
Flubendazole Plays an Important Anti-Tumor Role in Different Types of Cancers

 and
and
Abstract
:1. Introduction
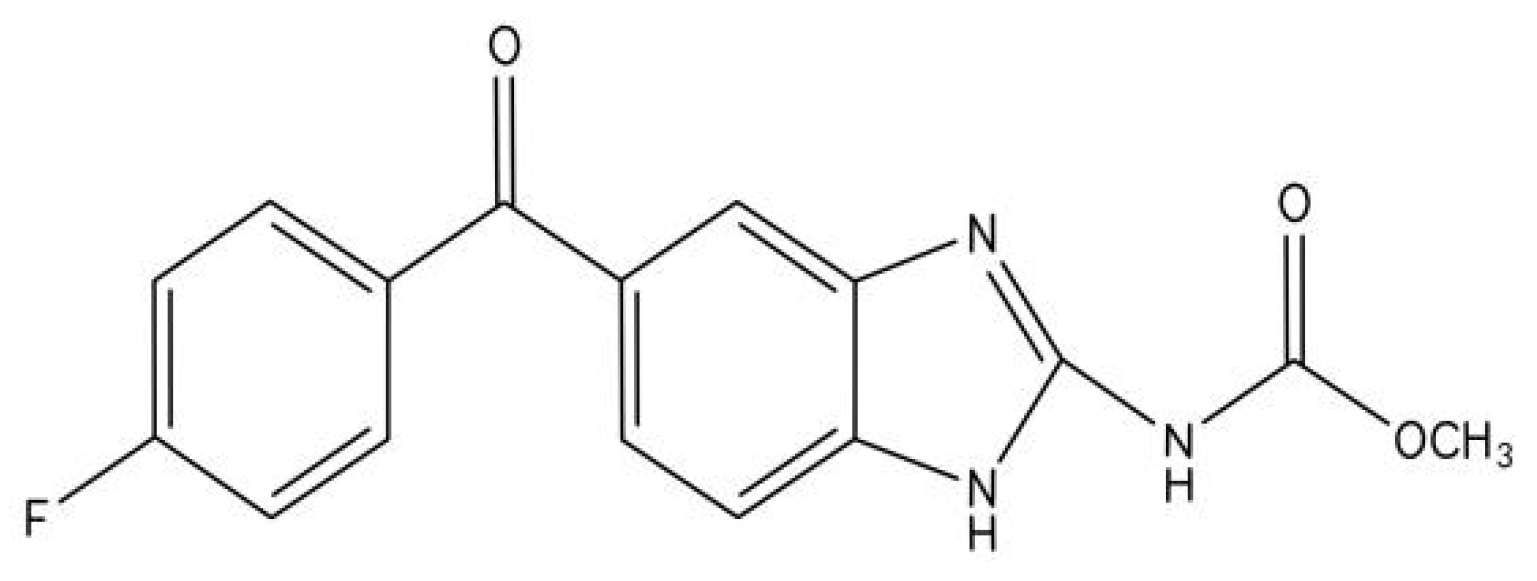
2. The Pharmacokinetics of Flubendazole
3. The Mechanism of Action of Flubendazole
4. Anti-Tumor Role of Flubendazole in Breast Cancer
4.1. Breast CS-like Cells Inhibition via Suppressing Cellcycle Progression and Tubulin Polymerization
4.2. Breast CS-like Cells Inhibition via Suppressing STAT3 Activation
4.3. Improvement of Drug Resistance by Inhibiting CS-like Cells Properties and HER2 Pathway, and Inducing Apoptosis and G2/M Phase Arrest in Breast Cancer
4.4. Promotion of Autophagic Cell Death of TNBC Cells through Upregulating EVA1A
5. Anti-Tumor Role of Flubendazole in Melanoma
5.1. Suppression of Mitosis and Induction of Apoptosis in Melanoma Cells
5.2. Silence of the Immunosuppressive Effects of PD-1 and MDSC in Melanoma Cells
6. Flubendazole Inhibits Prostate Cancer by Promoting P53 Signaling Pathway to Induce Ferroptosis and Cell Cycle Arrest
7. Flubendazole Inhibits Lung Cancer Proliferation by Promoting Autophagic Cell Death
8. Flubendazole Inhibits Colon Cancer by Suppressing Mitosis and Inducing Apoptosis and Cell Senescence
9. Flubendazole Inhibits Oral Squamous Cell Cancer by Suppressing Cell Migration and EMT
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, S.; Yang, L.; Yao, Y.; Xu, L.; Xiang, Y.; Zhao, H.; Wang, L.; Zuo, Z.; Huang, X.; Zhao, C. Flubendazole demonstrates valid antitumor effects by inhibiting STAT3 and activating autophagy. J. Exp. Clin. Cancer Res. 2019, 38, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, L.; Elissondo, M.; Bruni, S.S.; Denegri, G.; Alvarez, L.; Lanusse, C. Flubendazole in cystic echinococcosis therapy: Pharmaco-parasitological evaluation in mice. Parasitol. Int. 2009, 58, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, C.D.; Geary, T.G. Flubendazole: A candidate macrofilaricide for lymphatic filariasis and onchocerciasis field programs. Expert Rev. Anti-Infect. Ther. 2011, 9, 497–501. [Google Scholar] [CrossRef]
- Canova, K.; Rozkydalova, L.; Rudolf, E. Anthelmintic Flubendazole and Its Potential Use in Anticancer Therapy. Acta Med. 2017, 60, 5–11. [Google Scholar]
- Furtado, L.F.V.; de Paiva Bello, A.C.P.; Rabelo, E.M.L. Benzimidazole resistance in helminths: From problem to diagnosis. Acta Trop. 2016, 162, 95–102. [Google Scholar] [CrossRef]
- Ceballos, L.; Elissondo, C.; Sanchez Bruni, S.; Denegri, G.; Lanusse, C.; Alvarez, L. Comparative performances of flubendazole and albendazole in cystic echinococcosis: Ex vivo activity, plasma/cyst disposition, and efficacy in infected mice. Antimicrob. Agents Chemother. 2011, 55, 5861–5867. [Google Scholar] [CrossRef] [Green Version]
- Geary, T.G.; Mackenzie, C.D.; Silber, S.A. Flubendazole as a macrofilaricide: History and background. PLoS Negl. Trop. Dis. 2019, 13, e0006436. [Google Scholar] [CrossRef] [Green Version]
- Jasmer, D.P.; Yao, C.; Rehman, A.; Johnson, S. Multiple lethal effects induced by a benzimidazole anthelmintic in the anterior intestine of the nematode Haemonchus contortus. Mol. Biochem. Parasitol. 2000, 105, 81–90. [Google Scholar] [CrossRef]
