
Cholesterol in the Cell Membrane—An Emerging Player in Atherogenesis


{kind=link}
Abstract
:1. Introduction
2. Cholesterol Homeostasis
3. Cholesterol Transport
4. The Macrophage–Lipoprotein Interaction in Atherogenesis
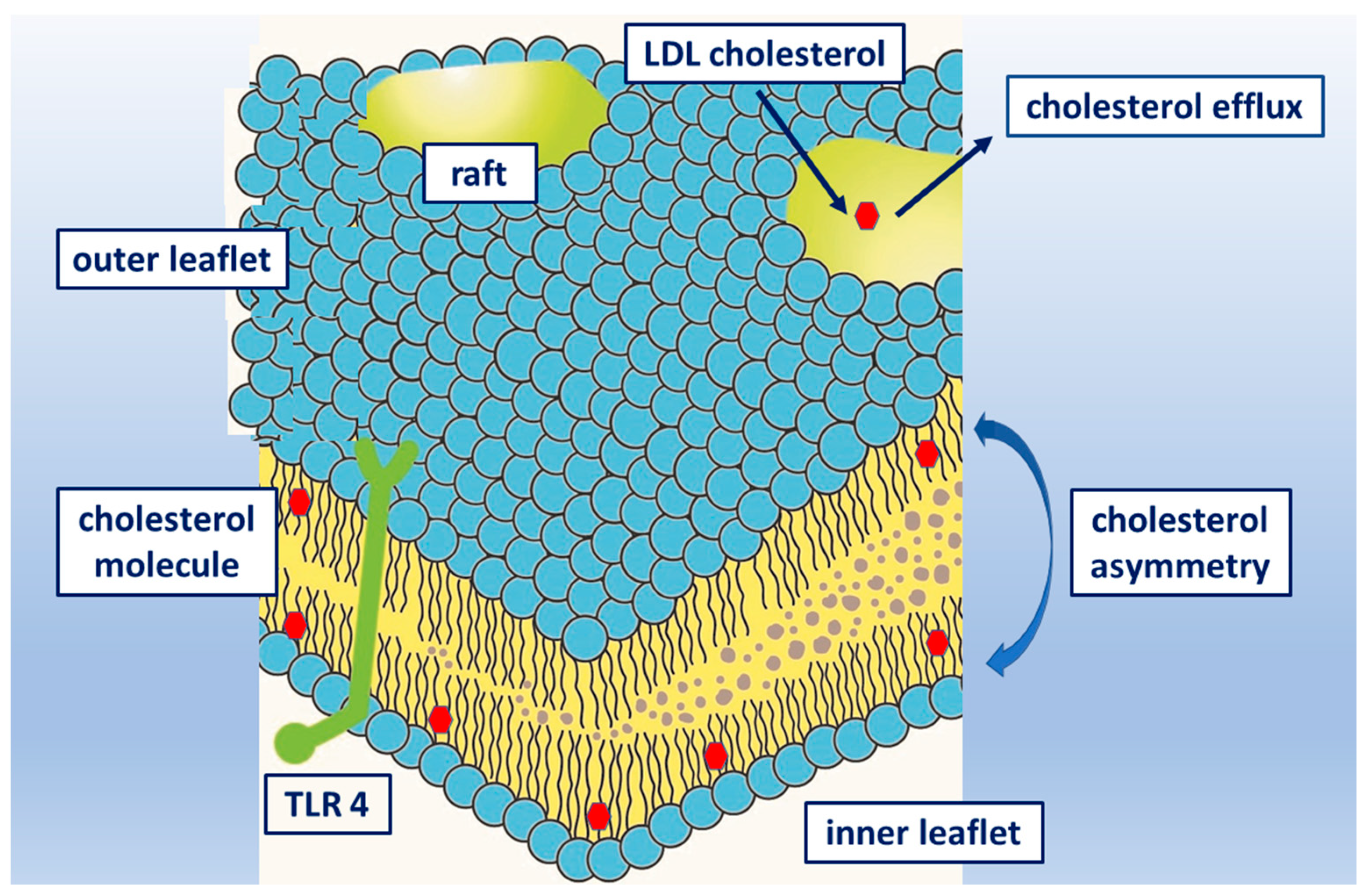
5. Cholesterol in Cell Membrane and Rafts
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| apoA-I | apolipoprotein A-1 |
| apoB | apolipoprotein B |
| FH | familial hypercholesterolemia |
| HDL | high-density lipoprotein |
| hsCRP | high-sensitivity C-reactive protein |
| IHD | ischemic heart disease |
| LDL | low-density lipoprotein |
| MHC | major histocompatibility complex |
| oxLDL | oxidized low-density lipoprotein |
| RCT | reverse cholesterol transport |
| SAFAs | saturated fatty acids |
| TLRs | toll-like receptors |
| VLDL | very-low-density lipoprotein |
References
- Anitschkow, N.N.; Chalatow, S. Über experimentelle Cholesterinsteatose und ihre Bedeutung für die Entstehung einiger pathologischer Prozesse. Zent. Fur. Allg. Pathol. Und Pathol. Anat. 1913, 24, 1–9. [Google Scholar]
- Keys, A.; Anderson, J.T.; Grande, F. Effect on Serum Cholesterol in Man of Mono-Ene Fatty Acid (Oleic Acid) in the Diet. Soc. Exp. Biol. Med. 2016, 98, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K. The Intermediary Metabolism of Cholesterol. Circulation 1950, 1, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gofman, J.W.; Glazier, F.; Tamplin, A.; Strisower, B.; Lalla, O.D. Lipoproteins, Coronary Heart Disease, and Atherosclerosis. Physiol. Rev. 1954, 34, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Herrick, J.B. An intimate account of my early experience with coronary thrombosis. Am. Heart J. 1944, 27, 1–18. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta-Biomembr. 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, D.; Perucha, E. Cholesterol metabolism: A new molecular switch to control inflammation. Clin. Sci. 2021, 135, 1389–1408. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Molecular medicine. The cholesterol quartet. Science 2001, 292, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.A.; Xu, L.; Chidambaram, A.A.; Soderberg, S.R.; Armstrong, E.J.; Wu, H.; Simon, S.I. CD11c/CD18 signals very late antigen-4 activation to initiate foamy monocyte recruitment during the onset of hypercholesterolemia. J. Immunol. 2015, 195, 5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sijbrands, E.J.G.; Westendorp, R.G.J.; Defesche, J.C.; Meier, P.H.E.M.d.; Smelt, A.H.M.; Kastelein, J.J.P. Mortality over two centuries in large pedigree with familial hypercholesterolaemia: Family tree mortality study. BMJ Br. Med. J. 2001, 322, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poledne, R.; Zicha, J. Human Genome Evolution and Development of Cardiovascular Risk Factors Through Natural Selection. Physiol. Res. 2018, 67, 155–163. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Demacker, P.N.; Kullberg, B.J.; Boerman, O.C.; Verschueren, I.; Stalenhoef, A.F.; Meer, J.W.V.d. Low-density lipoprotein receptor-deficient mice are protected against lethal endotoxemia and severe gram-negative infections. J. Clin. Investig. 1996, 97, 1366. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 437, 317–325. [Google Scholar] [CrossRef]
