
Integrative Study of the Structural and Dynamical Properties of a KirBac3.1 Mutant: Functional Implication of a Highly Conserved Tryptophan in the Transmembrane Domain

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
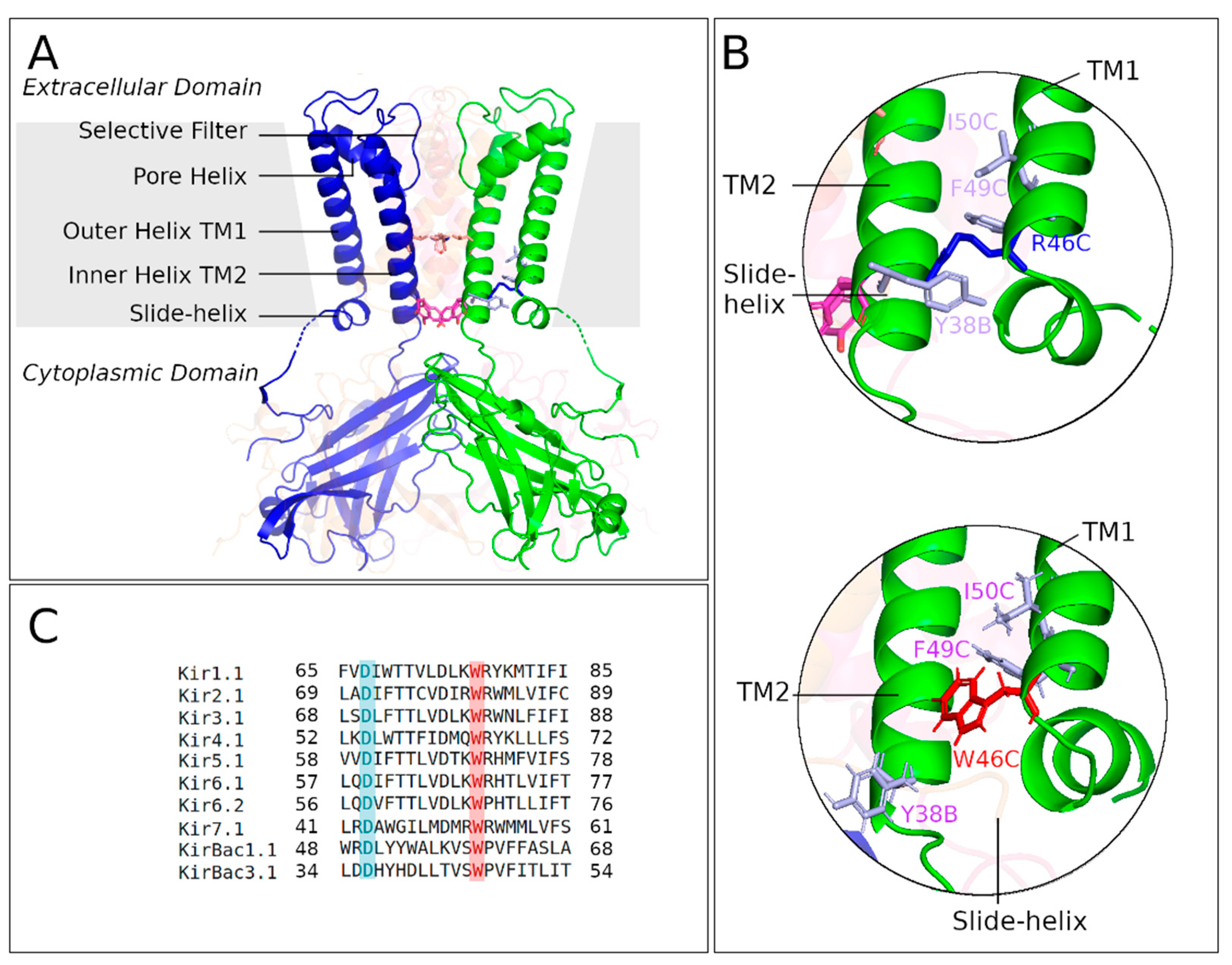
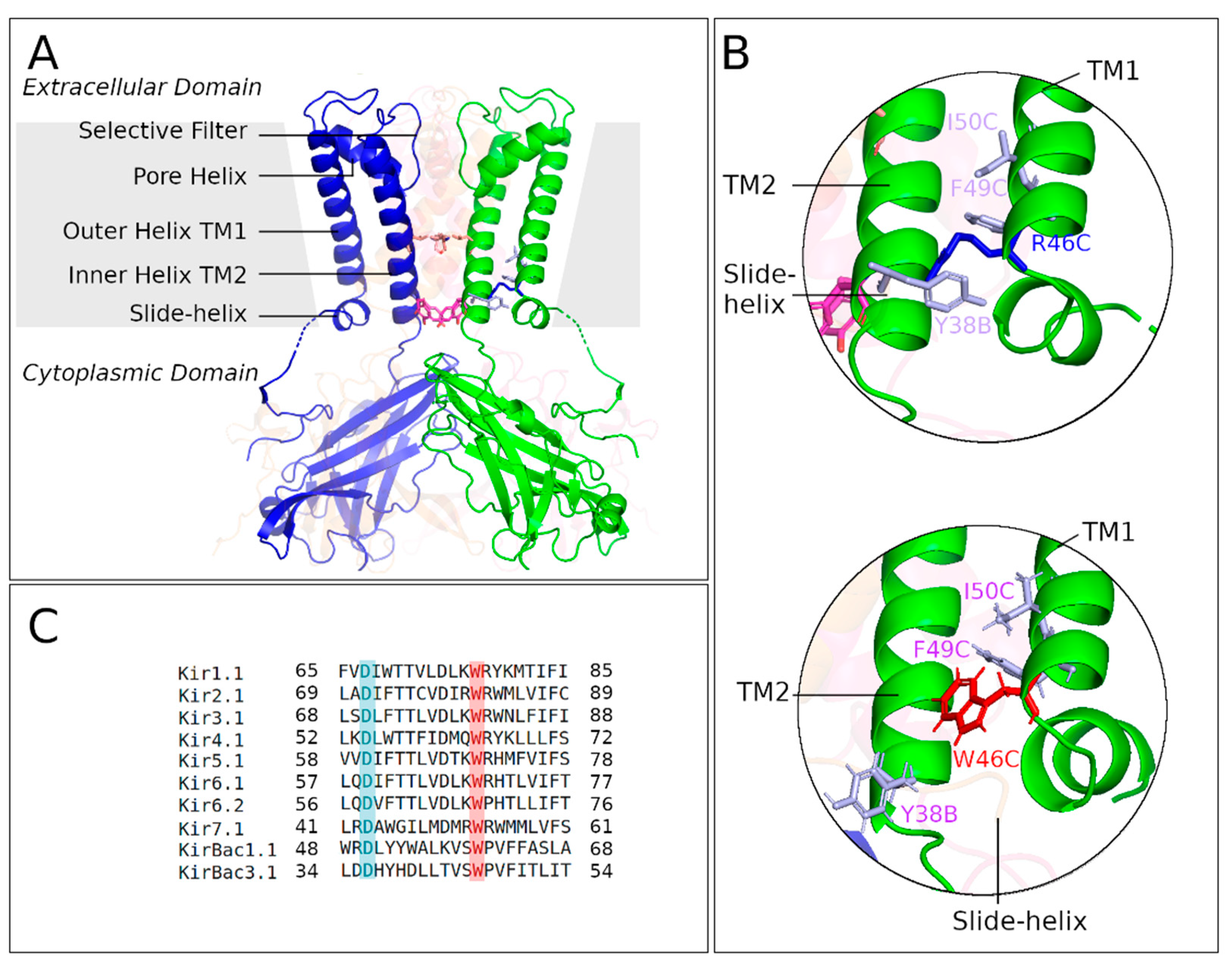
2.1. Crystal Structure of KirBac3.1 W46R
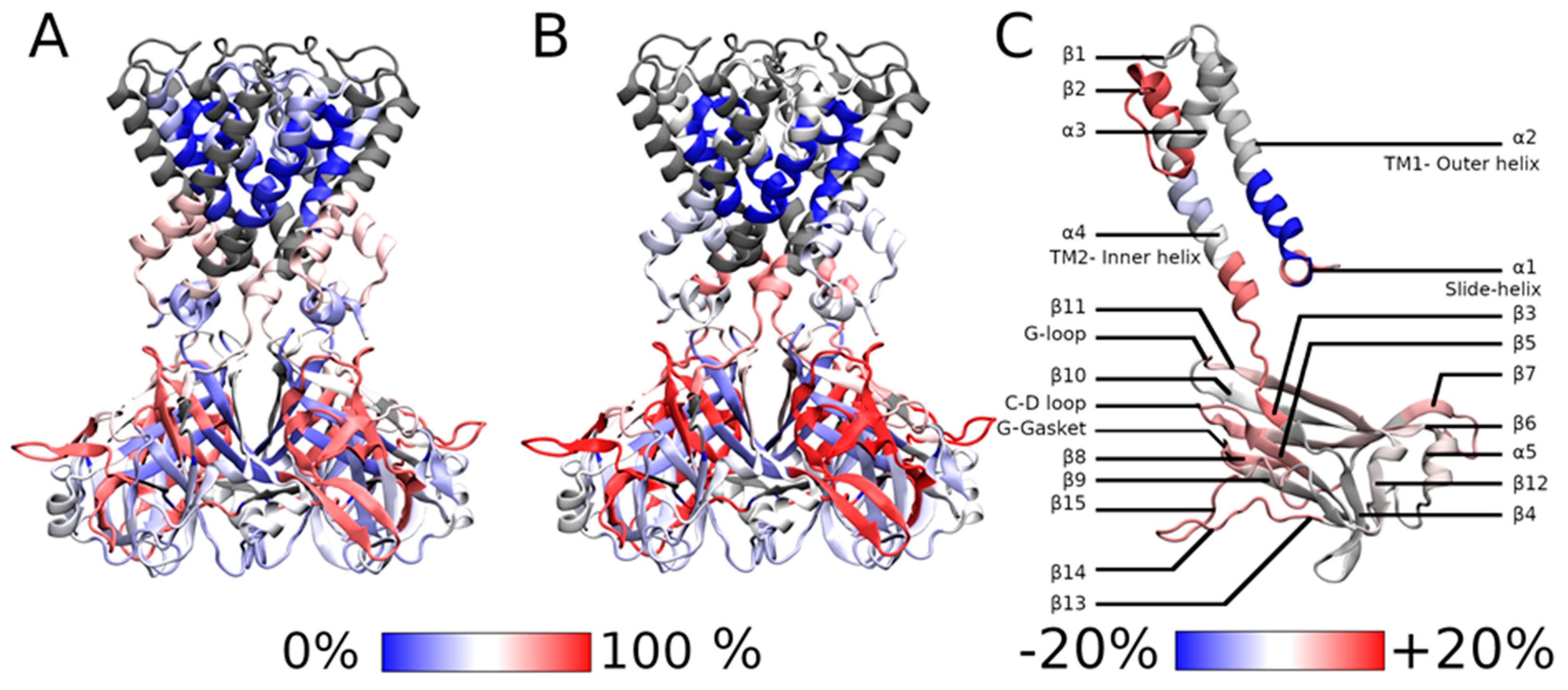
2.2. HDX-MS Measurements
2.3. Theoretical Results on KirBac3.1 W46R
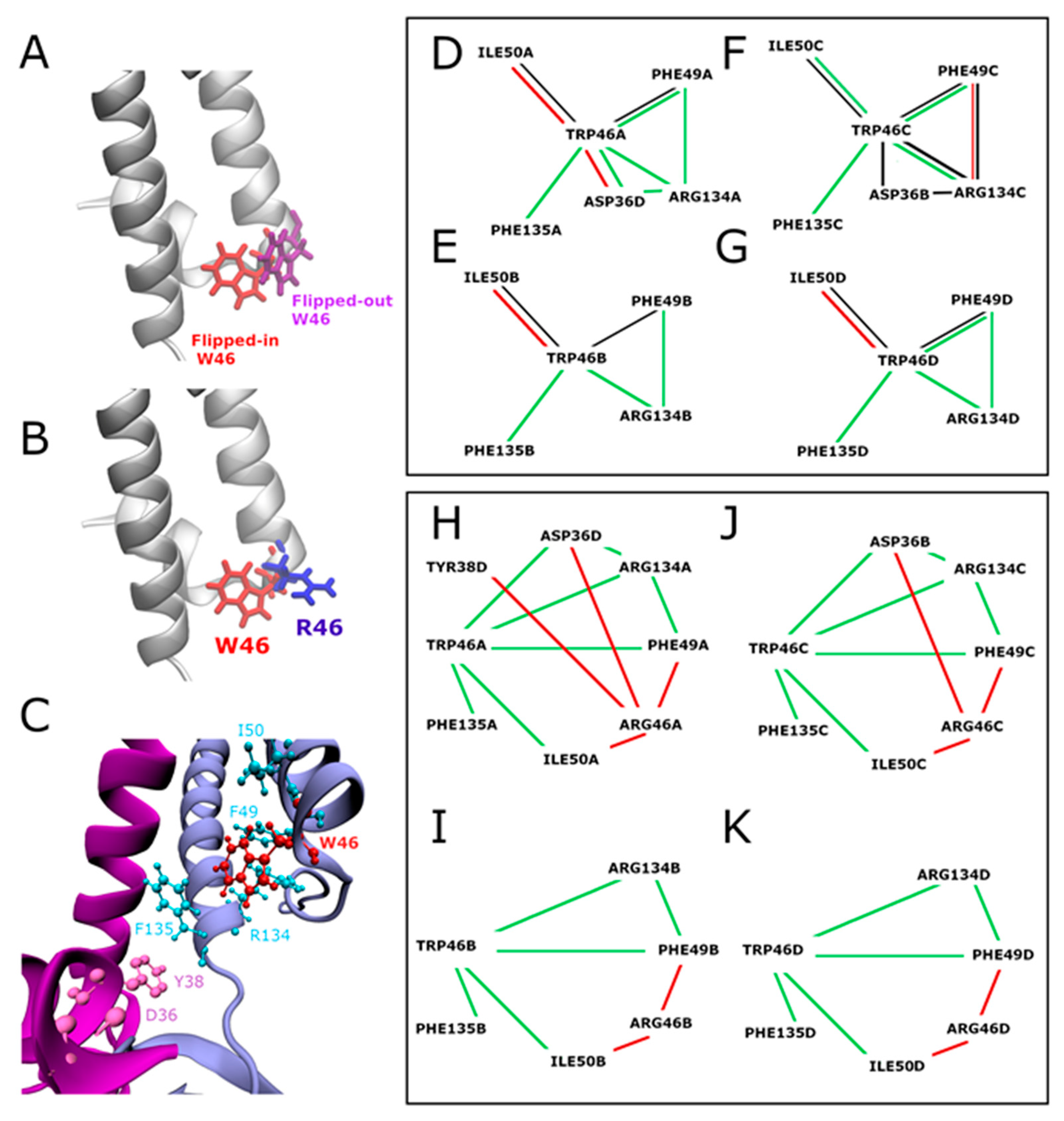
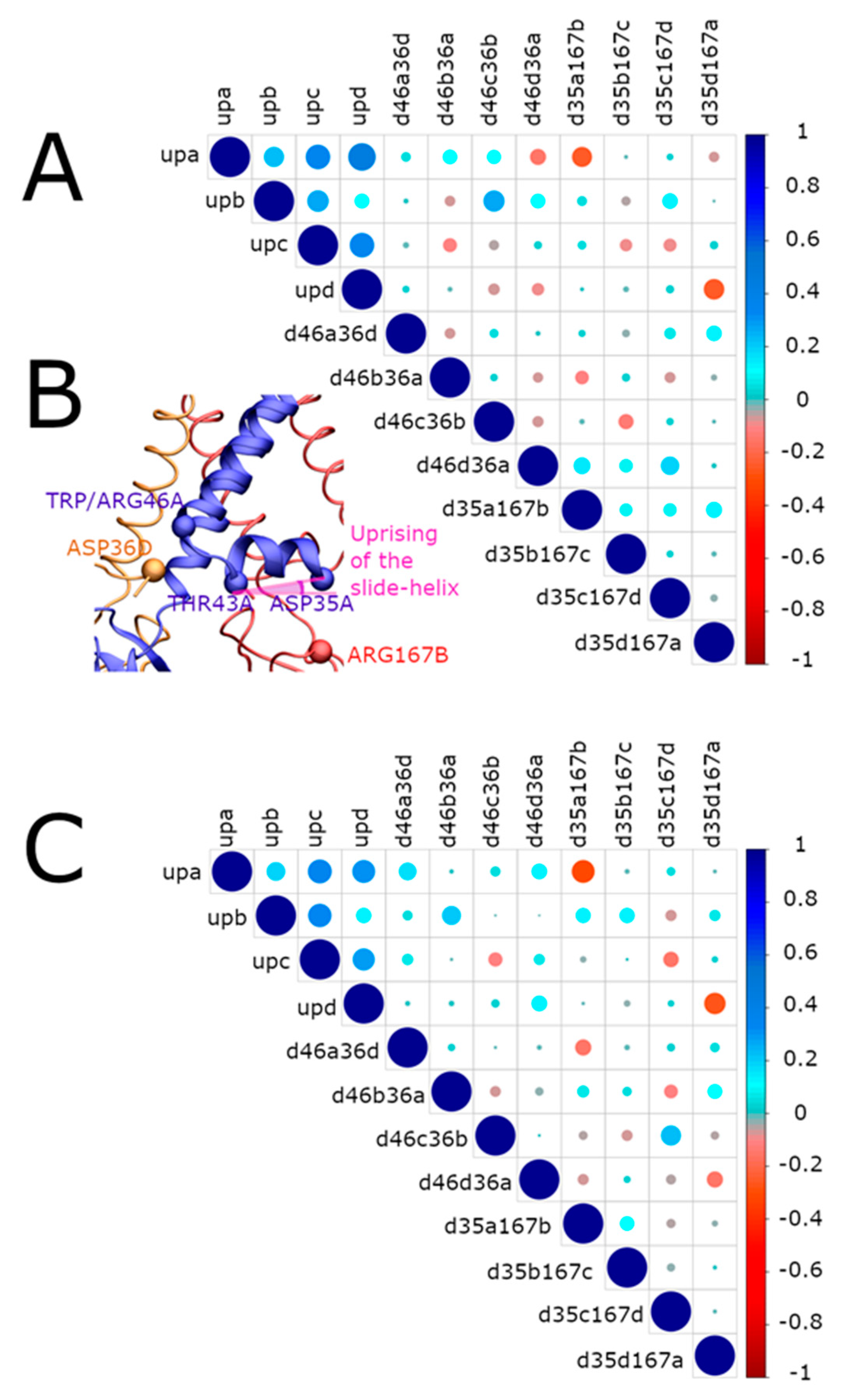
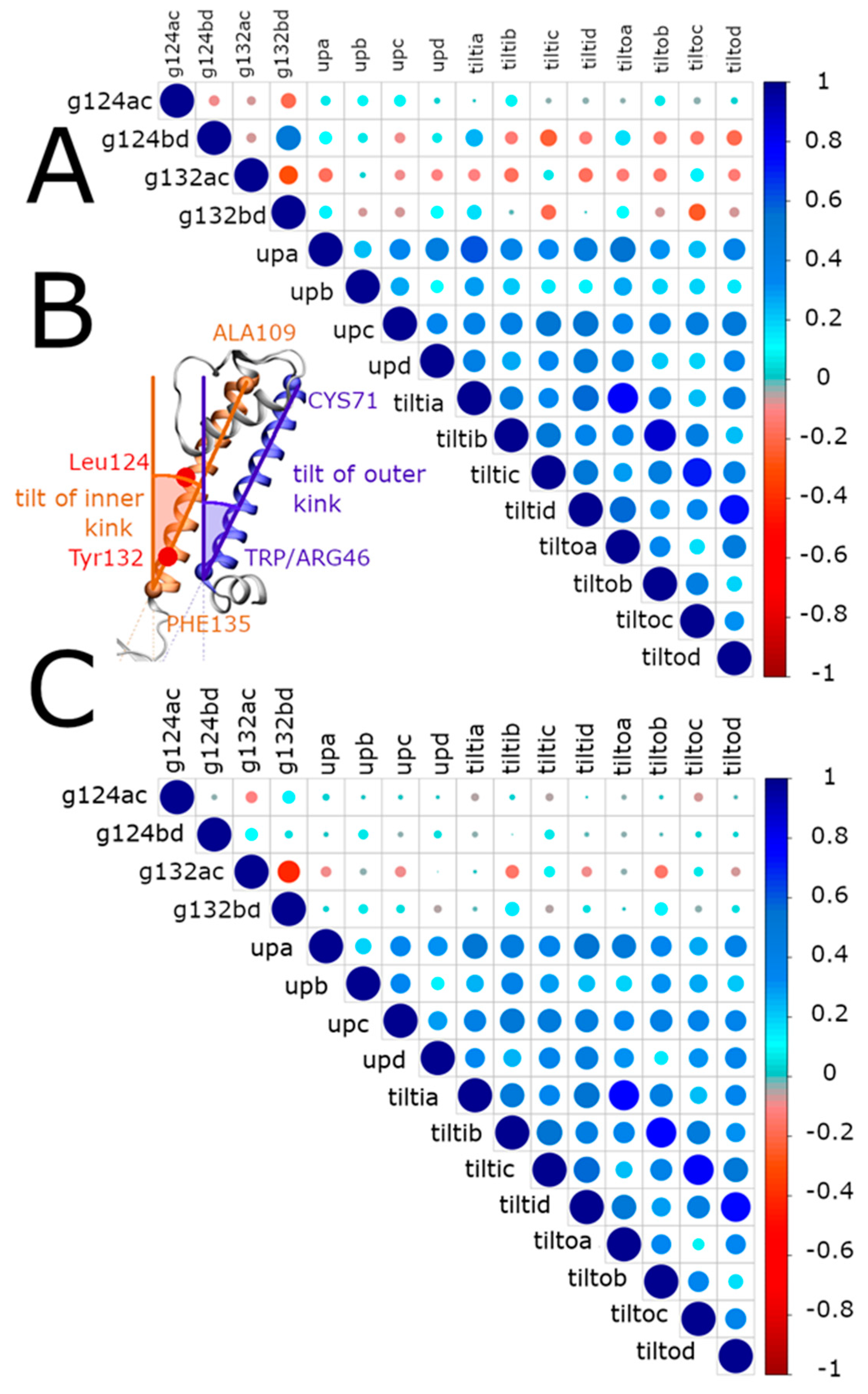
2.3.1. Interaction Networks
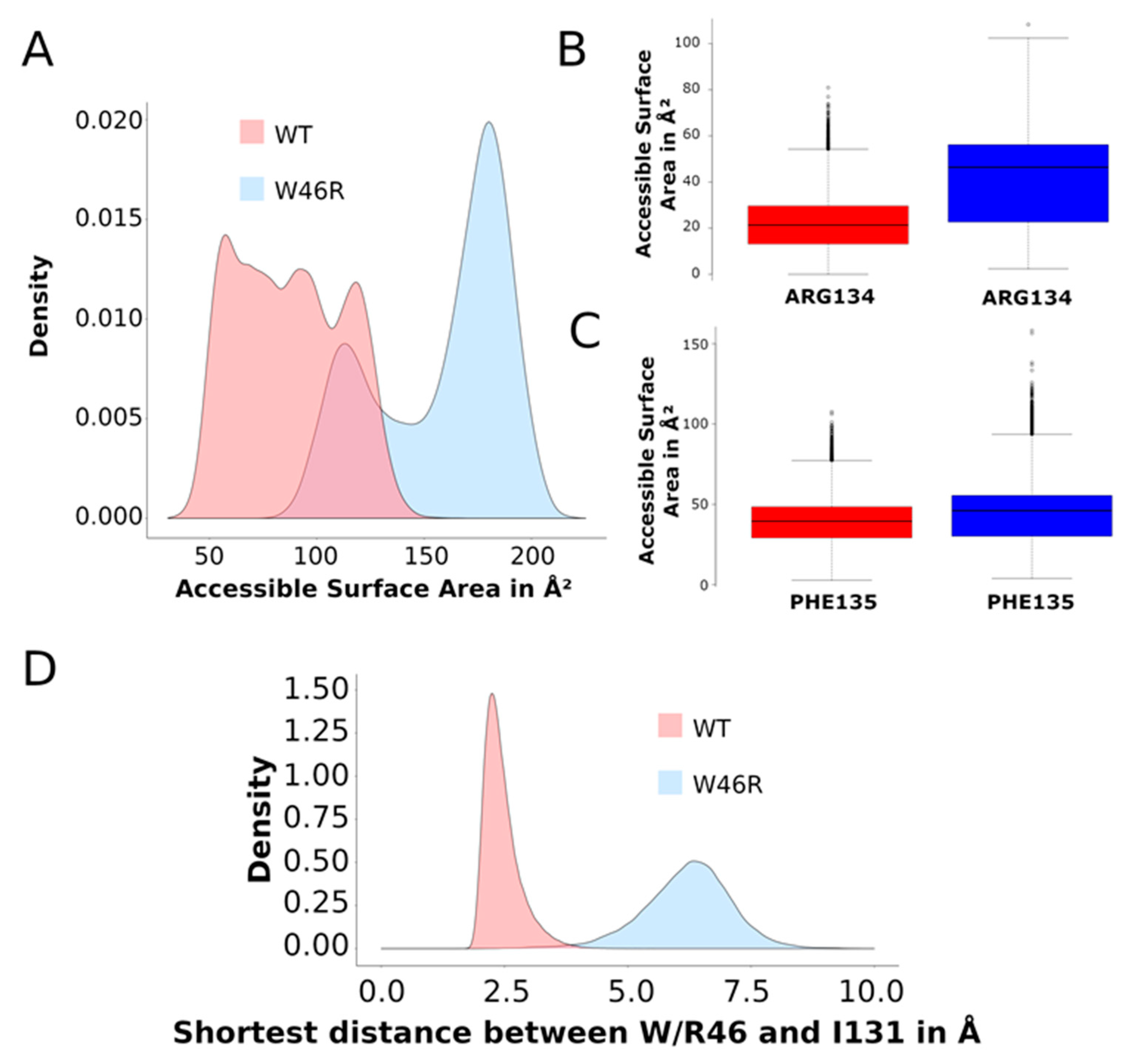
2.3.2. Contacts between Residues
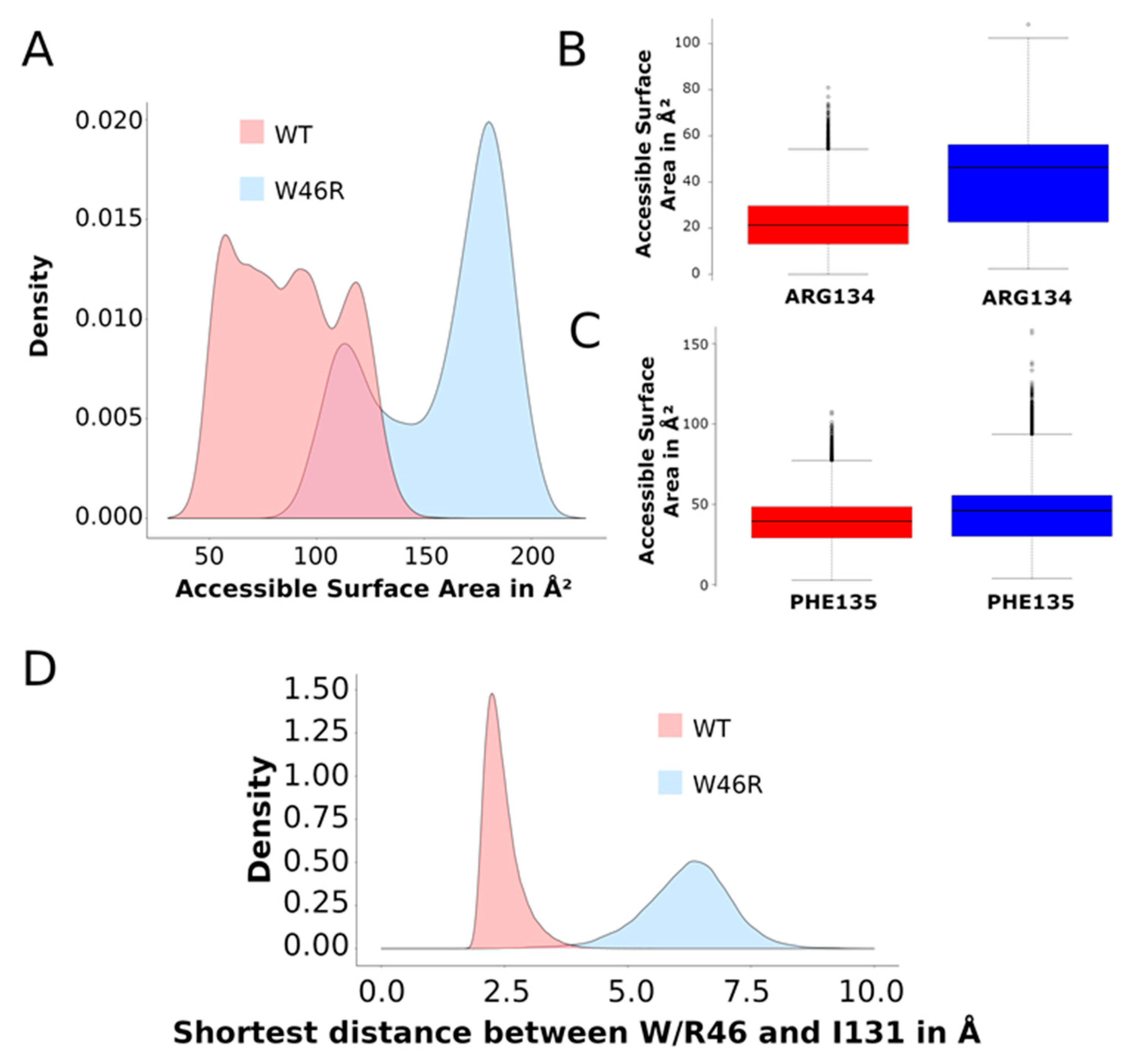
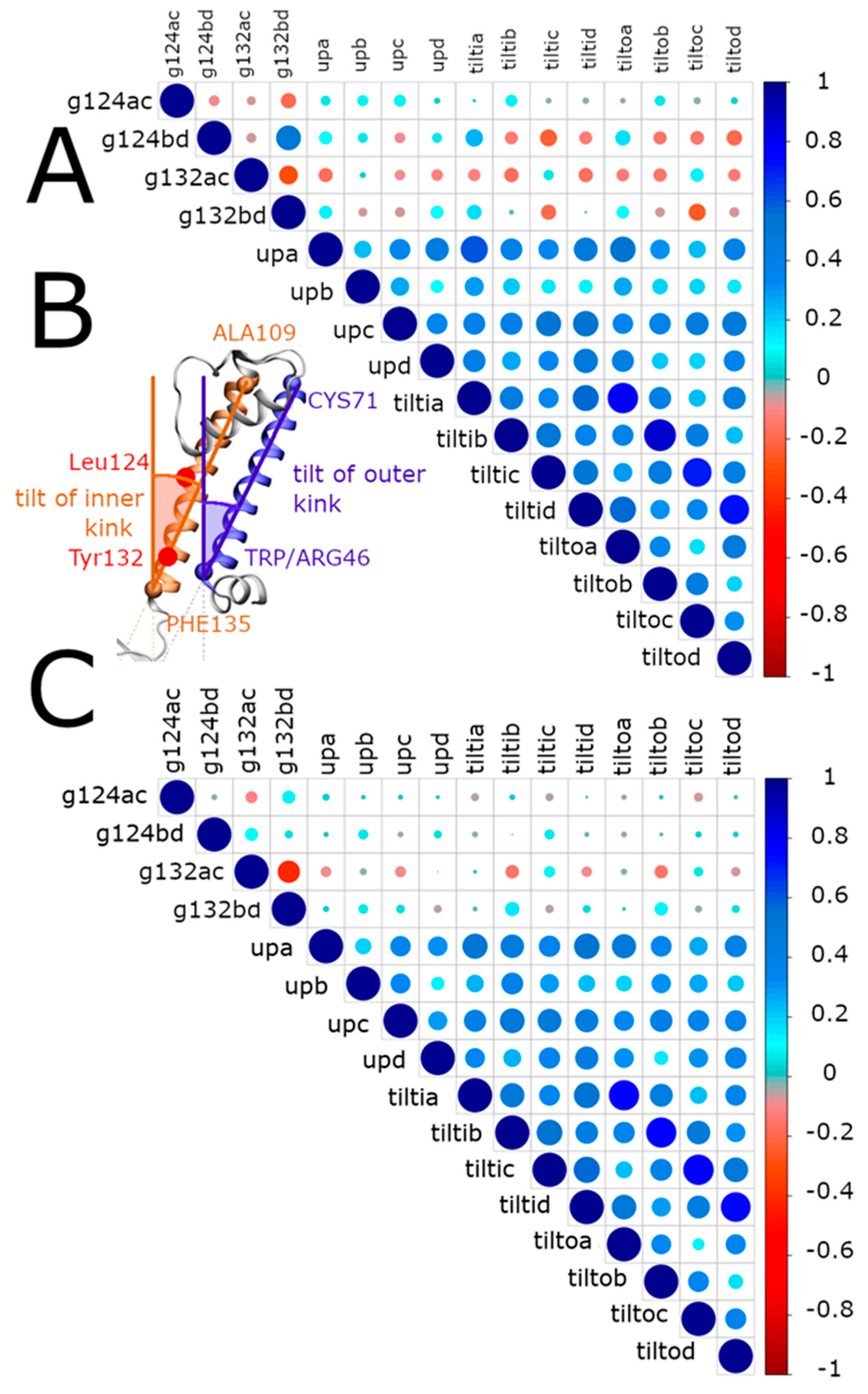
- Accessible surface of the residues W46 and R46
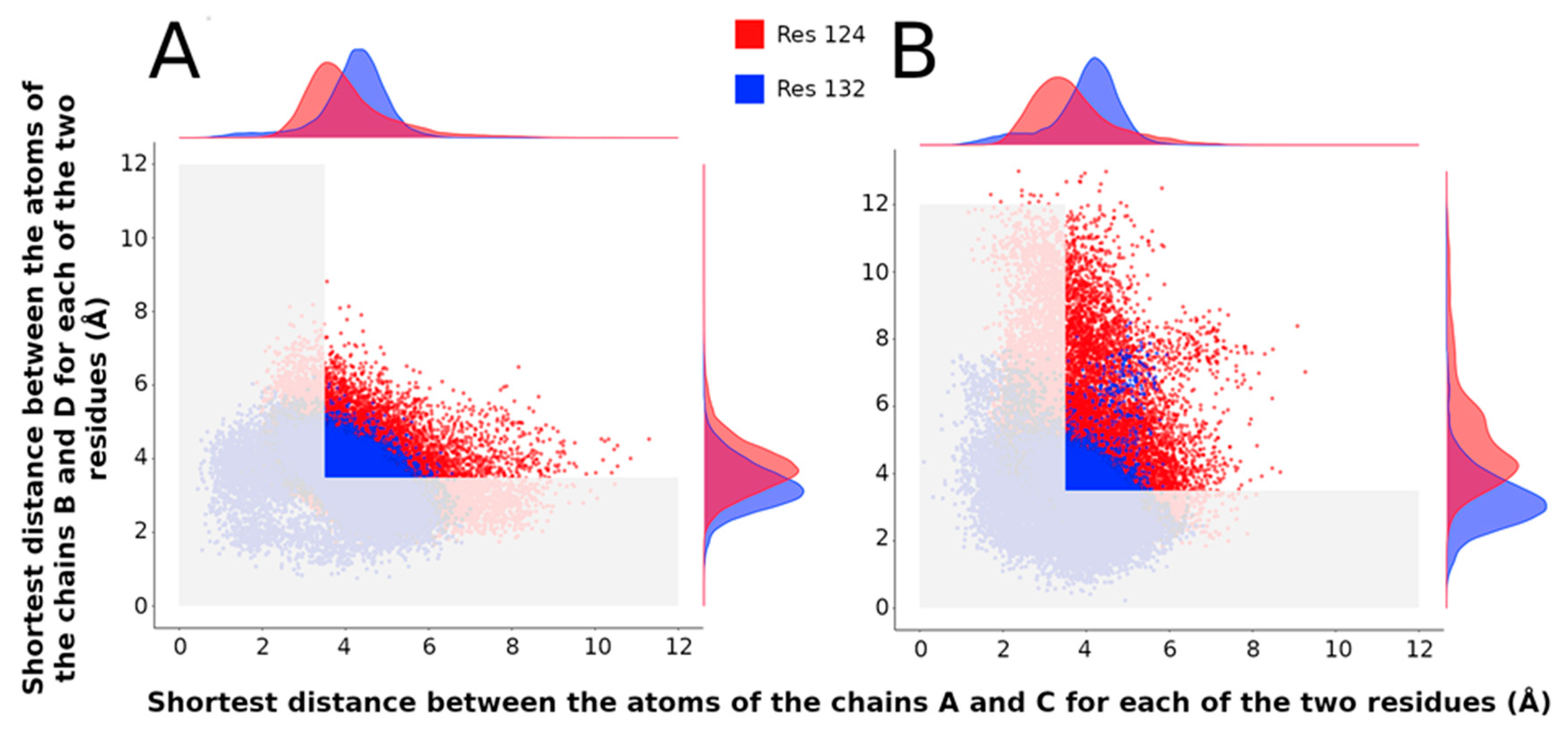
- Accessible surface of the residues R134 and F135 and contacts with I131
2.3.3. Impact of the Mutation on the Gating
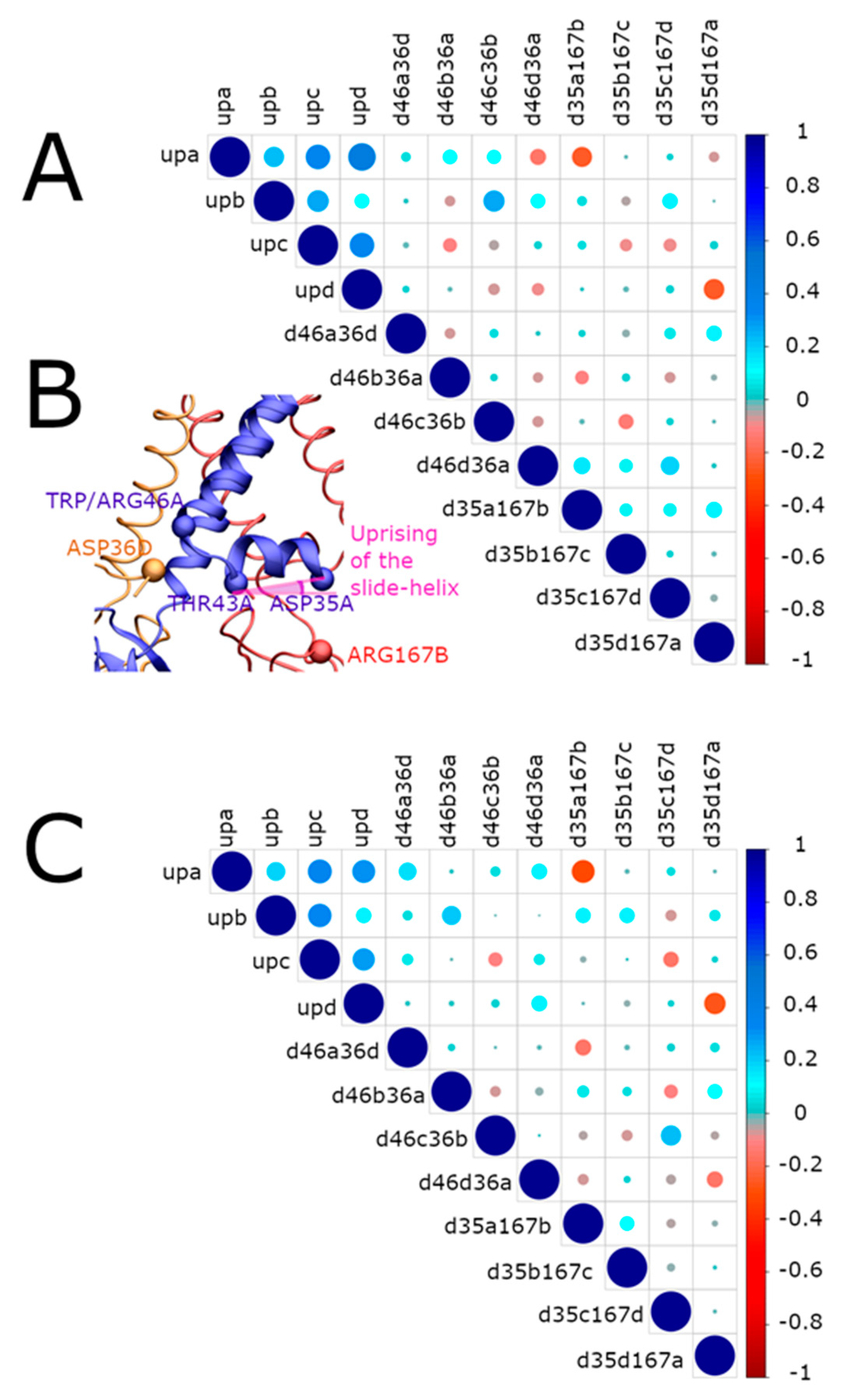
- Upward movement of the slide helix
- Movement of the cytoplasmic domain
- Movement of the transmembrane helices
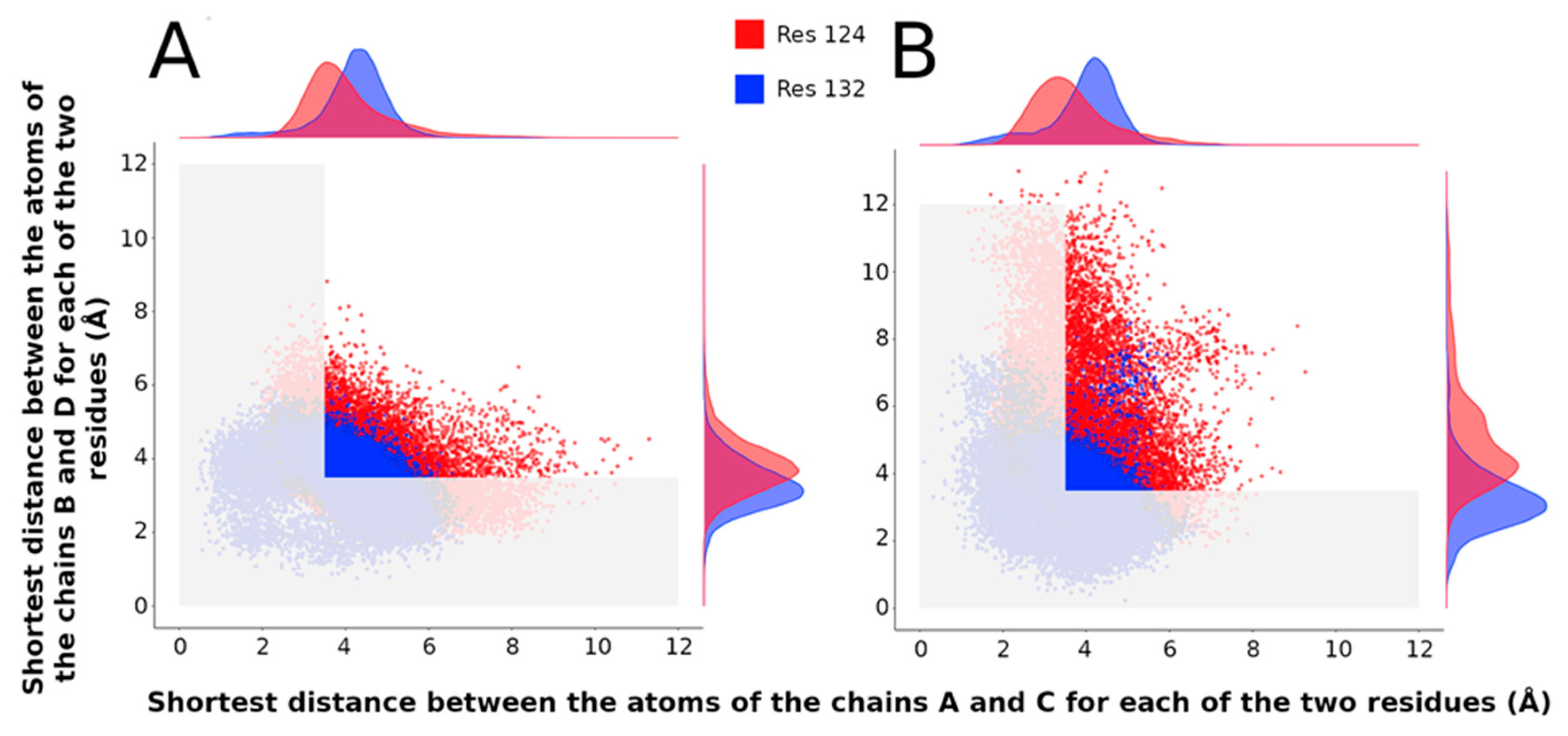