- Lacey, E. Mode of action of benzimidazoles. Parasitol. Today 1990, 6, 112–115. [Google Scholar] [CrossRef]
- Spagnuolo, P.A.; Hu, J.; Hurren, R.; Wang, X.; Gronda, M.; Sukhai, M.A.; Di Meo, A.; Boss, J.; Ashali, I.; Beheshti Zavareh, R.; et al. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood 2010, 115, 4824–4833. [Google Scholar] [CrossRef] [Green Version]
- Friedman, P.A.; Platzer, E.G. Interaction of anthelmintic benzimidazoles with Ascaris suum embryonic tubulin. Biochim. Biophys. Acta 1980, 630, 271–278. [Google Scholar] [CrossRef]
- Rudolf, K.; Rudolf, E. An analysis of mitotic catastrophe induced cell responses in melanoma cells exposed to flubendazole. Toxicol. Vitr. 2020, 68, 104930. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, M.; Li, J.; Zheng, Y.; Zhang, S.; Xie, T.; Liu, B. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol. Biosyst. 2015, 11, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Canova, K.; Rozkydalova, L.; Vokurkova, D.; Rudolf, E. Flubendazole induces mitotic catastrophe and apoptosis in melanoma cells. Toxicol. Vitr. 2018, 46, 313–322. [Google Scholar] [CrossRef]
- Tao, J.; Zhao, H.; Xie, X.; Luo, M.; Gao, Z.; Sun, H.; Huang, Z. The anthelmintic drug flubendazole induces cell apoptosis and inhibits NF-kappaB signaling in esophageal squamous cell carcinoma. Onco Targets Ther. 2019, 12, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Agha, B.; Rothweiler, F.; Loschmann, N.; Voges, Y.; Mittelbronn, M.; Starzetz, T.; Harter, P.N.; Abhari, B.A.; Fulda, S.; et al. Identification of flubendazole as potential anti-neuroblastoma compound in a large cell line screen. Sci. Rep. 2015, 5, 8202. [Google Scholar] [CrossRef] [PubMed]
- Hanusova, V.; Skalova, L.; Kralova, V.; Matouskova, P. The Effect of Flubendazole on Adhesion and Migration in SW480 and SW620 Colon Cancer Cells. Anticancer Agents Med. Chem. 2018, 18, 837–846. [Google Scholar] [CrossRef]
- Kralova, V.; Hanusova, V.; Rudolf, E.; Canova, K.; Skalova, L. Flubendazole induces mitotic catastrophe and senescence in colon cancer cells in vitro. J. Pharm. Pharmcol. 2016, 68, 208–218. [Google Scholar] [CrossRef]
- Kralova, V.; Hanusova, V.; Caltova, K.; Spacek, P.; Hochmalova, M.; Skalova, L.; Rudolf, E. Flubendazole and mebendazole impair migration and epithelial to mesenchymal transition in oral cell lines. Chem. Biol. Interact. 2018, 293, 124–132. [Google Scholar] [CrossRef]
- Li, Y.; Acharya, G.; Elahy, M.; Xin, H.; Khachigian, L.M. The anthelmintic flubendazole blocks human melanoma growth and metastasis and suppresses programmed cell death protein-1 and myeloid-derived suppressor cell accumulation. Cancer Lett. 2019, 459, 268–276. [Google Scholar] [CrossRef]