- Medbury, H.J.; Williams, H.; Li, S.; Fletcher, J.P. The Bidirectional Relationship between Cholesterol and Macrophage Polarization. J. Clin. Cell. Immunol. 2015, 6, 303. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-Reactive Protein and Low-Density Lipoprotein Cholesterol Levels in the Prediction of First Cardiovascular Events. N. Engl. J. Med. 2009, 347, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Cook, N. Clinical Usefulness of Very High and Very Low Levels of C-Reactive Protein Across the Full Range of Framingham Risk Scores. Circulation 2004, 109, 1955–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-Reactive Protein Levels and Outcomes after Statin Therapy. N. Engl. J. Med. 2009, 8, 8–9. [Google Scholar] [CrossRef]
- Aday, A.W.; Ridker, P.M. Targeting Residual Inflammatory Risk: A Shifting Paradigm for Atherosclerotic Disease. Front. Cardiovasc. Med. 2019, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ohtani, Y.; Irie, T.; Uekama, K.; Fukunaga, K.; Pitha, J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur. J. Biochem. 1989, 186, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Riottot, M.; Olivier, P.; Huet, A.; Caboche, J.-J.; Parquet, M.; Khallou, J.; Lutton, C. Hypolipidemic effects of β-cyclodextrin in the hamster and in the genetically hypercholesterolemic rico rat. Lipids 1993, 28, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Zhu, N.; Ao, B.X.; Liu, C.; Shi, Y.N.; Du, K.; Chen, J.X.; Zheng, X.L.; Liao, D.F. Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stöger, J.L.; Gijbels, M.J.J.; Velden, S.; Manca, M.; Loos, C.M.; Biessen, E.A.L.; Daemen, M.J.A.P.; Lutgens, E.; Winther, M.P.J.d. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012, 225, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkawa, R.; Low, H.; Mukhamedova, N.; Fu, Y.; Lai, S.-J.; Sasaoka, M.; Hara, A.; Yamazaki, A.; Kameda, T.; Horiuchi, Y.; et al. Cholesterol transport between red blood cells and lipoproteins contributes to cholesterol metabolism in blood. J. Lipid Res. 2020, 61, 1577–1588. [Google Scholar] [CrossRef]
- Turner, S.; Voogt, J.; Davidson, M.; Glass, A.; Killion, S.; Decaris, J.; Mohammed, H.; Minehira, K.; Boban, D.; Murphy, E.; et al. Measurement of Reverse Cholesterol Transport Pathways in Humans: In Vivo Rates of Free Cholesterol Efflux, Esterification, and Excretion. J. Am. Heart Assoc. 2012, 1, 1826. [Google Scholar] [CrossRef] [Green Version]
- Jakulj, L.; Dijk, T.H.; Boer, J.F.; Kootte, R.S.; Schonewille, M.; Paalvast, Y.; Boer, T.; Bloks, V.W.; Boverhof, R.; Nieuwdorp, M.; et al. Transintestinal Cholesterol Transport Is Active in Mice and Humans and Controls Ezetimibe-Induced Fecal Neutral Sterol Excretion. Cell Metab. 2016, 24, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Boer, J.F.; Schonewille, M.; Dikkers, A.; Koehorst, M.; Havinga, R.; Kuipers, F.; Tietge, U.J.F.; Groen, A.K. Transintestinal and Biliary Cholesterol Secretion Both Contribute to Macrophage Reverse Cholesterol Transport in Rats—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 643–646. [Google Scholar] [CrossRef] [Green Version]
