
Cancer Therapy-Induced Cardiotoxicity—A Metabolic Perspective on Pathogenesis, Diagnosis and Therapy


Abstract
:1. Introduction
2. Metabolic Dysfunction Leading to Cardiotoxicity of Specific Chemotherapies
2.1. Doxorubicin
2.2. Trastuzumab
2.3. Sunitinib
2.4. Imatinib
2.5. Ponatinib
2.6. Radiotherapy
3. Predicting Cardiotoxicity Using Metabolic Markers
3.1. Blood Biomarkers
3.2. 12F-FDG PET/CT
3.3. Hyperpolarised 13C MRI
4. Adjuvant Cardiotherapy
4.1. Metformin
4.2. SGLT2 Inhibitors
4.3. Resveratrol
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Sandri, M.T.; Lamantia, G.; Colombo, N.; Civelli, M.; Martinelli, G.; Veglia, F.; Fiorentini, C.; Cipolla, C.M. Prevention of high-dose chemotherapy-induced cardiotoxicity in high-risk patients by angiotensin-converting enzyme inhibition. Circulation 2006, 114, 2474–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Li, D.L.; Hill, J.A. Heart failure and loss of metabolic control. J. Cardiovasc. Pharmacol. 2014, 63, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Della Sala, A.; Tocchetti, C.G.; Porporato, P.E.; Ghigo, A. Metabolic Aspects of Anthracycline Cardiotoxicity. Curr. Treat. Options Oncol. 2021, 22, 18. [Google Scholar] [CrossRef]
- Mohan, N.; Jiang, J.; Dokmanovic, M.; Wu, W.J. Trastuzumab-mediated cardiotoxicity: Current understanding, challenges, and frontiers. Antib. Ther. 2018, 1, 13–17. [Google Scholar] [CrossRef]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaimomycin, a new antitumor antibiotic from S. Peucetius var. caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.; Cardoso, S.; Correia, S.; Oliveira, P.; Santos, M.; Moreira, P. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Kilickap, S.; Akgul, E.; Aksoy, S.; Aytemir, K.; Barista, I. Doxorubicin-induced second degree and complete atrioventricular block. Europace 2005, 7, 227–230. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrak, E.A.; Piťha, J.; Rosenheim, S.; Gottlieb, J.A. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 1973, 32, 302–314. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Takemura, G.; Fujiwara, H. Doxorubicin-Induced Cardiomyopathy. From the Cardiotoxic Mechanisms to Management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Šimůnek, T.; Štěrba, M.; Popelová, O.; Adamcová, M.; Hrdina, R.; Gerši, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 1–20. [Google Scholar] [CrossRef]
- Renu, K.; Abilash, V.G.; Tirupathi, T.P.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Dos Santos, D.S.; dos Santos Goldenberg, R.C. Doxorubicin-Induced Cardiotoxicity: From Mechanisms to Development of Efficient Therapy. In Cardiotoxicity; InTech: London, UK, 2018. [Google Scholar]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Zaugg, M.; Zuppinger, C.; Wallimann, T.; Schlattner, U. New insights into doxorubicin-induced cardiotoxicity: The critical role of cellular energetics. J. Mol. Cell. Cardiol. 2006, 41, 389–405. [Google Scholar] [CrossRef]
- Marcillat, O.; Zhang, Y.; Davies, K.J.A. Oxidative and non-oxidative mechanisms in the inactivation of cardiac mitochondrial electron transport chain components by doxorubicin. Biochem. J. 1989, 259, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, B.L.; Niederer, S. A Biophysical Systems Approach to Identifying the Pathways of Acute and Chronic Doxorubicin Mitochondrial Cardiotoxicity. PLoS Comput. Biol. 2016, 12, e1005214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timm, K.N.; Perera, C.; Ball, V.; Henry, J.A.; Miller, J.J.; Kerr, M.; West, J.A.; Sharma, E.; Broxholme, J.; Logan, A.; et al. Early detection of doxorubicin-induced cardiotoxicity in rats by its cardiac metabolic signature assessed with hyperpolarized MRI. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakasugi, S.; Fischman, A.J.; Babich, J.W.; Callahan, R.J.; Elmaleh, D.R.; Wilkinson, R.; Strauss, H.W. Myocardial substrate utilization and left ventricular function in adriamycin cardiomyopathy. J. Nucl. Med. 1993, 34, 1529–1535. [Google Scholar] [PubMed]
- Yuan, Y.; Fan, S.; Shu, L.; Huang, W.; Xie, L.; Bi, C.; Yu, H.; Wang, Y.; Li, Y. Exploration the Mechanism of Doxorubicin-Induced Heart Failure in Rats by Integration of Proteomics and Metabolomics Data. Front. Pharmacol. 2020, 11, 1988. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aleem, S.; El-Merzabani, M.M.; Sayed-Ahmed, M.; Taylor, D.A.; Lowe, J.E. Acute and chronic effects of adriamycin on fatty acid oxidation in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 1997, 29, 789–797. [Google Scholar] [CrossRef]