2.3.4. Population of Open and Closed States Corresponding to the MDeNM Relaxed Structures
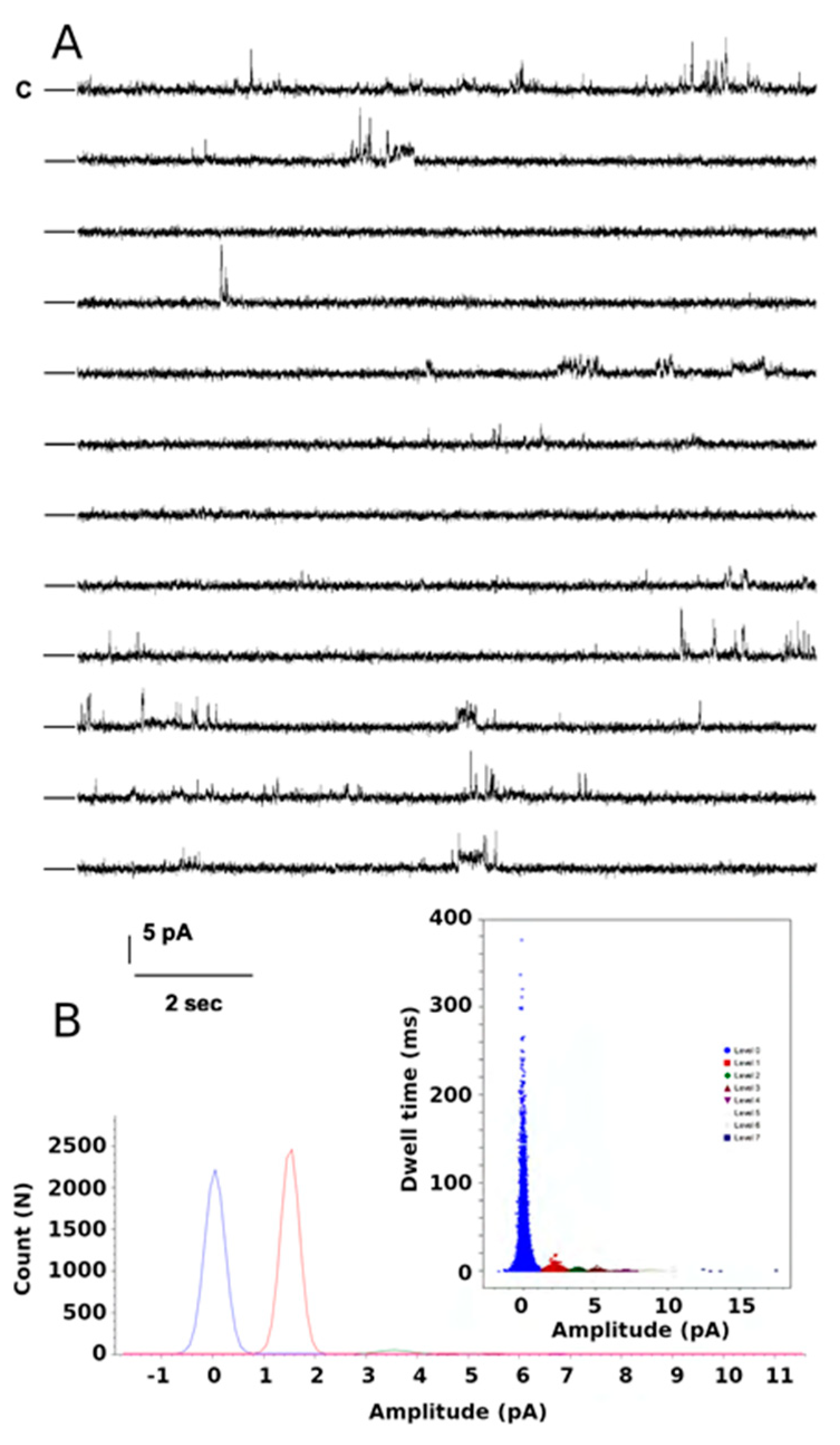
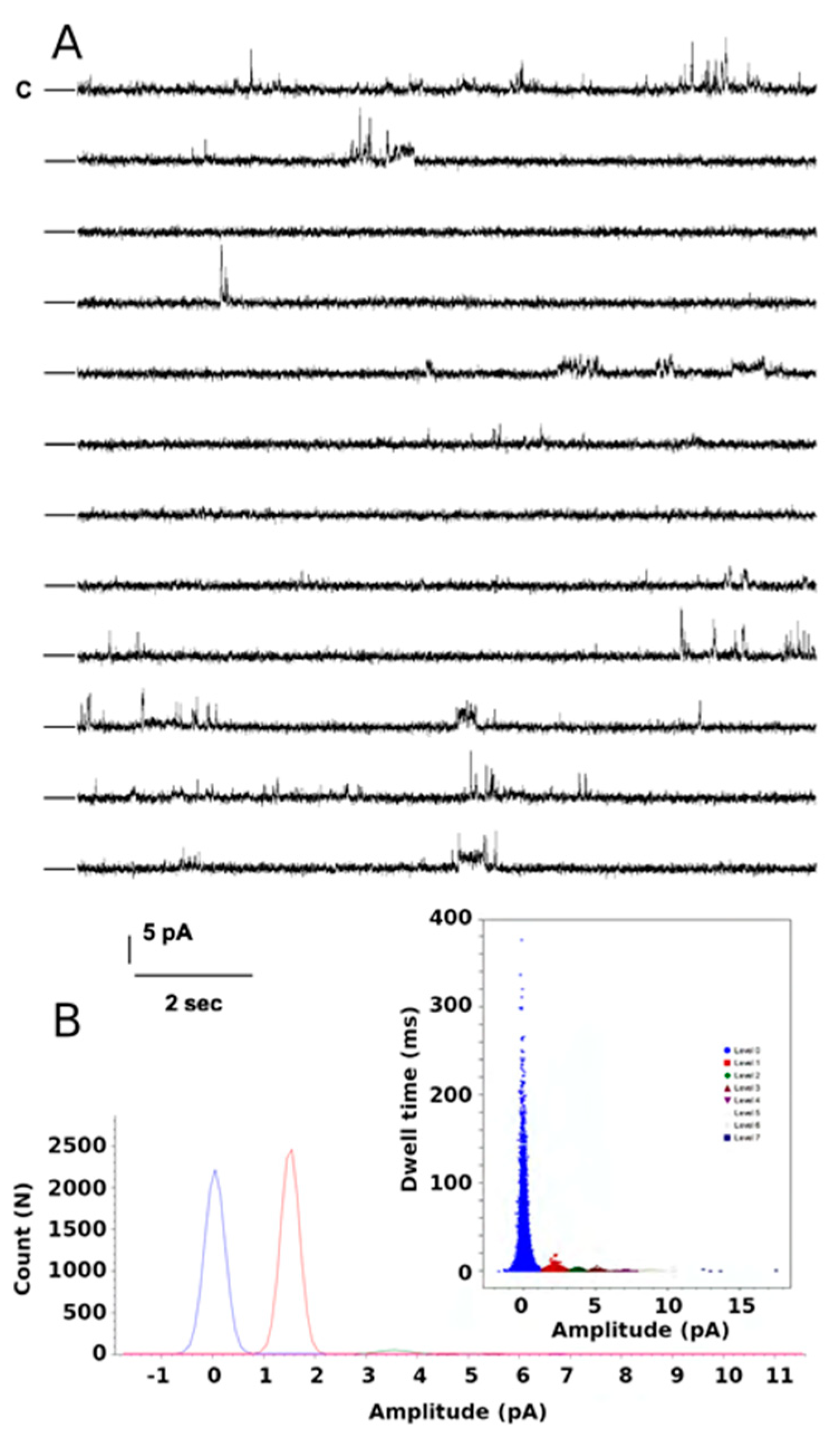
2.4. Functional Studies of the W46R Protein
3. Discussion
3.1. Characteristics of Mammalian Kir6.2 Channel
3.2. KirBac3.1 W46R: A Good Model for Studying Analogous W68R Mutation in Mammalian Kir6.2
3.3. Suggestions to Kir6.2 by Analogy
4. Materials and Methods
4.1. Experimental Data
4.1.1. Protein Expression and Purification
4.1.2. Protein Crystallization
- Data collection and structure determination
4.1.3. Pepsin Digestion, Hydrogen/Deuterium Exchange Approach Coupled to Mass Spectrometry (HDX-MS) and HPLC
4.1.4. Functional Studies of the KirBac W46R Protein in Black Lipid Membrane
4.2. Theoretical Data
4.2.1. Modeling and Dynamics
4.2.2. Normal Modes
4.2.3. Molecular Dynamics Using Excited Normal Modes
- Relaxation of the excited MDeNM structures
4.2.4. Interaction Networks
4.2.5. Structural Determinants
- Upward angle of the slide helices
- Tilt angle of the transmembrane helices
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aguilar-Bryan, L.; Bryan, J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 1999, 20, 101–135. [Google Scholar] [CrossRef]