- Hung, K.T.; Berisha, S.Z.; Ritchey, B.M.; Santore, J.; Smith, J.D. Red Blood Cells Play a Role in Reverse Cholesterol Transport. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1460. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.-J.; Ohkawa, R.; Horiuchi, Y.; Kubota, T.; Tozuka, M. Red blood cells participate in reverse cholesterol transport by mediating cholesterol efflux of high-density lipoprotein and apolipoprotein A-I from THP-1 macrophages. Biol. Chem. 2019, 400, 1593–1602. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of low-density lipoprotein receptors: Implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation 1987, 76, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Goldstein, J.L. Lipoprotein receptors in the liver. Control signals for plasma cholesterol traffic. J. Clin. Investig. 1983, 72, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Lipoprotein metabolism in the macrophage: Implications for Cholesterol Deposition in Atherosclerosis. Annu. Rev. Biochem. 2003, 52, 223–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36. [Google Scholar] [CrossRef]
- Cejkova, S.; Kubatova, H.; Thieme, F.; Janousek, L.; Fronek, J.; Poledne, R.; Kralova Lesna, I. The effect of cytokines produced by human adipose tissue on monocyte adhesion to the endothelium. Cell Adhes. Migr. 2019, 13, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, S.; Marcuzzi, A.; Piscianz, E.; Tommasini, A.; Fabris, B. The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 4058. [Google Scholar] [CrossRef] [Green Version]
- Poledne, R.; Králová Lesná, I.; Čejková, S. Adipose Tissue and Atherosclerosis. Physiol. Res. 2015, 64, 395–402. [Google Scholar] [CrossRef]
- Postea, O.; Vasina, E.M.; Cauwenberghs, S.; Projahn, D.; Liehn, E.A.; Lievens, D.; Theelen, W.; Kramp, B.K.; Butoi, E.D.; Soehnlein, O.; et al. Contribution of Platelet CX3CR1 to Platelet–Monocyte Complex Formation and Vascular Recruitment During Hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1186–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, A.B.; Cronstein, B.N. Regulation of Foam Cells by Adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojic, L.A.; Sawyez, C.G.; Telford, D.E.; Edwards, J.Y.; Hegele, R.A.; Huff, M.W. Activation of Peroxisome Proliferator-Activated Receptor δ Inhibits Human Macrophage Foam Cell Formation and the Inflammatory Response Induced by Very Low-Density Lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2919–2928. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Massberg, S. Atherosclerosis—Multiple Pathways to Lesional Macrophages. Sci. Transl. Med. 2014, 6, 8922. [Google Scholar] [CrossRef]
- Valledor, A.F.; Borràs, F.E.; Cullell-Young, M.; Celada, A. Transcription factors that regulate monocyte/macrophage differentiation. J. Leukoc. Biol. 1998, 63, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Sukhorukov, V.N.; Nikiforov, N.G.; Kubekina, M.V.; Sobenin, I.A.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; Kel, A.; et al. Signaling Pathways Potentially Responsible for Foam Cell Formation: Cholesterol Accumulation or Inflammatory Response-What is First? Int. J. Mol. Sci. 2020, 21, 2716. [Google Scholar] [CrossRef] [Green Version]
- Rudick, M.; Anderson, R.G.W. Multiple Functions of Caveolin-1. J. Biol. Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef] [Green Version]