- Carvalho, R.A.; Sousa, R.P.B.; Cadete, V.J.J.; Lopaschuk, G.D.; Palmeira, C.M.M.; Bjork, J.A.; Wallace, K.B. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology 2010, 270, 92–98. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Li, X.; Yang, T.; Jiang, Q. Effects of PPARα/PGC-1α on the myocardial energy metabolism during heart failure in the doxorubicin induced dilated cardiomyopathy in mice. Int. J. Clin. Exp. Med. 2014, 7, 2435. [Google Scholar]
- Tokarska-Schlattner, M.; Wallimann, T.; Schlattner, U. Multiple interference of anthracyclines with mitochondrial creatine kinases: Preferential damage of the cardiac isoenzyme and its implications for drug cardiotoxicity. Mol. Pharmacol. 2002, 61, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Maslov, M.Y.; Chacko, V.P.; Hirsch, G.A.; Akki, A.; Leppo, M.K.; Steenbergen, C.; Weiss, R.G. Reduced in vivo high-energy phosphates precede adriamycin-induced cardiac dysfunction. Am. J. Physiol. - Hear. Circ. Physiol. 2010, 299, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Gratia, S.; Kay, L.; Potenza, L.; Seffouh, A.; Novel-Chate, V.; Schnebelen, C.; Sestili, P.; Schlattner, U.; Tokarska-Schlattner, M. Inhibition of AMPK signalling by doxorubicin: At the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovasc. Res. 2012, 95, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R.B. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta—Mol. Basis Dis. 2016, 1862, 2199–2210. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Timm, K.N.; Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyck, J.R.B.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574, 95–112. [Google Scholar] [CrossRef]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y.J. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478. [Google Scholar] [CrossRef] [Green Version]
- Konishi, M.; Haraguchi, G.; Ohigashi, H.; Ishihara, T.; Saito, K.; Nakano, Y.; Isobe, M. Adiponectin protects against doxorubicin-induced cardiomyopathy by anti-apoptotic effects through AMPK up-regulation. Cardiovasc. Res. 2011, 89, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Asnani, A.; Shi, X.; Farrell, L.; Lall, R.; Sebag, I.A.; Plana, J.C.; Gerszten, R.E.; Scherrer-Crosbie, M. Changes in Citric Acid Cycle and Nucleoside Metabolism Are Associated with Anthracycline Cardiotoxicity in Patients with Breast Cancer. J. Cardiovasc. Transl. Res. 2020, 13, 349–356. [Google Scholar] [CrossRef]
- Laird-Fick, H.S.; Tokala, H.; Kandola, S.; Kehdi, M.; Pelosi, A.; Wang, L.; Grondahl, B. Early morphological changes in cardiac mitochondria after subcutaneous administration of trastuzumab in rabbits: Possible prevention with oral selenium supplementation. Cardiovasc. Pathol. 2020, 44, 107159. [Google Scholar] [CrossRef]
- Kitani, T.; Ong, S.G.; Lam, C.K.; Rhee, J.W.; Zhang, J.Z.; Oikonomopoulos, A.; Ma, N.; Tian, L.; Lee, J.; Telli, M.L.; et al. Human-Induced Pluripotent Stem Cell Model of Trastuzumab-Induced Cardiac Dysfunction in Patients with Breast Cancer. Circulation 2019, 139, 2451–2465. [Google Scholar] [CrossRef]
- Necela, B.M.; Axenfeld, B.C.; Serie, D.J.; Kachergus, J.M.; Perez, E.A.; Thompson, E.A.; Norton, N. The antineoplastic drug, trastuzumab, dysregulates metabolism in iPSC-derived cardiomyocytes. Clin. Transl. Med. 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Cho, S.G.; Kang, S.R.; Yoo, S.W.; Kwon, S.Y.; Min, J.J.; Bom, H.S.; Song, H.C. Association between FDG uptake in the right ventricular myocardium and cancer therapy-induced cardiotoxicity. J. Nucl. Cardiol. 2020, 27, 2154–2163. [Google Scholar] [CrossRef]
- Kerkela, R.; Woulfe, K.C.; Durand, J.B.; Vagnozzi, R.; Kramer, D.; Chu, T.F.; Beahm, C.; Chen, M.H.; Force, T. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin. Transl. Sci. 2009, 2, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, N.; Chen, T.; Zhang, C.; Liu, L.; Qi, Y.; Bu, P. Trimetazidine ameliorates sunitinib-induced cardiotoxicity in mice via the AMPK/mTOR/autophagy pathway. Pharm. Biol. 2019, 57, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Li, X.; Song, J.; Guo, M.; Cai, H.; Wu, Z.; Wu, P.; Li, L.; Yang, M.; Wang, Y.; et al. Metabolic changes precede radiation-induced cardiac remodeling in beagles: Using noninvasive18f-fdg (18f-fludeoxyglucose) and13n-ammonia positron emission tomography/computed tomography scans. J. Am. Heart Assoc. 2020, 9, e016875. [Google Scholar] [CrossRef] [PubMed]
- Unal, K.; Mustafa, U.; Akdemir, O.; Akmansu, M. 18F-FDG PET/CT findings of radiotherapy-related myocardial changes in patients with thoracic malignancies. Nucl. Med. Commun. 2013, 34, 855–859. [Google Scholar] [CrossRef]