- Raisova Stuchlikova, L.; Kralova, V.; Lnenickova, K.; Zarybnicky, T.; Matouskova, P.; Hanusova, V.; Ambroz, M.; Subrt, Z.; Skalova, L. The metabolism of flubendazole in human liver and cancer cell lines. Drug Test. Anal. 2018, 10, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Sung, D.; Oh, E.; Cho, Y.; Cho, T.M.; Farrand, L.; Seo, J.H.; Kim, J.Y. Flubendazole overcomes trastuzumab resistance by targeting cancer stem-like properties and HER2 signaling in HER2-positive breast cancer. Cancer Lett. 2018, 412, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Kim, Y.J.; An, H.; Sung, D.; Cho, T.M.; Farrand, L.; Jang, S.; Seo, J.H.; Kim, J.Y. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int. J. Cancer 2018, 143, 1978–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.J.; Luo, X.; Zhang, W.; Peng, F.; Cui, B.; Wu, S.J.; Zheng, F.M.; Xu, J.; Xu, L.Z.; Long, Z.J.; et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget 2015, 6, 6326–6340. [Google Scholar] [CrossRef] [Green Version]
- Kralova, V.; Hanusova, V.; Stankova, P.; Knoppova, K.; Canova, K.; Skalova, L. Antiproliferative effect of benzimidazole anthelmintics albendazole, ricobendazole, and flubendazole in intestinal cancer cell lines. Anticancer Drugs 2013, 24, 911–919. [Google Scholar] [CrossRef]
- Son, D.S.; Lee, E.S.; Adunyah, S.E. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw. 2020, 20, e29. [Google Scholar] [CrossRef]
- Dong, T.; Lu, Z.; Li, J.; Liu, Y.; Wen, J. Flubendazole Inhibits the Proliferation of A549 and H460 Cells and Promotes Autophagy. Zhongguo Fei Ai Za Zhi 2020, 23, 306–313. [Google Scholar]
- Else, K.J.; Keiser, J.; Holland, C.V.; Grencis, R.K.; Sattelle, D.B.; Fujiwara, R.T.; Bueno, L.L.; Asaolu, S.O.; Sowemimo, O.A.; Cooper, P.J. Whipworm and roundworm infections. Nat. Rev. Dis. Primers 2020, 6, 44. [Google Scholar] [CrossRef]
- Feldmeier, H.; Bienzle, U.; Dohring, E.; Dietrich, M. Flubendazole versus mebendazole in intestinal helminthic infections. Acta Trop. 1982, 39, 185–189. [Google Scholar]
- Dominguez-Vazquez, A.; Taylor, H.R.; Greene, B.M.; Ruvalcaba-Macias, A.M.; Rivas-Alcala, A.R.; Murphy, R.P.; Beltran-Hernandez, F. Comparison of flubendazole and diethylcarbamazine in treatment of onchocerciasis. Lancet 1983, 1, 139–143. [Google Scholar] [CrossRef]
- Kubicek, V.; Skalova, L.; Skarka, A.; Kralova, V.; Holubova, J.; Stepankova, J.; Subrt, Z.; Szotakova, B. Carbonyl Reduction of Flubendazole in the Human Liver: Strict Stereospecificity, Sex Difference, Low Risk of Drug Interactions. Front. Pharmacol. 2019, 10, 600. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Risinger, A.L.; Giles, F.J.; Mooberry, S.L. Microtubule dynamics as a target in oncology. Cancer Treat. Rev. 2009, 35, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Westermann, S.; Weber, K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Jasra, N.; Sanyal, S.N.; Khera, S. Effect of thiabendazole and fenbendazole on glucose uptake and carbohydrate metabolism in Trichuris globulosa. Vet. Parasitol. 1990, 35, 201–209. [Google Scholar] [CrossRef]
- Cumino, A.C.; Elissondo, M.C.; Denegri, G.M. Flubendazole interferes with a wide spectrum of cell homeostatic mechanisms in Echinococcus granulosus protoscoleces. Parasitol. Int. 2009, 58, 270–277. [Google Scholar] [CrossRef]
