
NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. OxLDL-Activated Monocytes Promote VSMC Phenotypic Switch
2.2. Monocytes Promote VSMC NLRP3 Inflammasome Activation
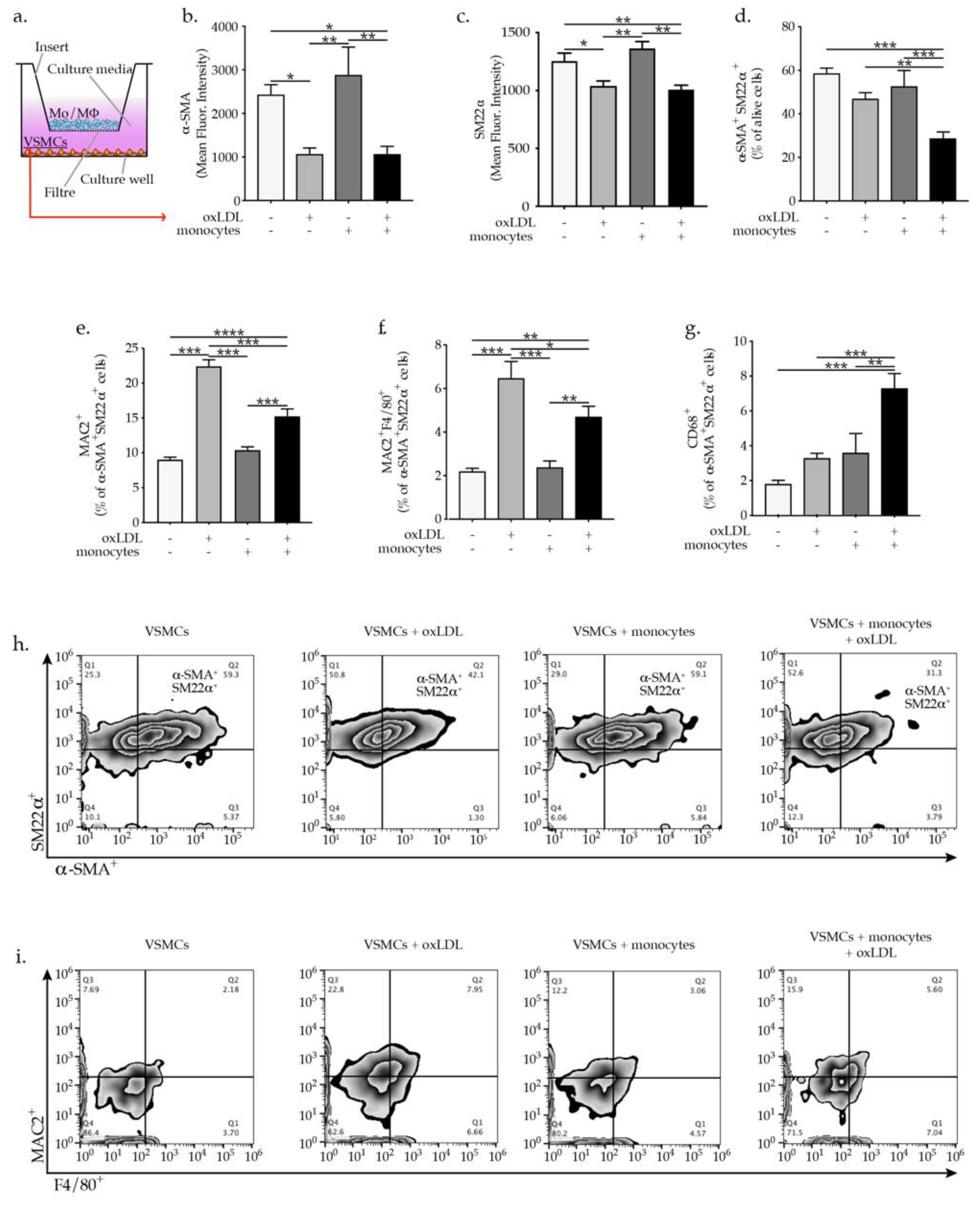

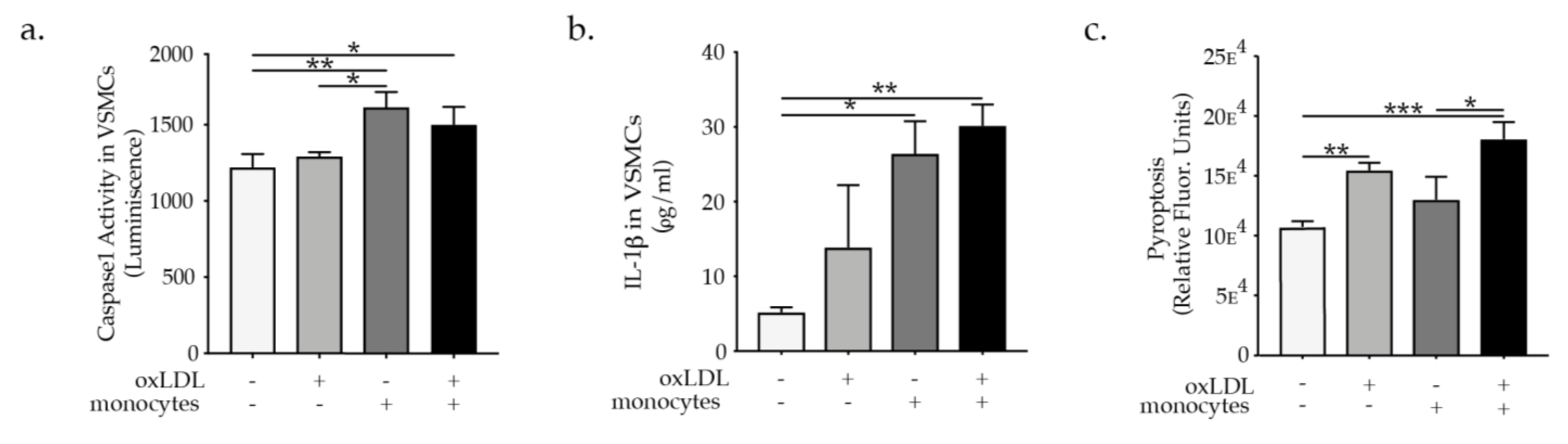
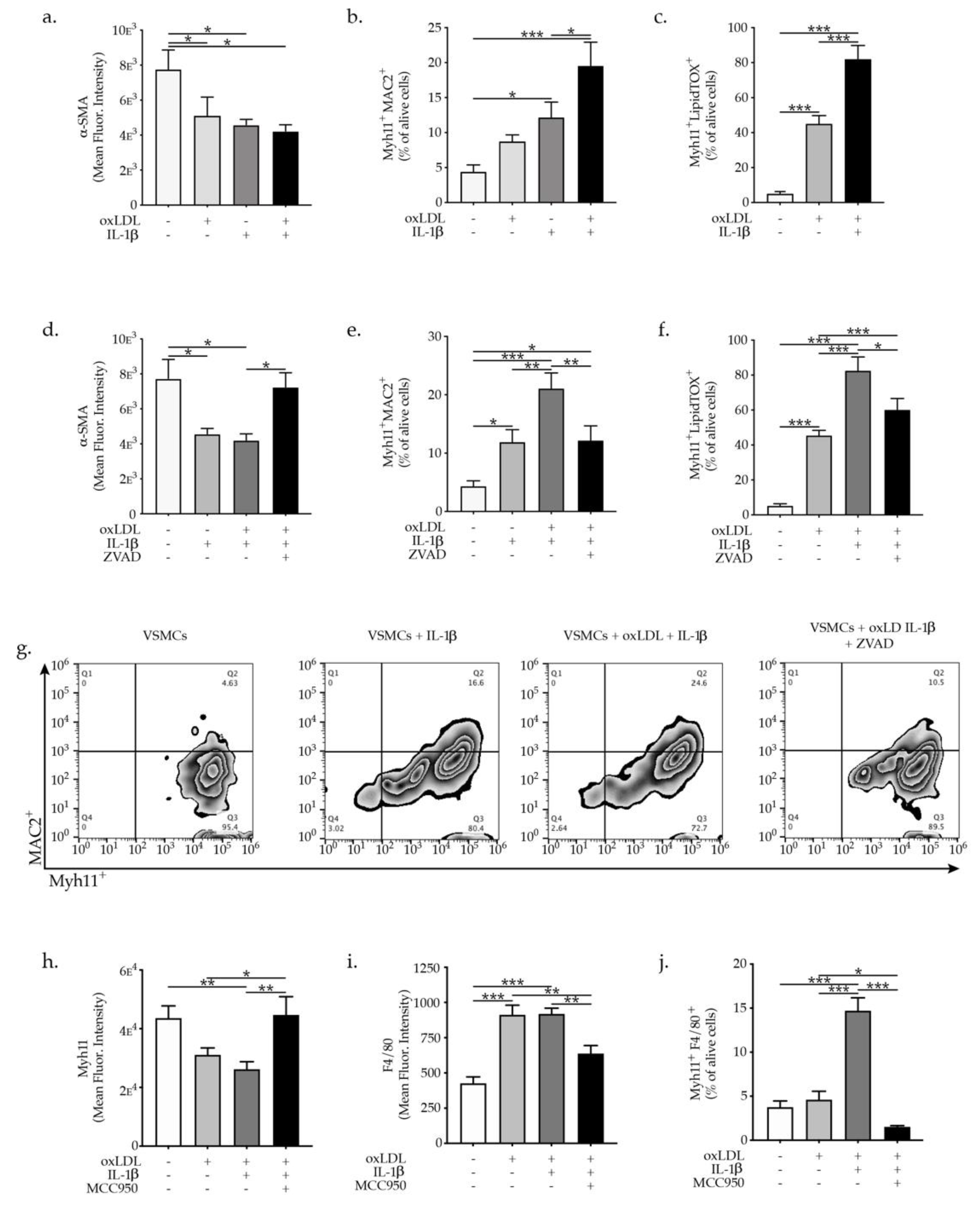
2.3. IL-1β Promotes VSMC Phenotypic Switch and Transdifferentiation to Macrophages-Like Cells
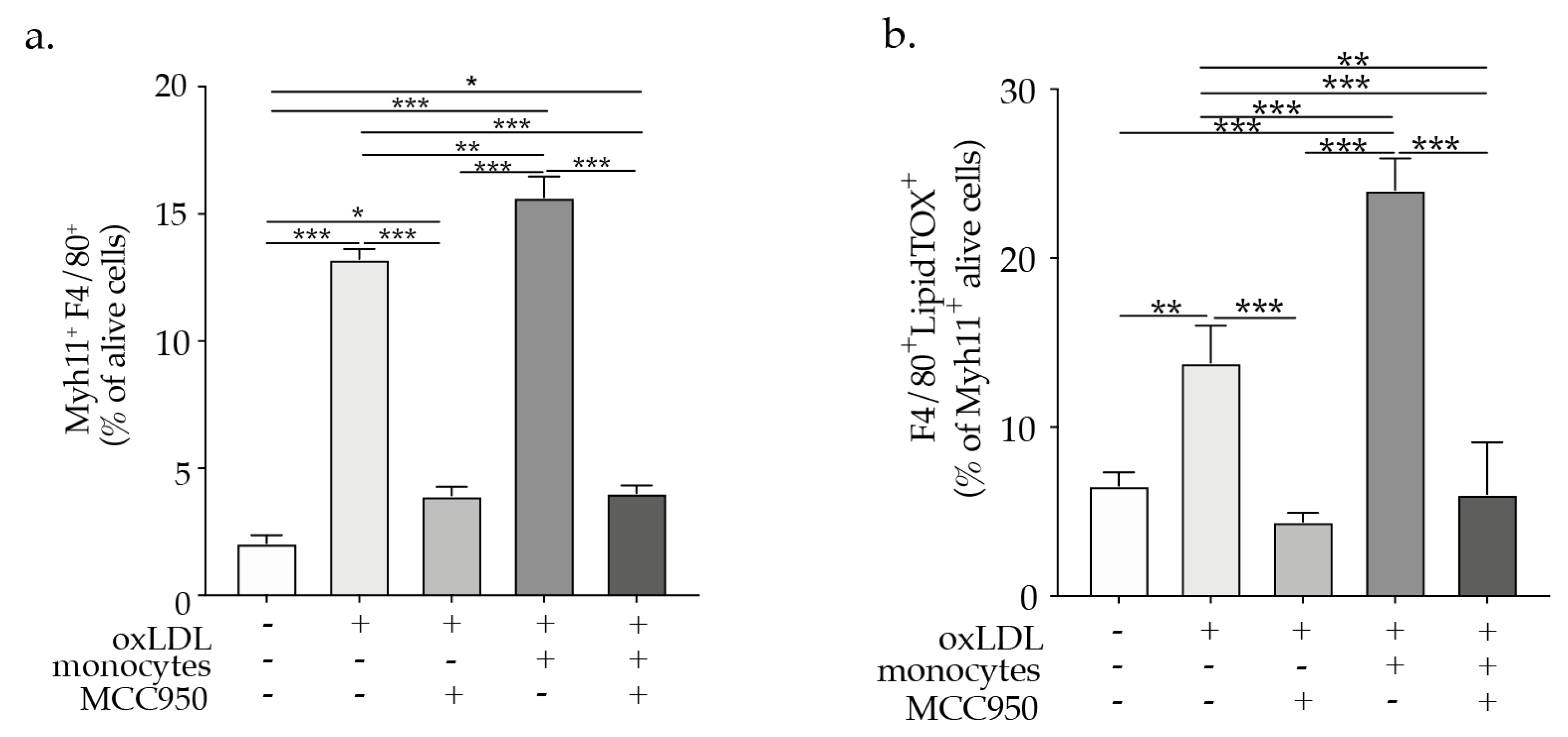
2.4. NLRP3 Inflammasome Inhibition Abrogates VSMCs Phenotypic Switch

2.5. Hypercholesteremia In Vivo Promotes NLRP3 Inflammasome Activation in VSMCs Associated with VSMCs Phenotypic Switch
2.6. NLRP3-Inflammasome Activation in VSMCs Is Associated with Plaque Rupture in Human Carotid Artery Disease
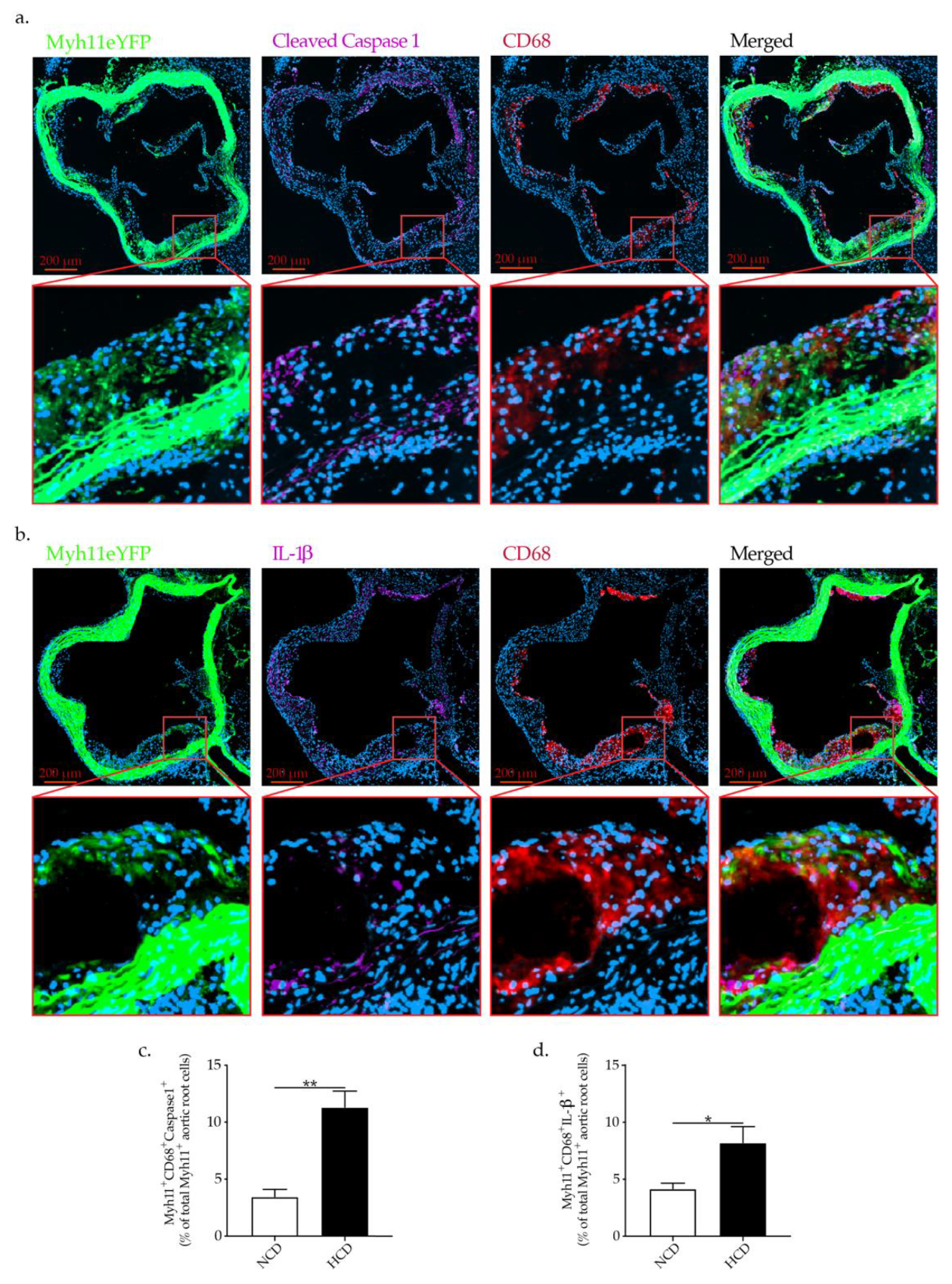
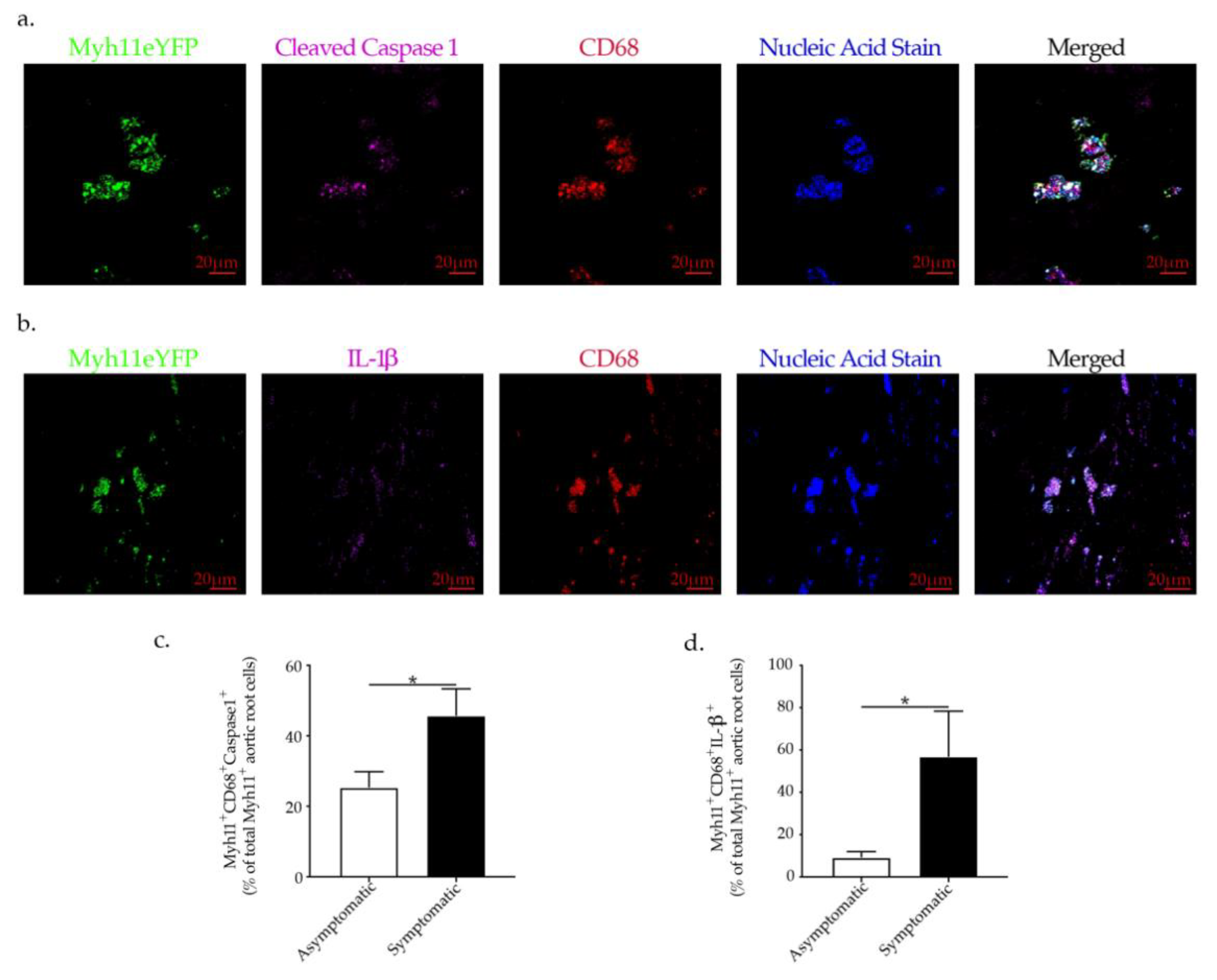
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Human Samples
4.3. Cells Isolation
4.4. Flow Cytometry Analysis of Vascular Smooth Muscle Cells Phenotypic Switch
4.5. Quantitative Real-Time PCR
4.6. Immunofluorescent Staining and Quantification
4.7. Caspase-1 Activity Assay and Pyroptosis/Caspase-1 Assay
4.8. IL-1β ELISA
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Şahin, B.; Ilgün, G. Risk factors of deaths related to cardiovascular diseases in World Health Organization (WHO) member countries. Health Soc. Care Community 2020, 30, 73–80. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.M.; Lee-Kim, V.S.; Libby, P. The March of Monocytes in Atherosclerosis. Circ. Res. 2020, 126, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Woollard, K.J.; Geissmann, F. Monocytes in atherosclerosis: Subsets and functions. Nat. Rev. Cardiol. 2010, 7, 77–86. [Google Scholar] [CrossRef]
- Jacinto, T.A.; Meireles, G.S.; Dias, A.T.; Aires, R.; Porto, M.L.; Gava, A.L.; Vasquez, E.C.; Pereira, T.M.C.; Campagnaro, B.P.; Meyrelles, S.S. Increased ROS production and DNA damage in monocytes are biomarkers of aging and atherosclerosis. Biol. Res. 2018, 51, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined Inhibition of CCL2, CX3CR1, and CCR5 Abrogates Ly6C hi and Ly6C lo Monocytosis and Almost Abolishes Atherosclerosis in Hypercholesterolemic Mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Fu, J. Novel Insights into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Hear. Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.J.; Auger, K.R.; Libby, P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J. Exp. Med. 1987, 165, 1316–1331. [Google Scholar] [CrossRef]
- Sahni, A.; Sahni, S.K.; Francis, C.W. Endothelial Cell Activation by IL-1β in the Presence of Fibrinogen Requires αVβArter. Thromb. Vasc. Biol. 2005, 25, 2222–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotzius, P.; Thams, S.; Soehnlein, O.; Kenne, E.; Tseng, C.