- Jingu, K.; Kaneta, T.; Nemoto, K.; Ichinose, A.; Oikawa, M.; Takai, Y.; Ogawa, Y.; Nakata, E.; Sakayauchi, T.; Takai, K.; et al. The utility of 18F-fluorodeoxyglucose positron emission tomography for early diagnosis of radiation-induced myocardial damage. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 845–851. [Google Scholar] [CrossRef]
- Nahta, R.; Esteva, F.J. Trastuzumab: Triumphs and tribulations. Oncogene 2007, 26, 3637–3643. [Google Scholar] [CrossRef] [Green Version]
- Farolfi, A.; Melegari, E.; Aquilina, M.; Scarpi, E.; Ibrahim, T.; Maltoni, R.; Sarti, S.; Cecconetto, L.; Pietri, E.; Ferrario, C.; et al. Trastuzumab-induced cardiotoxicity in early breast cancer patients: A retrospective study of possible risk and protective factors. Heart 2013, 99, 634–639. [Google Scholar] [CrossRef]
- Tarantini, L.; Cioffi, G.; Gori, S.; Tuccia, F.; Boccardi, L.; Bovelli, D.; Lestuzzi, C.; Maurea, N.; Oliva, S.; Russo, G.; et al. Trastuzumab adjuvant chemotherapy and cardiotoxicity in real-world women with breast cancer. J. Card. Fail. 2012, 18, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Calvillo-Argüelles, O.; Abdel-Qadir, H.; Suntheralingam, S.; Michalowska, M.; Amir, E.; Thavendiranathan, P. Trastuzumab-Related Cardiotoxicity and Cardiac Care in Patients With HER2 Positive Metastatic Breast Cancer. Am. J. Cardiol. 2020, 125, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.C.; Bouras, S.; Sawaya, H.; Sebag, I.A.; Cohen, V.; Picard, M.H.; Passeri, J.; Kuter, I.; Scherrer-Crosbie, M. Time trends of left ventricular ejection fraction and myocardial deformation indices in a cohort of women with breast cancer treated with anthracyclines, taxanes, and trastuzumab. J. Am. Soc. Echocardiogr. 2015, 28, 509–514. [Google Scholar] [CrossRef] [PubMed]
- De Keulenaer, G.W.; Doggen, K.; Lemmens, K. The vulnerability of the heart as a pluricellular paracrine organ: Lessons from unexpected triggers of heart failure in targeted ErbB2 anticancer therapy. Circ. Res. 2010, 106, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, B.T.; Varga, Z.V.; Wu, W.J.; Pacher, P. Trastuzumab cardiotoxicity: From clinical trials to experimental studies. Br. J. Pharmacol. 2017, 174, 3727–3748. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.I.; Burke, M.A.; Singh, A.T.K.; Prachand, S.; Lieberman, E.D.; Sun, L.; Naik, T.J.; Naga Prasad, S.V.; Ardehali, H. Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J. Biol. Chem. 2009, 284, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Spector, N.L.; Yarden, Y.; Smith, B.; Lyass, L.; Trusk, P.; Pry, K.; Hill, J.E.; Xia, W.; Seger, R.; Bacus, S.S. Activation of AMP-activated protein kinase by human EGF receptor 2/EGF receptor tyrosine kinase inhibitor protects cardiac cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10607–10612. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bu, P. Progress on the cardiotoxicity of sunitinib: Prognostic significance, mechanism and protective therapies. Chem. Biol. Interact. 2016, 257, 125–131. [Google Scholar] [CrossRef]
- Chu, T.F.; Rupnick, M.A.; Kerkela, R.; Dallabrida, S.M.; Zurakowski, D.; Nguyen, L.; Woulfe, K.; Pravda, E.; Cassiola, F.; Desai, J.; et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007, 370, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Ewer, M.S.; Suter, T.M.; Lenihan, D.J.; Niculescu, L.; Breazna, A.; Demetri, G.D.; Motzer, R.J. Cardiovascular events among 1090 cancer patients treated with sunitinib, interferon, or placebo: A comprehensive adjudicated database analysis demonstrating clinically meaningful reversibility of cardiac events. Eur. J. Cancer 2014, 50, 2162–2170. [Google Scholar] [CrossRef]
- Greineder, C.F.; Kohnstamm, S.; Ky, B. Heart failure associated with sunitinib: Lessons learned from animal models. Curr. Hypertens. Rep. 2011, 13, 436–441. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Kerkelä, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atallah, E.; Durand, J.-B.; Kantarjian, H.; Cortes, J. Congestive heart failure is a rare event in patients receiving imatinib therapy. Blood 2007, 110, 1233–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, T.P.; Santiesteban, L.; Gomez, D.; Chambers, J.W. Sab mediates mitochondrial dysfunction involved in imatinib mesylate-induced cardiotoxicity. Toxicology 2017, 382, 24–35. [Google Scholar] [CrossRef]