- Hanusova, V.; Skalova, L.; Kralova, V.; Matouskova, P. Potential anti-cancer drugs commonly used for other indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef]
- Yadav, S.; Narasimhan, B.; Kaur, H. Perspectives of Benzimidazole Derivatives as Anticancer Agents in the New Era. Anticancer Agents Med. Chem. 2016, 16, 1403–1425. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Ganz, P.A.; Goodwin, P.J. Breast Cancer Survivorship: Where Are We Today? Adv. Exp. Med. Biol. 2015, 862, 1–8. [Google Scholar]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef]
- Libson, S.; Lippman, M. A review of clinical aspects of breast cancer. Int. Rev. Psychiatry 2014, 26, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, J. Breast cancer stem cells: Features, key drivers and treatment options. Semin. Cancer Biol. 2018, 53, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Xu, X.; Lin, S.; Zhang, Y.; Liu, H.; Zhang, C.; Mo, R. A nanotherapeutic strategy to overcome chemotherapeutic resistance of cancer stem-like cells. Nat. Nanotechnol. 2021, 16, 104–113. [Google Scholar] [CrossRef]
- Phan, N.L.; Pham, K.D.; Le Minh, P.; Nguyen, M.T.; Kim, N.P.; Truong, K.D.; Van Pham, P. Hopea odorata Extract Can Efficiently Kill Breast Cancer Cells and Cancer Stem-Like Cells in Three-Dimensional Culture More Than in Monolayer Cell Culture. Adv. Exp. Med. Biol. 2020, 1292, 145–155. [Google Scholar] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Ther. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef]
- Ko, E.S.; Lee, B.H.; Kim, H.A.; Noh, W.C.; Kim, M.S.; Lee, S.A. Triple-negative breast cancer: Correlation between imaging and pathological findings. Eur. Radiol. 2010, 20, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J. Triple-negative breast cancer: Role of specific chemotherapy agents. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Karantza, V.; Aktan, G.; Lala, M. Current treatment landscape for patients with locally recurrent inoperable or metastatic triple-negative breast cancer: A systematic literature review. Breast Cancer Res. 2019, 21, 143. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.B.; Zhang, H.; Damelin, M.; Geles, K.G.; Grindley, J.C.; Dirks, P.B. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov. 2009, 8, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loibl, S.; Gianni, L. HER2-positive breast cancer. Lancet 2017, 389, 2415–2429. [Google Scholar] [CrossRef]
- Figueroa-Magalhaes, M.C.; Jelovac, D.; Connolly, R.; Wolff, A.C. Treatment of HER2-positive breast cancer. Breast 2014, 23, 128–136. [Google Scholar] [CrossRef]
- Montemurro, F.; Di Cosimo, S.; Arpino, G. Human epidermal growth factor receptor 2 (HER2)-positive and hormone receptor-positive breast cancer: New insights into molecular interactions and clinical implications. Ann. Oncol. 2013, 24, 2715–2724. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothe, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Wehrman, T.S.; Raab, W.J.; Casipit, C.L.; Doyonnas, R.; Pomerantz, J.H.; Blau, H.M. A system for quantifying dynamic protein interactions defines a role for Herceptin in modulating ErbB2 interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 19063–19068. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.S.; Nam, K.S.; Kim, S. Tamoxifen overcomes the trastuzumab-resistance of SK-BR-3 tumorspheres by targeting crosstalk between cytoplasmic estrogen receptor alpha and the EGFR/HER2 signaling pathway. Biochem. Pharmacol. 2021, 190, 114635. [Google Scholar] [CrossRef]