- Antcliff, J.; Haider, S.; Proks, P.; Sansom, M.; Ashcroft, F.M. Functional analysis of a structural model of the ATP-binding site of the K-ATP channel Kir6.2 subunit. EMBO J. 2005, 24, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Nichols, C.G.; Singh, G.K.; Grange, D.K. K-ATP Channels and Cardiovascular Disease Suddenly a Syndrome. Circ. Res. 2013, 112, 1059–1072. [Google Scholar] [CrossRef] [Green Version]
- Nichols, C. K-ATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef]
- Vedovato, N.; Ashcroft, F.M.; Puljung, M.C. The Nucleotide-Binding Sites of SUR1: A Mechanistic Model. Biophys. J. 2015, 109, 2452–2460. [Google Scholar] [CrossRef] [Green Version]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P.J.; Ashcroft, F.M. New insights into KATP channel gene mutations and neonatal diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M. ATP-sensitive K+ channels and disease: From molecule to malady. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, E880–E889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babenko, A.P.; Polak, M.; Cave, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006, 355, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.; Pearson, E.; Antcliff, J.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.; Molnes, J.; et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 2004, 350, 1838–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattersley, A.; Ashcroft, F. Activating mutations in Kir6.2 and neonatal diabetes—New clinical syndromes, new scientific insights, and new therapy. Diabetes 2005, 54, 2503–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proks, P.; Antcliff, J.; Gloyn, A.; Hattersley, A.T.; Ashcroft, F.M. Molecular basis of Kir6.2 mutations causing neonatal diabetes and neonatal diabetes with neurological features. Biophys. J. 2005, 88, 181A. [Google Scholar]
- Ashcroft, F. Adenosine 5’-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 1988, 11, 97–118. [Google Scholar] [CrossRef] [PubMed]
- Maennikkoe, R.; Stansfeld, P.J.; Ashcroft, A.S.; Hattersley, A.T.; Sansom, M.S.P.; Ellard, S.; Ashcroft, F.M. A conserved tryptophan at the membrane-water interface acts as a gatekeeper for Kir6.2/SUR1 channels and causes neonatal diabetes when mutated. J. Physiol. 2011, 589, 3071–3083. [Google Scholar] [CrossRef]