- Endemannl, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. THE JOURNAL OF BIOLOGICAL CHEMISTRV CD36 Is a Receptor for Oxidized Low Density Lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [CrossRef]
- Tsai, T.H.; Chen, S.F.; Huang, T.Y.; Tzeng, C.F.; Chiang, A.S.; Kou, Y.R.; Lee, T.S.; Shyue, S.K. Impaired Cd14 and Cd36 expression, bacterial clearance, and Toll-like receptor 4-Myd88 signaling in caveolin-1-deleted macrophages and mice. Shock 2011, 35, 92–99. [Google Scholar] [CrossRef]
- Heit, B.; Kim, H.; Cosío, G.; Castaño, D.; Collins, R.; Lowell, C.A.; Kain, K.C.; Trimble, W.S.; Grinstein, S. Multimolecular signaling complexes enable Syk-mediated signaling of CD36 internalization. Dev. Cell 2013, 24, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, A.; Le Lay, S.; Pohl, J.; Verkade, P.; Stremmel, W. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2006, 1761, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049. [Google Scholar] [CrossRef] [Green Version]
- Thomas-Ecker, S.; Lindecke, A.; Hatzmann, W.; Kaltschmidt, C.; Zänker, K.S.; Dittmar, T. Alteration in the gene expression pattern of primary monocytes after adhesion to endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 5539. [Google Scholar] [CrossRef] [Green Version]
- Gils, J.M.; Derby, M.C.; Fernandes, L.R.; Ramkhelawon, B.; Ray, T.D.; Rayner, K.J.; Parathath, S.; Distel, E.; Feig, J.L.; Alvarez-Leite, J.I.; et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting macrophage emigration from plaques. Nat. Immunol. 2012, 13, 136. [Google Scholar] [CrossRef] [Green Version]
- Ramkhelawon, B.; Hennessy, E.J.; Menager, M.; Ray, T.D.; Sheedy, F.J.; Hutchison, S.; Wanschel, A.; Oldebeken, S.; Geoffrion, M.; Spiro, W.; et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med. 2014, 20, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Javadifar, A.; Rastgoo, S.; Banach, M.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 2529. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N. Desialylated low density lipoprotein--naturally occurring modified lipoprotein with atherogenic potency. Atherosclerosis 1991, 86, 153–161. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N. Characterization of desialylated low-density lipoproteins which cause intracellular lipid accumulation. Int. J. Tissue React. 1992, 14, 155–162. [Google Scholar] [PubMed]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Jaakkola, O.; Solakivi, T.; Nikkari, T.; Smirnov, V.N.; Orekhov, A.N. Multiple-modified desialylated low density lipoproteins that cause intracellular lipid accumulation. Isolation, fractionation and characterization. Lab. Investig. 1992, 67, 665–675. [Google Scholar] [PubMed]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Orekhov, A.N. Low-density lipoprotein modification occurring in human plasma possible mechanism of in vivo lipoprotein desialylation as a primary step of atherogenic modification. Atherosclerosis 1998, 138, 183–195. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Tertov, V.V.; Orekhov, A.N.; Smirnov, V.N. Synergetic effect of desialylated and glycated low density lipoproteins on cholesterol accumulation in cultured smooth muscle intimal cells. Atherosclerosis 1991, 89, 151–154. [Google Scholar] [CrossRef]