- Wu, Y.; Sarkissyan, M.; Ogah, O.; Kim, J.; Vadgama, J.V. Expression of MALAT1 Promotes Trastuzumab Resistance in HER2 Overexpressing Breast Cancers. Cancers 2020, 12, 1918. [Google Scholar] [CrossRef]
- Huang, Y.; Fu, P.; Fan, W. Novel targeted therapies to overcome trastuzumab resistance in HER2-overexpressing metastatic breast cancer. Curr. Drug Targets 2013, 14, 889–898. [Google Scholar] [CrossRef]
- Wang, L.; Yu, C.; Lu, Y.; He, P.; Guo, J.; Zhang, C.; Song, Q.; Ma, D.; Shi, T.; Chen, Y. TMEM166, a novel transmembrane protein, regulates cell autophagy and apoptosis. Apoptosis 2007, 12, 1489–1502. [Google Scholar] [CrossRef]
- Chang, Y.; Li, Y.; Hu, J.; Guo, J.; Xu, D.; Xie, H.; Lv, X.; Shi, T.; Chen, Y. Adenovirus vector-mediated expression of TMEM166 inhibits human cancer cell growth by autophagy and apoptosis in vitro and in vivo. Cancer Lett. 2013, 328, 126–134. [Google Scholar] [CrossRef]
- Xie, H.; Hu, J.; Pan, H.; Lou, Y.; Lv, P.; Chen, Y. Adenovirus vector-mediated FAM176A overexpression induces cell death in human H1299 non-small cell lung cancer cells. BMB Rep. 2014, 47, 104–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, Y.; Zhao, R.; Wang, M.; Jiang, X.; Gao, F.; Fu, L.; Zhang, L.; Zhou, X.L. Flubendazole elicits anti-cancer effects via targeting EVA1A-modulated autophagy and apoptosis in Triple-negative Breast Cancer. Theranostics 2020, 10, 8080–8097. [Google Scholar] [CrossRef] [PubMed]
- Haass, N.K.; Smalley, K.S. Melanoma biomarkers: Current status and utility in diagnosis, prognosis, and response to therapy. Mol. Diagn. Ther. 2009, 13, 283–296. [Google Scholar] [CrossRef]
- Niehues, N.B.; Evanson, B.; Smith, W.A.; Fiore, C.T.; Parekh, P. Melanoma patient notification and treatment timelines. Dermatol. Online J. 2019, 25, 13. [Google Scholar] [CrossRef]
- Moser, J.C.; Grossman, K.F. Adjuvant therapy for resected high-risk melanoma. Semin. Cutan. Med. Surg. 2018, 37, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Abbas, O.; Miller, D.D.; Bhawan, J. Cutaneous malignant melanoma: Update on diagnostic and prognostic biomarkers. Am. J. Dermatol. 2014, 36, 363–379. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D.P. Understanding p53 functions through p53 antibodies. J. Mol. Cell Biol. 2019, 11, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Derech-Haim, S.; Friedman, Y.; Hizi, A.; Bakhanashvili, M. p53 regulates its own expression by an intrinsic exoribonuclease activity through AU-rich elements. J. Mol. Med. 2020, 98, 437–449. [Google Scholar] [CrossRef]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef]