- Chan, O.; Talati, C.; Isenalumhe, L.; Shams, S.; Nodzon, L.; Fradley, M.; Sweet, K.; Pinilla-Ibarz, J. Side-effects profile and outcomes of ponatinib in the treatment of chronic myeloid leukemia. Blood Adv. 2020, 4, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Van Hasselt, J.G.C.; Rahman, R.; Hansen, J.; Stern, A.; Shim, J.V.; Xiong, Y.; Pickard, A.; Jayaraman, G.; Hu, B.; Mahajan, M.; et al. Transcriptomic profiling of human cardiac cells predicts protein kinase inhibitor-associated cardiotoxicity. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Cirmi, S.; El Abd, A.; Letinier, L.; Navarra, M.; Salvo, F. Cardiovascular toxicity of tyrosine kinase inhibitors used in chronic myeloid leukemia: An analysis of the FDA adverse event reporting system database (FAERS). Cancers 2020, 12, 826. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Umbarkar, P.; Tousif, S.; Lal, H. Cardiotoxicity of the BCR-ABL1 tyrosine kinase inhibitors: Emphasis on ponatinib. Int. J. Cardiol. 2020, 316, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Gover-Proaktor, A.; Granot, G.; Shapira, S.; Raz, O.; Pasvolsky, O.; Nagler, A.; Lev, D.L.; Inbal, A.; Lubin, I.; Raanani, P.; et al. Ponatinib reduces viability, migration, and functionality of human endothelial cells. Leuk. Lymphoma 2017, 58, 1455–1467. [Google Scholar] [CrossRef]
- Latifi, Y.; Moccetti, F.; Wu, M.; Xie, A.; Packwood, W.; Qi, Y.; Ozawa, K.; Shentu, W.; Brown, E.; Shirai, T.; et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood 2019, 133, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.P.; Glennon, M.S.; Umbarkar, P.; Gupte, M.; Galindo, C.L.; Zhang, Q.; Force, T.; Becker, J.R.; Lal, H. Ponatinib-induced cardiotoxicity: Delineating the signalling mechanisms and potential rescue strategies. Cardiovasc. Res. 2019, 115, 966–977. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Mingard, C.; Paech, F.; Bouitbir, J.; Krähenbühl, S. Mechanisms of toxicity associated with six tyrosine kinase inhibitors in human hepatocyte cell lines. J. Appl. Toxicol. 2018, 38, 418–431. [Google Scholar] [CrossRef]
- Hancock, S.L.; Hoppe, R.T.; Tucker, M.A. Factors Affecting Late Mortality From Heart Disease After Treatment of Hodgkin’s Disease. JAMA J. Am. Med. Assoc. 1993, 270, 1949–1955. [Google Scholar] [CrossRef]
- Darby, S.C.; Ewertz, M.; McGale, P.; Bennet, A.M.; Blom-Goldman, U.; Brønnum, D.; Correa, C.; Cutter, D.; Gagliardi, G.; Gigante, B.; et al. Risk of Ischemic Heart Disease in Women after Radiotherapy for Breast Cancer. N. Engl. J. Med. 2013, 368, 987–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Bogaard, V.A.B.; Ta, B.D.P.; Van Der Schaaf, A.; Bouma, A.B.; Middag, A.M.H.; Bantema-Joppe, E.J.; Van Dijk, L.V.; Van Dijk-Peters, F.B.J.; Marteijn, L.A.W.; De Bock, G.H.; et al. Validation and modification of a prediction model for acute cardiac events in patients with breast cancer treated with radiotherapy based on three-dimensional dose distributions to cardiac substructures. J. Clin. Oncol. 2017, 35, 1171–1178. [Google Scholar] [CrossRef]
- Wang, H.; Wei, J.; Zheng, Q.; Meng, L.; Xin, Y.; Yin, X.; Jiang, X. Radiation-induced heart disease: A review of classification, mechanism and prevention. Int. J. Biol. Sci. 2019, 15, 2128–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borde, C. Enhanced myocardial fluorodeoxyglucose uptake following Adriamycin-based therapy: Evidence of early chemotherapeutic cardiotoxicity? World J. Radiol. 2012, 4, 220. [Google Scholar] [CrossRef]
- Park, J.M.; Reed, G.D.; Liticker, J.; Putnam, W.C.; Chandra, A.; Yaros, K.; Afzal, A.; MacNamara, J.; Raza, J.; Hall, R.G.; et al. Effect of Doxorubicin on Myocardial Bicarbonate Production from Pyruvate Dehydrogenase in Women with Breast Cancer. Circ. Res. 2020, 127, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Ky, B.; Putt, M.; Sawaya, H.; French, B.; Januzzi, J.L.; Sebag, I.A.; Plana, J.C.; Cohen, V.; Banchs, J.; Carver, J.R.; et al. Early increases in multiple biomarkers predict subsequent cardiotoxicity in patients with breast cancer treated with doxorubicin, taxanes, and trastuzumab. J. Am. Coll. Cardiol. 2014, 63, 809–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirinos, J.A.; Orlenko, A.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Spires, T.E.; Yarde, M.; Wang, Z.; Seiffert, D.A.; et al. Multiple Plasma Biomarkers for Risk Stratification in Patients With Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2020, 75, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Kawada, K.; Iwamoto, M.; Sakai, Y. Mechanisms underlying 18 F-fluorodeoxyglucose accumulation in colorectal cancer. World J. Radiol. 2016, 8, 880. [Google Scholar] [CrossRef] [PubMed]
- Early Diagnosis of Therapy-associated Cardiotoxicity Basing on Multi-tracer Multimodality PET/MRI—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04555642 (accessed on 26 October 2021).
