. Sci. 2020, 21, 2257. [Google Scholar] [CrossRef]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Brück, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Muhoberac, B.B.; Vidal, R. Abnormal iron homeostasis and neurodegeneration. Front. Aging Neurosci. 2013, 5, 32. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2015, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A Novel Form of Programmed Cell Death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Tiziani, S.; Park, G.; Kaul, M.; Paternostro, G. Cellular protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat. Commun. 2014, 5, 3672. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.; Pringle, A.; McManus, T.; Ellard, J.; Bradley, M.; Signorelli, F.; Iannotti, F.; E Sundstrom, L. L -Arginyl-3,4-Spermidine is neuroprotective in several in vitro models of neurodegeneration and in vivo ischaemia without suppressing synaptic transmission. Br. J. Pharmacol. 2002, 137, 1255–1268. [Google Scholar] [CrossRef]
- Sundstrom, L.; Pringle, A.; Morrison, B.; Bradley, M. Organotypic cultures as tools for functional screening in the CNS. Drug Discov. Today 2005, 10, 993–1000. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Y.; Huang, X.; Can, L.; Zhao, X.; Wang, Y.; Xue, J.; Cheng, M.; Zhu, L. Thymosin beta 4 alleviates non-alcoholic fatty liver by inhibiting ferroptosis via up-regulation of GPX4. Eur. J. Pharmacol. 2021, 908, 174351. [Google Scholar] [CrossRef]
- Alghrably, M.; Dudek, D.; Emwas, A.-H.; Jaremko, M.; Rowińska-Żyrek, M. Copper(II) and Amylin Analogues: A Complicated Relationship. Inorg. Chem. 2020, 59, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Sharfalddin, A.A.; Emwas, A.H.; Jaremko, M.; Hussien, M.A. Transition metal complexes of 6-mercaptopurine: Characterization, Theoretical calculation, DNA-Binding, molecular docking, and anticancer activity. Appl. Organomet. Chem. 2021, 35, e6041. [Google Scholar] [CrossRef]
- Glasoe, P.K.; Long, F.A. USE OF GLASS ELECTRODES TO MEASURE ACIDITIES IN DEUTERIUM OXIDE1,2. J. Phys. Chem. 1960, 64, 188–190. [Google Scholar] [CrossRef]
- Willker, W.; Wollborn, U.; Leibfritz, D. Exact Measurement of 3JCH Coupling Constants Using Proton-Detected Editing and Selection Sequences. J. Magn. Reson. Ser. B 1993, 101, 83–86. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- MacroModel; Schrödinger LLC: New York, NY, USA, 2020.
- Chang, G.; Guida, W.C.; Still, W.C. An internal-coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar] [CrossRef]
- Polak, E.; Ribiere, G. Note sur la convergence de méthodes de directions conjuguées. ESAIM Math. Model. Numer. Anal. 1969, 3, 35–43. [Google Scholar] [CrossRef]
- Ponder, J.W.; Richards, F.M. An efficient newton-like method for molecular mechanics energy minimization of large mole-cules. J. Comput. Chem. 1987, 8, 1016–1024. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Best, R.B.; Hummer, G. Optimized Molecular Dynamics Force Fields Applied to the Helix−Coil Transition of Polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Song, L.F.; Merz, K.M., Jr. Parameterization of highly charged metal ions using the 12-6-4 LJ-type nonbonded model in explicit water. J. Phys. Chem. B 2015, 119, 883–895. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Feenstra, K.A.; Hess, B.; Berendsen, H.J. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999, 20, 786–798. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Durzyńska, J.; Barton, E. IGF expression in HPV-related and HPV-unrelated human cancer cells. Oncol. Rep. 2014, 32, 893–900. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Region | I | II | III | IV | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe (II) | S1 | K3 | P4 | - | M6 | - | - | K11 | F12 | D13 | S15 | L17 | T22 | P27 | L28 | P29 | S30 | T33 | I34 | - | - | A40 | S43 |
| Fe (III) | S1 | K3 | P4 | - | M6 | I9 | - | - | F12 | D13 | S15 | - | T22 | - | L28 | - | - | T33 | I34 | Q35 | E36 | A40 | - |
| Al (III) | - | - | P4 | D5 | M6 | I9 | E10 | - | F12 | D13 | - | - | - | - | L28 | - | - | - | I34 | Q35 | E36 | - | - |
| TB4N-C | TB4N-mid | TB4N-N | ||||||
|---|---|---|---|---|---|---|---|---|
| Donor | Acceptor | Occ. (%) | Donor | Acceptor | Occ. (%) | Donor | Acceptor | Occ. (%) |
| GLU35-Nbb | THR33-Osc | 27.6 | LYS38-Nbb | GLU35-Obb | 26.0 | THR22-Nbb | LYS19-Obb | 23.8 |
| LEU17-Nbb | LYS14-Obb | 25.8 | LEU28-Nbb | ASN26-Osc | 28.2 | GLN23-Nbb | LYS18-Obb | 22.2 |
| LEU28-Nbb | ASN26-Osc | 36.6 | ILE34-Nbb | LYS31-Obb | 26.7 | GLU21-Nbb | LEU17-Obb | 23.4 |
| THR33-Nbb | GLN36-Obb | 32.0 | LYS31-Nbb | LEU28-Obb | 21.1 | ILE9-Nbb | MET6-Obb | 53.0 |
| LYS31-Nbb | LEU28-Obb | 22.5 | - | - | - | GLU8-Nbb | ASP5-Obb | 51.5 |
| - | - | - | - | - | - | LYS38-Nbb | GLU35-Obb | 20.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachowicz, J.I.; Pichiri, G.; Piludu, M.; Fais, S.; Orrù, G.; Congiu, T.; Piras, M.; Faa, G.; Fanni, D.; Dalla Torre, G.; et al. Thymosin β4 Is an Endogenous Iron Chelator and Molecular Switcher of Ferroptosis. Int. J. Mol. Sci. 2022, 23, 551. https://doi.org/10.3390/ijms23010551
Lachowicz JI, Pichiri G, Piludu M, Fais S, Orrù G, Congiu T, Piras M, Faa G, Fanni D, Dalla Torre G, et al. Thymosin β4 Is an Endogenous Iron Chelator and Molecular Switcher of Ferroptosis. International Journal of Molecular Sciences. 2022; 23(1):551. https://doi.org/10.3390/ijms23010551
Chicago/Turabian StyleLachowicz, Joanna I., Giusi Pichiri, Marco Piludu, Sara Fais, Germano Orrù, Terenzio Congiu, Monica Piras, Gavino Faa, Daniela Fanni, Gabriele Dalla Torre, and et al. 2022. "Thymosin β4 Is an Endogenous Iron Chelator and Molecular Switcher of Ferroptosis" International Journal of Molecular Sciences 23, no. 1: 551. https://doi.org/10.3390/ijms23010551
APA StyleLachowicz, J. I., Pichiri, G., Piludu, M., Fais, S., Orrù, G., Congiu, T., Piras, M., Faa, G., Fanni, D., Dalla Torre, G., Lopez, X., Chandra, K., Szczepski, K., Jaremko, L., Ghosh, M., Emwas, A.-H., Castagnola, M., Jaremko, M., Hannappel, E., & Coni, P. (2022). Thymosin β4 Is an Endogenous Iron Chelator and Molecular Switcher of Ferroptosis. International Journal of Molecular Sciences, 23(1), 551. https://doi.org/10.3390/ijms23010551