- Clarke, O.B.; Caputo, A.; Hills, A.P.; Vandenberg, J.I.; Smith, B.J.; Gulbis, J.M. Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir Potassium Channels. Cell 2010, 141, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Forest, E.; Man, P. Conformational Dynamics and Interactions of Membrane Proteins by Hydrogen/Deuterium Mass Spectrometry. In Heterologous Expression of Membrane Proteins: Methods and Protocoles, 2nd ed.; MusVeteau, I., Ed.; Humana Press: New York, NY, USA, 2016; pp. 269–279. [Google Scholar]
- Oganesyan, I.; Lento, C.; Wilson, D.J. Contemporary hydrogen deuterium exchange mass spectrometry. Methods 2018, 144, 27–42. [Google Scholar] [CrossRef]
- Vadas, O.; Jenkins, M.L.; Dornan, G.L.; Burke, J.E. Using Hydrogen-Deuterium Exchange Mass Spectrometry to Examine Protein-Membrane Interactions. Methods Enzymol. 2017, 583, 143–172. [Google Scholar]
- Fagnen, C.; Bannwarth, L.; Oubella, I.; Forest, E.; De Zorzi, R.; De Araujo, A.; Mhoumadi, Y.; Bendahhou, S.; Perahia, D.; Vénien-Bryan, C. New Structural insights into Kir channel gating from molecular simulations, HDX-MS and functional studies. Sci. Rep. 2020, 10, 8392. [Google Scholar] [CrossRef]