-N.; Björkström, N.; Malmberg, K.-J.; Lindbom, L.; Eriksson, E.E. Distinct Infiltration of Neutrophils in Lesion Shoulders in ApoE−/− Mice. Am. J. Pathol. 2010, 177, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O. Multiple Roles for Neutrophils in Atherosclerosis. Circ. Res. 2012, 110, 875–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, D.; Baylis, R.A.; Durgin, B.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; Hilaire, C.S.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zheng, B.; Zhang, Y.; Zhang, X.-H.; Wang, C.; Yang, Z.; Sun, Y.; Wu, X.-L.; Wen, J.-K. KLF4 mediates the link between TGF-β1-induced gene transcription and H3 acetylation in vascular smooth muscle cells. FASEB J. 2015, 29, 4059–4070. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sinha, S.; McDonald, O.G.; Shang, Y.; Hoofnagle, M.H.; Owens, G.K. Kruppel-like Factor 4 Abrogates Myocardin-induced Activation of Smooth Muscle Gene Expression. J. Biol. Chem. 2005, 280, 9719–9727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Lei, S.; Zhao, B.; Wu, Y.; Su, W.; Liu, M.; Meng, Q.; Zhou, B.; Leng, Y.; Xia, Z.-Y. NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxidative Med. Cell. Longev. 2017, 2017, 9743280. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.; Robertson, A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Diaz, R.; Maggioni, A.P.; Pinto, F.J. The COLchicine Cardiovascular Outcomes Trial (COLCOT). In Circulation; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2019; Volume 140, p. E966. Available online: https://www.acc.org/latest-in-cardiology/clinical-trials/2019/11/15/17/23/colcot (accessed on 15 September 2021). (In English)
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Wirth, A.; Benyó, Z.; Lukasova, M.; Leutgeb, B.; Wettschureck, N.; Gorbey, S.; Örsy, P.; Horváth, B.; Maser-Gluth, C.; Greiner, E.; et al. G12-G13–LARG–mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008, 14, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Jessup, W.; Kritharides, L. Metabolism of oxidized LDL by macrophages. Curr. Opin. Lipidol. 2000, 11, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Grodzinsky, A.J.; Libby, P.; Clinton, S.K.; Lark, M.W.; Lee, R.T. Human Vascular Smooth Muscle Cell–Monocyte Interactions and Metalloproteinase Secretion in Culture. Arter. Thromb. Vasc. Biol. 1995, 15, 2284–2289. [Google Scholar] [CrossRef]
- Zhu, Y.; Hojo, Y.; Ikeda, U.; Takahashi, M.; Shimada, K. Interaction between Monocytes and Vascular Smooth Muscle Cells Enhances Matrix Metalloproteinase-1 Production. J. Cardiovasc. Pharmacol. 