- Grauers Wiktorin, H.; Nilsson, M.S.; Kiffin, R.; Sander, F.E.; Lenox, B.; Rydstrom, A.; Hellstrand, K.; Martner, A. Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of PD-1/PD-L1 checkpoint blockade. Cancer Immunol. Immunother. 2019, 68, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-beta1 production. Sci. Transl. Med. 2018, 10, eaar8356. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Deng, W.W.; Chen, L.; Li, Y.C.; Wu, L.; Ma, S.R.; Zhang, W.F.; Bu, L.L.; Sun, Z.J. Inhibition of JAK2/STAT3 reduces tumor-induced angiogenesis and myeloid-derived suppressor cells in head and neck cancer. Mol. Carcinog. 2018, 57, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer statistics, 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.; Haskins, C.; Siddiqui, M.M.; Hussain, A.; D’Adamo, C. The evolving role of diet in prostate cancer risk and progression. Curr. Opin. Oncol. 2019, 31, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Scott, E. Prostate cancer. Sci. World J. 2011, 11, 749–750. [Google Scholar]
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015, 6, 35542–35555. [Google Scholar] [CrossRef] [Green Version]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zou, L.; Chen, W.; Yang, T.; Luo, J.; Wu, K.; Shu, F.; Tan, X.; Yang, Y.; Cen, S.; et al. Flubendazole, FDA-approved anthelmintic, elicits valid antitumor effects by targeting P53 and promoting ferroptosis in castration-resistant prostate cancer. Pharmacol. Res. 2021, 164, 105305. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Hoy, H.; Lynch, T.; Beck, M. Surgical Treatment of Lung Cancer. Crit. Care Nurs. Clin. N. Am. 2019, 31, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY facilitates colon cancer cell metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Labianca, R.; Beretta, G.D.; Kildani, B.; Milesi, L.; Merlin, F.; Mosconi, S.; Pessi, M.A.; Prochilo, T.; Quadri, A.; Gatta, G.; et al. Colon cancer. Crit. Rev. Oncol. Hematol. 2010, 74, 106–133. [Google Scholar] [CrossRef]
- Tierno, M.B.; Kitchens, C.A.; Petrik, B.; Graham, T.H.; Wipf, P.; Xu, F.L.; Saunders, W.S.; Raccor, B.S.; Balachandran, R.; Day, B.W.; et al. Microtubule binding and disruption and induction of premature senescence by disorazole C(1). J. Pharmacol. Exp. Ther. 2009, 328, 715–722. [Google Scholar] [CrossRef]
- Dan, H.; Liu, S.; Liu, J.; Liu, D.; Yin, F.; Wei, Z.; Wang, J.; Zhou, Y.; Jiang, L.; Ji, N.; et al. RACK1 promotes cancer progression by increasing the M2/M1 macrophage ratio via the NF-kappaB pathway in oral squamous cell carcinoma. Mol. Oncol. 2020, 14, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.P.; Xu, K.; Cui, J.; Yuan, D.Y.; Zou, B.; Li, J.; Liu, J.L.; Li, K.Y.; Meng, Z.; Zhang, B. Cancerassociated fibroblastderived exosomal miR3825p promotes the migration and invasion of oral squamous cell carcinoma. Oncol. Rep. 2019, 42, 1319–1328. [Google Scholar]
- Dave, K.; Ali, A.; Magalhaes, M. Increased expression of PD-1 and PD-L1 in oral lesions progressing to oral squamous cell carcinoma: A pilot study. Sci. Rep. 2020, 10, 9705. [Google Scholar] [CrossRef]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]

{kind=link}