- Giladi, M.; Almagor, L.; van Dijk, L.; Hiller, R.; Man, P.; Forest, E.; Khananshvili, D. Asymmetric Preorganization of Inverted Pair Residues in the Sodium-Calcium Exchanger. Sci. Rep. 2016, 6, 20753. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, S.; Domene, C.; Forest, E.; Jault, J.-M. Dynamics of a bacterial multidrug ABC transporter in the inward- and outward-facing conformations. Proc. Natl. Acad. Sci. USA 2012, 109, 10832–10836. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Bavro, V.N.; D’Mello, R.; Tucker, S.J.; Venien-Bryan, C.; Chance, M.R. Conformational changes during the gating of a potassium channel revealed by structural mass spectrometry. Struct. Lond Engl. 2010, 18, 839–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.G.S.; Batista, P.R.; Bisch, P.M.; Perahia, D. Exploring Free Energy Landscapes of Large Conformational Changes: Molecular Dynamics with Excited Normal Modes. J. Chem. Theory Comput. 2015, 11, 2755–2767. [Google Scholar] [CrossRef]
- Fagnen, C.; Bannwarth, L.; Zuniga, D.; Oubella, I.; De Zorzi, R.; Forest, E.; Scala, R.; Guilbault, S.; Bendahhou, S.; Perahia, D.; et al. Unexpected Gating Behaviour of an Engineered Potassium Channel Kir. Front. Mol. Biosci. 2021, 8, 691901. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M. Ion Channels and Diseases; Academic Press: New York, NY, USA, 2000. [Google Scholar]