2000, 36, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Tintut, Y.; Patel, J.; Territo, M.; Saini, T.; Parhami, F.; Demer, L.L. Monocyte/Macrophage Regulation of Vascular Calcification In Vitro. Circulation 2002, 105, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Weinert, S.; Poitz, D.M.; Auffermann-Gretzinger, S.; Eger, L.; Herold, J.; Medunjanin, S.; Schmeisser, A.; Strasser, R.H.; Braun-Dullaeus, R.C. The lysosomal transfer of LDL/cholesterol from macrophages into vascular smooth muscle cells induces their phenotypic alteration. Cardiovasc. Res. 2012, 97, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, D.; Fitzsimmons, C.; Torzewski, J.; Bowyer, D. Inhibition of human arterial smooth muscle cell growth by human monocyte/macrophages: A co-culture study. Atherosclerosis 1999, 145, 157–165. [Google Scholar] [CrossRef]
- Boyle, J.J.; Bowyer, D.E.; Weissberg, P.L.; Bennett, M.R. Human Blood-Derived Macrophages Induce Apoptosis in Human Plaque-Derived Vascular Smooth Muscle Cells by Fas-Ligand/Fas Interactions. Arter. Thromb. Vasc. Biol. 2001, 21, 1402–1407. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, S.S.; Lopes, N.H.M.; Seshiah, P.N.; Wang, T.; Marsh, C.B.; Kereiakes, D.J.; Dong, C.; Goldschmidt-Clermont, P.J. Mac-1 and Fas activities are concurrently required for execution of smooth muscle cell death by M-CSF-stimulated macrophages. Cardiovasc. Res. 2003, 59, 723–733. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, R.; Tan, H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int. J. Biol. Sci. 2019, 15, 1345–1357. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.C.; Talib, S.; Figg, N.L.; Bennett, M.R. Vascular Smooth Muscle Cell Apoptosis Induces Interleukin-1–Directed Inflammation. Circ. Res. 2010, 106, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A.; Chusid, M.J.; Fauci, A.S.; Gallin, J.I.; Dale, D.C.; Wolff, S.M. Effect of prophylactic colchicine therapy on leukocyte function in patients with familial mediterranean fever. Arthritis Rheum. 1976, 19, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Cooper, L.; Torre-Amione, G.; Van Linthout, S. Management of Myocarditis-Related Cardiomyopathy in Adults. Circ. Res. 2019, 124, 1568–1583. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Celermajer, D.; Patel, S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef]
- Thompson, P.L.; Nidorf, S.M. Colchicine. Curr. Opin. Lipidol. 2018, 29, 467–473. [Google Scholar] [CrossRef]
- Montecucco, F.; Vuilleumier, N.; Pagano, S.; Lenglet, S.; Bertolotto, M.B.; Braunersreuther, V.; Pelli, G.; Kovari, E.; Pane, B.; Spinella, G.; et al. Anti-Apolipoprotein A-1 auto-antibodies are active mediators of atherosclerotic plaque vulnerability. Eur. Hear. J. 2011, 32, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Braunersreuther, V.; Zernecke, A.; Arnaud, C.; Liehn, E.A.; Steffens, S.; Shagdarsuren, E.; Bidzhekov, K.; Burger, F.; Pelli, G.; Luckow, B.; et al. Ccr5 But Not Ccr1 Deficiency Reduces Development of Diet-Induced Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2007, 27, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Lenglet, S.; Gayet-Ageron, A.; Bertolotto, M.; Pelli, G.; Palombo, D.; Pane, B.; Spinella, G.; Steffens, S.; Raffaghello, L.; et al. Systemic and Intraplaque Mediators of Inflammation Are Increased in Patients Symptomatic for Ischemic Stroke. Stroke 2010, 41, 1394–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecucco, F.; Matias, I.; Lenglet, S.; Petrosino, S.; Burger, F.; Pelli, G.; Braunersreuther, V.; Mach, F.; Steffens, S.; Di Marzo, V. Regulation and possible role of endocannabinoids and related mediators in hypercholesterolemic mice with atherosclerosis. Atherosclerosis 2009, 205, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Halliday, A.; MRC Asymptomatic Carotid Surgery Trial (ACST) Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: Randomised controlled trial. Lancet 2004, 363, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- European Carotid Surgery Trialists’ Collaborative Group. Randomised trial of endarterectomy for recently symptomatic carotid stenosis: Final results of the MRC European Carotid Surgery Trial (ECST). Lancet 1998, 351, 1379–1387. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9593407 (accessed on 9 October 2021). [CrossRef]
- Wang, K.; Li, Y.; Luo, C.; Chen, Y. Dynamic AFM detection of the oxidation-induced changes in size, stiffness, and stickiness of low-density lipoprotein. J. Nanobiotechnol. 2020, 18, 167. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burger, F.; Baptista, D.; Roth, A.; da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 340. https://doi.org/10.3390/ijms23010340
Burger F, Baptista D, Roth A, da Silva RF, Montecucco F, Mach F, Brandt KJ, Miteva K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(1):340. https://doi.org/10.3390/ijms23010340
Chicago/Turabian StyleBurger, Fabienne, Daniela Baptista, Aline Roth, Rafaela Fernandes da Silva, Fabrizio Montecucco, François Mach, Karim J. Brandt, and Kapka Miteva. 2022. "NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis" International Journal of Molecular Sciences 23, no. 1: 340. https://doi.org/10.3390/ijms23010340
APA StyleBurger, F., Baptista, D., Roth, A., da Silva, R. F., Montecucco, F., Mach, F., Brandt, K. J., & Miteva, K. (2022). NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. International Journal of Molecular Sciences, 23(1), 340. https://doi.org/10.3390/ijms23010340