- Ardenkjær-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.H.; Lau, J.Y.C.; Chen, A.P.; Geraghty, B.J.; Perks, W.J.; Roifman, I.; Wright, G.A.; Connelly, K.A. Hyperpolarized 13C Metabolic MRI of the Human Heart: Initial Experience. Circ. Res. 2016, 119, 1177–1182. [Google Scholar] [CrossRef]
- Agger, P.; Hyldebrandt, J.A.; Hansen, E.S.S.; Omann, C.; Bøgh, N.; Waziri, F.; Nielsen, P.M.; Laustsen, C. Magnetic resonance hyperpolarization imaging detects early myocardial dysfunction in a porcine model of right ventricular heart failure. Eur. Hear. J.-Cardiovasc. Imaging 2020, 21, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Rider, O.J.; Apps, A.; Miller, J.J.J.J.; Lau, J.Y.C.; Lewis, A.J.M.; Peterzan, M.A.; Dodd, M.S.; Lau, A.Z.; Trumper, C.; Gallagher, F.A.; et al. Noninvasive in vivo assessment of cardiac metabolism in the healthy and diabetic human heart using hyperpolarized 13C MRI. Circ. Res. 2020, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Effect of Cardiotoxic Anticancer Chemotherapy on the Metabolism of [1-13C]Pyruvate in Cardiac Mitochondria—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03685175 (accessed on 26 October 2021).
- Hyperpolarized Carbon 13-Based Metabolic Imaging to Detect Radiation-Induced Cardiotoxicity—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04044872 (accessed on 26 October 2021).
- Wang, Z.J.; Ohliger, M.A.; Larson, P.E.Z.; Gordon, J.W.; Bok, R.A.; Slater, J.; Villanueva-Meyer, J.E.; Hess, C.P.; Kurhanewicz, J.; Vigneron, D.B. Hyperpolarized 13C MRI: State of the art and future directions. Radiology 2019, 291, 273–284. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [Green Version]
- Inzucchi, S.E. Metformin and heart failure: Innocent until proven guilty. Diabetes Care 2005, 28, 2585–2587. [Google Scholar] [CrossRef] [Green Version]
- Halabi, A.; Sen, J.; Huynh, Q.; Marwick, T.H. Metformin treatment in heart failure with preserved ejection fraction: A systematic review and meta-regression analysis. Cardiovasc. Diabetol. 2020, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-M.; Hsieh, M.-C.; Qin, L.; Zhang, J.; Wu, S.-Y. Metformin reduces radiation-induced cardiac toxicity risk in patients having breast cancer. Am. J. Cancer Res. 2019, 9, 1017–1026. [Google Scholar] [PubMed]
- Argun, M.; Üzüm, K.; Sönmez, M.F.; Özyurt, A.; Karabulut, D.; Soyersarıca, Z.; Çilenk, K.T.; Unalmış, S.; Pamukcu, Ö.; Baykan, A.; et al. Cardioprotective effect of metformin against doxorubicin cardiotoxicity in rats. Anatol. J. Cardiol. 2016, 16, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, L.C.; Xu, Y.C.; Padbury, J.F.; Tseng, Y.T.; Yano, N. Metformin protects cardiomyocyte from doxorubicin induced cytotoxicity through an AMP-activated protein kinase dependent signaling pathway: An in Vitro study. PLoS One 2014, 9, e104888. [Google Scholar] [CrossRef] [Green Version]