- Mezentsev, A.; Bezsonov, E.; Kashirskikh, D.; Baig, M.S.; Eid, A.H.; Orekhov, A. Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside. Biomedicines 2021, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Lange, Y.; Swaisgood, M.H.; Ramos, B.V.; Steck, T.L. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J. Biol. Chem. 1989, 264, 3786–3793. [Google Scholar] [CrossRef]
- Yang, S.-T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The Role of Cholesterol in Membrane Fusion. Chem. Phys. Lipids 2016, 199, 136. [Google Scholar] [CrossRef] [Green Version]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and physiological relevance of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361. [Google Scholar] [CrossRef] [Green Version]
- Brachet, A.; Norwood, S.; Brouwers, J.F.; Palomer, E.; Helms, J.B.; Dotti, C.G.; Esteban, J.A. LTP-triggered cholesterol redistribution activates Cdc42 and drives AMPA receptor synaptic delivery. J. Cell Biol. 2015, 208, 791. [Google Scholar] [CrossRef] [Green Version]
- Pagler, T.A.; Wang, M.; Mondal, M.; Murphy, A.J.; Westerterp, M.; Moore, K.J.; Maxfield, F.R.; Tall, A.R. Deletion of ABCA1 and ABCG1 Impairs Macrophage Migration Because of Increased Rac1 Signaling. Circ. Res. 2011, 108, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frechin, M.; Stoeger, T.; Daetwyler, S.; Gehin, C.; Battich, N.; Damm, E.M.; Stergiou, L.; Riezman, H.; Pelkmans, L. Cell-intrinsic adaptation of lipid composition to local crowding drives social behaviour. Nature 2015, 523, 88–91. [Google Scholar] [CrossRef]
- Liu, S.-L.; Sheng, R.; Jung, J.H.; Wang, L.; Stec, E.; O’Connor, M.J.; Song, S.; Bikkavilli, R.K.; Winn, R.A.; Lee, D.; et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nat. Chem. Biol. 2017, 13, 268. [Google Scholar] [CrossRef] [Green Version]
- Buwaneka, P.; Ralko, A.; Liu, S.-L.; Cho, W. Evaluation of the available cholesterol concentration in the inner leaflet of the plasma membrane of mammalian cells. J. Lipid Res. 2021, 62, 84. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the Keystone symposium on lipid rafts and cell function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Hryniewicz-Jankowska, A.; Augoff, K.; Sikorski, A.F. Highlight article: The role of cholesterol and cholesterol-driven membrane raft domains in prostate cancer. Exp. Biol. Med. 2019, 244, 1053. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Honsho, M.; Ekroos, K.; Shevchenko, A.; Simons, K. Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. USA 2003, 100, 5795. [Google Scholar] [CrossRef] [Green Version]
- Parton, R.G.; Simons, K. The multiple faces of caveolae. Nat. Rev. Mol. Cell. Biol. 2007, 8, 185–194. [Google Scholar] [CrossRef]
- Medina, F.A.; Almeida, C.J.d.; Dew, E.; Li, J.; Bonuccelli, G.; Williams, T.M.; Cohen, A.W.; Pestell, R.G.; Frank, P.G.; Tanowitz, H.B.; et al. Caveolin-1-Deficient Mice Show Defects in Innate Immunity and Inflammatory Immune Response during Salmonella enterica Serovar Typhimurium Infection. Infect. Immun. 2006, 74, 6665. [Google Scholar] [CrossRef] [Green Version]
- Cammarota, E.; Soriani, C.; Taub, R.; Morgan, F.; Sakai, J.; Veatch, S.L.; Bryant, C.E.; Cicuta, P. Criticality of plasma membrane lipids reflects activation state of macrophage cells. J. R. Soc. Interface 2020, 17, 803. [Google Scholar] [CrossRef] [Green Version]