| Type of Cancer | Antitumor Mechanism of Flubendazole | Experimental Model | Reference |
|---|---|---|---|
| CS-like cells-enriched breast cancer | Inhibition of breast CS-like cells via suppressing cellcycle progression and tubulin polymerization in breast cancer cells | Breast cancer cell(MDA-MB-231, BT-549, MCF-7 and SK-BR-3 cells) and mouse allograft breast cancer model | [24] |
| Triple-negative breast cancers (TNBC) | Inhibition of proliferation and metastasis maily by suppressing breast CS-like cells via suppressing STAT3 activation | TNBC cell lines: (MDA-MB-231,Hs578T, BT-549 and 4T1-Luc) and mouse allograft breast cancer model | [23] |
| Human epidermal growth factor receptor-2 (HER2-positive breast cancer) | Improvement in drug resistance through suppressing CS-like cells properties and HER2 pathway, and inducing apoptosis and G2/M phase arrest | Human breast cancer cell lines (BT474,SKBR3, MDA-MB-453) and mouse allograft breast cancer model | [22] |
| Triple-negative breast cancers (TNBC) | Promotion of autophagic cell death of TNBC cells by promoting EVA1A | TNBC cell lines and mouse allograft breast cancer model | [70] |
| Melanoma | Inhibition of mitosis and induction of apoptosis | Human melanoma cell lines | [14] |
| Melanoma | Silencing of immunosuppressive effects of PD-1 and MDSC | Melanoma cell line (MDA-MB-435) and mouse xenograft melanoma model | [20] |
| Castration-resistant human prostate cancer (CRPC) | Promotion of p53 signaling pathway to induce ferroptosis and cell cycle arrest | human CRPC cell lines (PC3, DU145) | [87] |
| Non-small cell lung cancer (NSCLC) | Induction of autophagic cell death (needs to be further elucidated) | NSCLC cell lines (A549, H460) | [27] |
| Colon cancer | Inhibition of mitosis and induction of apoptosis and cells senescence | Human colon cell lines (SW480 and SW620) | [18] |
| Oral squamous cell cancer(OSCC) | Inhibition of cell migration and epithelial–mesenchymal transition (EMT) | oral squamous cancer cells (PE/CA-PJ15 cells and H376 cells) and precancerous oral keratinocytes | [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Ding, Y.; Liu, H.; Sun, M.; Wang, H.; Wu, D. Flubendazole Plays an Important Anti-Tumor Role in Different Types of Cancers. Int. J. Mol. Sci. 2022, 23, 519. https://doi.org/10.3390/ijms23010519
Chen C, Ding Y, Liu H, Sun M, Wang H, Wu D. Flubendazole Plays an Important Anti-Tumor Role in Different Types of Cancers. International Journal of Molecular Sciences. 2022; 23(1):519. https://doi.org/10.3390/ijms23010519
Chicago/Turabian StyleChen, Chaoran, Yueming Ding, Huiyang Liu, Mengyao Sun, Honggang Wang, and Dongdong Wu. 2022. "Flubendazole Plays an Important Anti-Tumor Role in Different Types of Cancers" International Journal of Molecular Sciences 23, no. 1: 519. https://doi.org/10.3390/ijms23010519
APA StyleChen, C., Ding, Y., Liu, H., Sun, M., Wang, H., & Wu, D. (2022). Flubendazole Plays an Important Anti-Tumor Role in Different Types of Cancers. International Journal of Molecular Sciences, 23(1), 519. https://doi.org/10.3390/ijms23010519