- Ajzashokouhi, A.H.; Bostan, H.B.; Jomezadeh, V.; Hayes, A.W.; Karimi, G. A review on the cardioprotective mechanisms of metformin against doxorubicin. Hum. Exp. Toxicol. 2020, 39, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Puntoni, M.; Heckman-Stoddard, B.M.; Dunn, B.K.; Ford, L.; DeCensi, A.; Szabo, E. Metformin and cancer risk and mortality: A systematic review and meta-analysis taking into account biases and confounders. Cancer Prev. Res. 2014, 7, 867–885. [Google Scholar] [CrossRef] [Green Version]
- Martin-Castillo, B.; Pernas, S.; Dorca, J.; Álvarez, I.; Martínez, S.; Pérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; Amillano, K.; Domínguez, S.; et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: The METTEN study. Oncotarget 2018, 9, 35687–35704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Kosiborod, M.; Fitchett, D.; Wanner, C.; Hehnke, U.; Kaspers, S.; George, J.T.; Zinman, B. Improvement in cardiovascular outcomes with empaglifozin is independent of glycemic control. Circulation 2018, 138, 1904–1907. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatino, J.; De Rosa, S.; Tammè, L.; Iaconetti, C.; Sorrentino, S.; Polimeni, A.; Mignogna, C.; Amorosi, A.; Spaccarotella, C.; Yasuda, M.; et al. Empagliflozin prevents doxorubicin-induced myocardial dysfunction. Cardiovasc. Diabetol. 2020, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Barış, V.Ö.; Dinçsoy, A.B.; Gedikli, E.; Zırh, S.; Müftüoğlu, S.; Erdem, A. Empagliflozin Significantly Prevents the Doxorubicin-induced Acute Cardiotoxicity via Non-antioxidant Pathways. Cardiovasc. Toxicol. 2021, 21, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Chen, C.C.; Lin, M.H.; Su, H.T.; Ho, M.Y.; Yeh, J.K.; Tsai, M.L.; Hsieh, I.C.; Wen, M.S. TLR9 binding to beclin 1 and mitochondrial SIRT3 by a sodium-glucose co-transporter 2 inhibitor protects the heart from doxorubicin toxicity. Biology 2020, 9, 369. [Google Scholar] [CrossRef]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef]
- Oh, C.M.; Cho, S.; Jang, J.Y.; Kim, H.; Chun, S.; Choi, M.; Park, S.; Ko, Y.G. Cardioprotective potential of an SGLT2 inhibitor against doxorubicin-induced heart failure. Korean Circ. J. 2019, 49, 1183–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yu, Y.; Zhang, Y.; Zhang, Z.; An, W.; Zhao, X. Treatment with D-β-hydroxybutyrate protects heart from ischemia/reperfusion injury in mice. Eur. J. Pharmacol. 2018, 829, 121–128. [Google Scholar] [CrossRef]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.; Botti, G.; Maurea, N. SGLT2 inhibitor dapagliflozin against anthracycline and trastuzumab-induced cardiotoxicity: The role of MYD88, NLRP3, Leukotrienes/Interleukin 6 axis and mTORC1 /Fox01/3a mediated apoptosis. Eur. Heart J. 2020, 41, ehaa946.3253. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Magyar, K.; Halmosi, R.; Palfi, A.; Feher, G.; Czopf, L.; Fulop, A.; Battyany, I.; Sumegi, B.; Toth, K.; Szabados, E. Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc. 2012, 50, 179–187. [Google Scholar] [CrossRef]