- Varshney, P.; Yadav, V.; Saini, N. Lipid rafts in immune signalling: Current progress and future perspective. Immunology 2016, 149, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Poledne, R.; Malinska, H.; Kubatova, H.; Fronek, J.; Thieme, F.; Kauerova, S.; Lesna, I.K. Polarization of Macrophages in Human Adipose Tissue is Related to the Fatty Acid Spectrum in Membrane Phospholipids. Nutrients 2019, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Kralova Lesna, I.; Petras, M.; Cejkova, S.; Kralova, A.; Fronek, J.; Janousek, L.; Thieme, F.; Tyll, T.; Poledne, R. Cardiovascular disease predictors and adipose tissue macrophage polarization: Is there a link? Eur. J. Prev. Cardiol. 2018, 25, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Poledne, R.; Kralova Lesna, I. Adipose tissue macrophages and atherogenesis—A synergy with cholesterolaemia. Physiol. Res. 2021, 12, 88. [Google Scholar]
- Molfetta, R.; Gasparrini, F.; Peruzzi, G.; Vian, L.; Piccoli, M.; Frati, L.; Santoni, A.; Paolini, R. Lipid Raft-Dependent FcεRI Ubiquitination Regulates Receptor Endocytosis through the Action of Ubiquitin Binding Adaptors. PLoS ONE 2009, 4, e5604. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Roy, K.; Mukherjee, S.; Mukhopadhyay, R.; Roy, S. Restoration of IFNγR Subunit Assembly, IFNγ Signaling and Parasite Clearance in Leishmania donovani Infected Macrophages: Role of Membrane Cholesterol. PLoS Pathog. 2011, 7, e1002229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, L.; Kim, J.; Adam, R.M.; Solomon, K.R.; Freeman, M.R. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Investig. 2005, 115, 959. [Google Scholar] [CrossRef] [Green Version]
- Lemaire-Ewing, S.; Lagrost, L.; Néel, D. Lipid rafts: A signalling platform linking lipoprotein metabolism to atherogenesis. Atherosclerosis 2012, 221, 303–310. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; Macfadyen, J.G.; et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: A prospective study of the JUPITER trial. Lancet 2009, 373, 1175–1182. [Google Scholar] [CrossRef]
- Wang, S.h.; Yuan, S.g.; Peng, D.q.; Zhao, S.p. HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis 2012, 225, 105–114. [Google Scholar] [CrossRef]
- Pirillo, A.; Bonacina, F.; Norata, G.D.; Catapano, A.L. The Interplay of Lipids, Lipoproteins, and Immunity in Atherosclerosis. Curr. Atheroscler. Rep. 2018, 20, 7150. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paukner, K.; Králová Lesná, I.; Poledne, R. Cholesterol in the Cell Membrane—An Emerging Player in Atherogenesis. Int. J. Mol. Sci. 2022, 23, 533. https://doi.org/10.3390/ijms23010533
Paukner K, Králová Lesná I, Poledne R. Cholesterol in the Cell Membrane—An Emerging Player in Atherogenesis. International Journal of Molecular Sciences. 2022; 23(1):533. https://doi.org/10.3390/ijms23010533
Chicago/Turabian StylePaukner, Karel, Ivana Králová Lesná, and Rudolf Poledne. 2022. "Cholesterol in the Cell Membrane—An Emerging Player in Atherogenesis" International Journal of Molecular Sciences 23, no. 1: 533. https://doi.org/10.3390/ijms23010533
APA StylePaukner, K., Králová Lesná, I., & Poledne, R. (2022). Cholesterol in the Cell Membrane—An Emerging Player in Atherogenesis. International Journal of Molecular Sciences, 23(1), 533. https://doi.org/10.3390/ijms23010533