- Riba, A.; Deres, L.; Sumegi, B.; Toth, K.; Szabados, E.; Halmosi, R. Cardioprotective Effect of Resveratrol in a Postinfarction Heart Failure Model. Oxid. Med. Cell. Longev. 2017, 2017, 6819281. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, I.; Grant, M.; Zordoky, B. Leveraging the Cardio-Protective and Anticancer Properties of Resveratrol in Cardio-Oncology. Nutrients 2019, 11, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, N.; Zordoky, B.N.; Robertson, I.M.; Hamza, S.M.; Parajuli, N.; Soltys, C.L.M.; Beker, D.L.; Grant, M.K.; Razzoli, M.; Bartolomucci, A.; et al. Co-administration of resveratrol with doxorubicin in youngmice attenuates detrimental late-occurring cardiovascular changes. Cardiovasc. Res. 2018, 114, 1350–1359. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Rogan, K.J.; Sung, M.M.; Zordoky, B.N.; Haykowsky, M.J.; Young, M.E.; Jones, L.W.; Dyck, J.R.B. Both aerobic exercise and resveratrol supplementation attenuate doxorubicin-induced cardiac injury in mice. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, E243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, G.; Mishra, S.; Suman, S.; Shukla, Y. Resveratrol improves the anticancer effects of doxorubicin in vitro and in vivo models: A mechanistic insight. Phytomedicine 2016, 23, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.H.; Lin, X.L.; Guo, D.M.; Zhang, Y.; Yuan, C.; Tan, T.P.; Chen, Y.D.; Wu, S.J.; Ye, Z.F.; He, J. Resveratrol protects cardiomyocytes from doxorubicin-induced apoptosis through the AMPK/P53 pathway. Mol. Med. Rep. 2016, 13, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Brookins Danz, E.D.; Skramsted, J.; Henry, N.; Bennett, J.A.; Keller, R.S. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic. Biol. Med. 2009, 46, 1589–1597. [Google Scholar] [CrossRef]
- De Freitas, R.B.; Boligon, A.A.; Rovani, B.T.; Piana, M.; De Brum, T.F.; Da Silva Jesus, R.; Rother, F.C.; Alves, N.M.; Da Rocha, J.B.T.; Athayde, M.L.; et al. Effect of black grape juice against heart damage from acute gamma TBI in rats. Molecules 2013, 18, 12154–12167. [Google Scholar] [CrossRef] [Green Version]
- Gramatyka, M.; Widłak, P.; Gabryś, D.; Kulik, R.; Sokół, M. Resveratrol administration prevents radiation-related changes in metabolic profles of hearts 20 weeks after irradiation of mice with a single 2 Gy dose. Acta Biochim. Pol. 2020, 67, 629–632. [Google Scholar] [CrossRef] [PubMed]
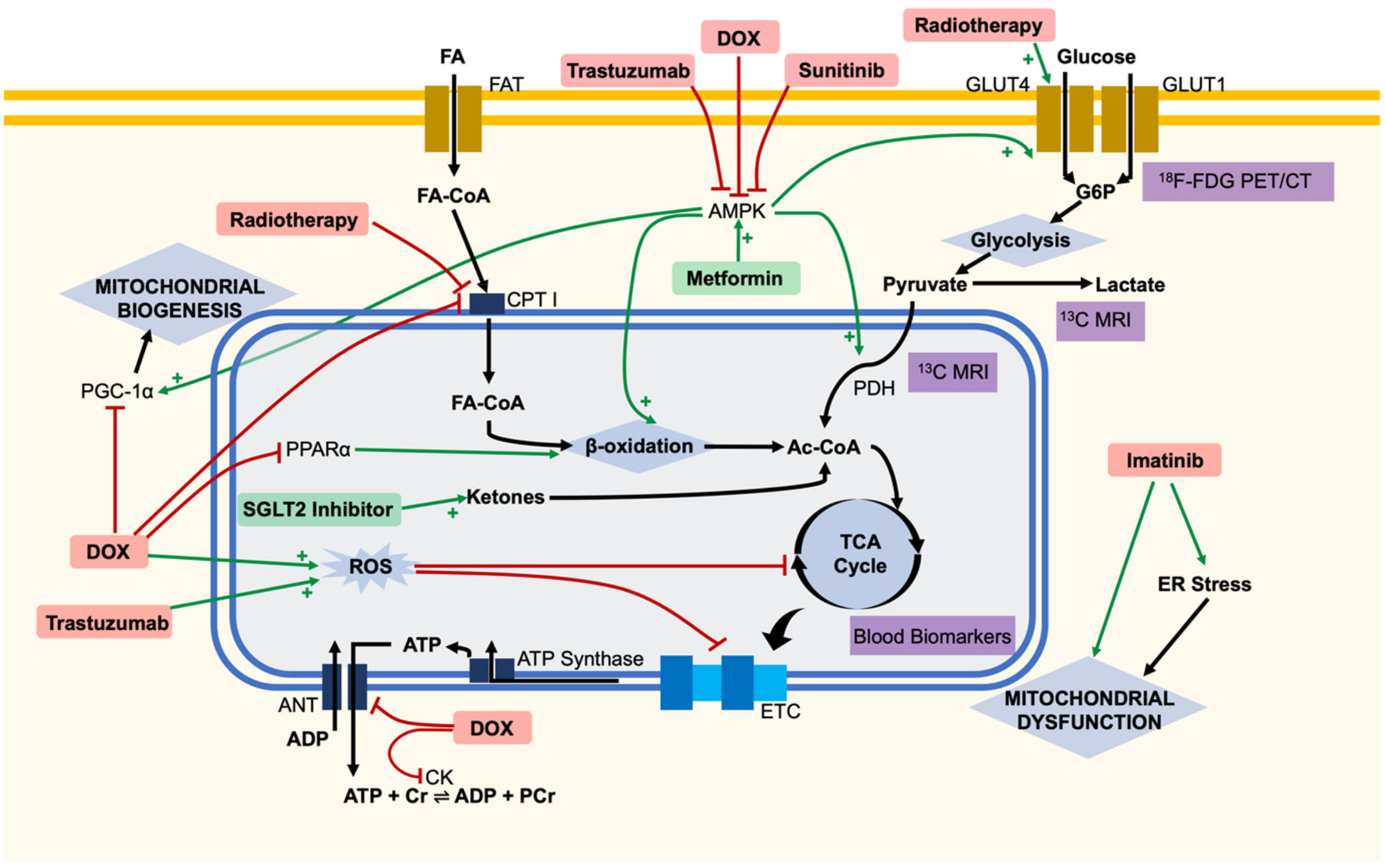

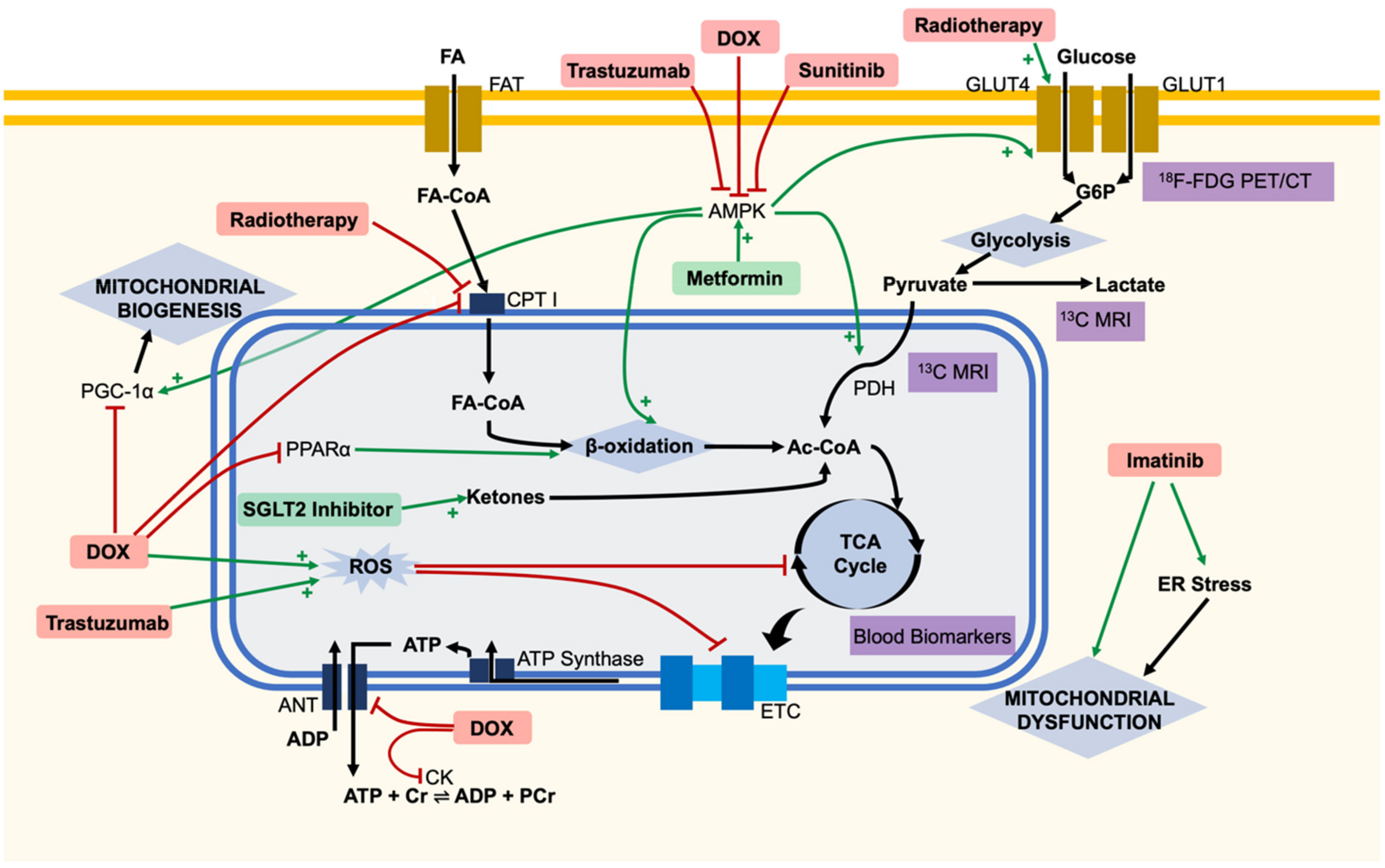{kind=link}
| Model of Disease | Metabolic Alteration | Ref. |
|---|---|---|
| Doxorubicin | ||
| Rat | Decrease in long-chain fatty acid oxidation Inhibition of CPT I | [28] |
| Rat | Decrease in long-chain fatty acid oxidation | [29] |
| Rat | Abnormal amino acid metabolism Abnormal fatty acid metabolism Abnormal glycerol phospholipid metabolism | [27] |
| Mouse | Decrease in expression of PPARα Decrease in expression of PGC-1α Inhibition of adenine nucleotide translocator Decrease in availability of cytosolic ATP | [30] |
| Mouse | Decrease in PCr to ATP ratio Loss of high energy phosphate buffering | [32] |
| Rat | Inhibition of AMPK Inhibition of acetyl-CoA carboxylase | [33] |
| Rat | Inhibition of carbohydrate oxidation Decrease in mitochondrial number | [24] |
| Human | Alterations in citric acid and aconitic acid levels Alterations in purine and pyrimidine metabolism | [40] |
| Trastuzumab | ||
| Rabbit | Mitochondrial dysfunction, likely due to ROS | [41] |
| Cardiac iPSCs | Inhibition of AMPK Decrease in cellular ATP Alterations in mTOR signalling pathway Decrease in glucose uptake | [42] |
| Cardiac iPSCs | Decrease in expression of small molecule metabolism genes Decrease in expression of cholesterol and sterol processing genes Decrease in glucose uptake | [43] |
| Human | Increase in glucose uptake | [44] |
| Sunitinib | ||
| Mouse | Inhibition of AMPK Loss of mitochondrial membrane potential | [45] |
| Mouse | Inhibition of AMPK Inhibition of mTOR | [46] |
| Imatinib | ||
| Lack of clinically relevant studies | ||
| Ponatinib | ||
| Lack of clinically relevant studies | ||
| Radiotherapy | ||
| Beagle | Increase in glucose uptake Increase in GLUT4 expression Decrease in CPT I expression | [47] |
| Human | Increase in glucose uptake | [48] |
| Human | Increase in glucose uptake | [49] |
| Patient Population | Type of Study | Cancer Therapy | Sample Size | Findings | Ref. |
|---|---|---|---|---|---|
| Blood Biomarkers | |||||
| HER2+ Breast Cancer | Case-control | Anthracyclines, Taxanes, Trastuzumab | 38 | LC-MS metabolomics Heart failure preceded by: ● Decreased citric acid:isocitric acid ratio ● Altered purine acid & pyrimidine metabolites | [40] |
| 18F-FDG PET/CT | |||||
| Breast Cancer | Retrospective Logistic Regression | Anthracyclines, Trastuzumab | 121 | SUV in right ventricular wall post therapy associated with development of cardiotoxicity | [44] |
| Lymphoma | Retrospective Analysis | Doxorubicin | 18 | Unclear alterations in 18F-FDG uptake | [81] |
| Hyperpolarised 13C MRI | |||||
| Breast Cancer | Prospective | Doxorubicin | 9 | Early decline in [13C]bicarbonate/total 13C signal | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choksey, A.; Timm, K.N. Cancer Therapy-Induced Cardiotoxicity—A Metabolic Perspective on Pathogenesis, Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 441. https://doi.org/10.3390/ijms23010441
Choksey A, Timm KN. Cancer Therapy-Induced Cardiotoxicity—A Metabolic Perspective on Pathogenesis, Diagnosis and Therapy. International Journal of Molecular Sciences. 2022; 23(1):441. https://doi.org/10.3390/ijms23010441
Chicago/Turabian StyleChoksey, Anurag, and Kerstin N. Timm. 2022. "Cancer Therapy-Induced Cardiotoxicity—A Metabolic Perspective on Pathogenesis, Diagnosis and Therapy" International Journal of Molecular Sciences 23, no. 1: 441. https://doi.org/10.3390/ijms23010441
APA StyleChoksey, A., & Timm, K. N. (2022). Cancer Therapy-Induced Cardiotoxicity—A Metabolic Perspective on Pathogenesis, Diagnosis and Therapy. International Journal of Molecular Sciences, 23(1), 441. https://doi.org/10.3390/ijms23010441