- Ding, D.; Wang, M.; Wu, J.-X.; Kang, Y.; Chen, L. The Structural Basis for the Binding of Repaglinide to the Pancreatic K-ATP Channel. Cell Rep. 2019, 27, 1848–1857.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavro, V.N.; De Zorzi, R.; Schmidt, M.R.; Muniz, J.; Zubcevic, L.; Sansom, M.; Vénien-Bryan, C.; Tucker, S.J. Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat. Struct. Mol. Biol. 2012, 19, 158–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernsteiner, H.; Zangerl-Plessl, E.-M.; Chen, X.; Stary-Weinzinger, A. Conduction through a narrow inward-rectifier K+ channel pore. J. Gen. Physiol. 2019, 151, 1231–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Jin, T.; Gazgalis, D.; Cui, M.; Logothetis, D.E. On the mechanism of GIRK2 channel gating by phosphatidylinositol bisphosphate, sodium, and the Gβγ dimer. J. Biol. Chem. 2019, 294, 18934–18948. [Google Scholar] [CrossRef] [PubMed]
- Whorton, M.R.; MacKinnon, R. Crystal Structure of the Mammalian GIRK2 K(+) Channel and Gating Regulation by G Proteins, PIP(2), and Sodium. Cell 2011, 147, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Cooper, P.E.; McClenaghan, C.; Chen, X.; Weinzinger, A.; Nichols, C.G. Conserved functional consequences of disease-associated mutations in the slide helix of Kir6.1 and Kir6.2 subunits of the ATP-sensitive potassium channel. J. Biol. Chem. 2017, 292, 17387–17398. [Google Scholar] [CrossRef] [Green Version]
- Puljung, M.C. Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. J. Gen. Physiol. 2018, 150, 653–669. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.K.; Chen, J.; MacKinnon, R. Molecular structure of human KATP in complex with ATP and ADP. eLife 2017, 6, e32481. [Google Scholar] [CrossRef]
- Shimomura, K.; Hoerster, F.; de Wet, H.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Wolf, N.I.; Ashcroft, F.; Ebinger, F. A novel mutation causing DEND syndrome—A treatable channelopathy of pancreas and brain. Neurology 2007, 69, 1342–1349. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Girard, C.A.; Larkin, B.; Tammaro, P.; Flanagan, S.E.; Ellard, S.; Ashcroft, F.M. Functional analysis of two Kir6.2 (KCNJ11) mutations, K170T and E322K, causing neonatal diabetes. Diabetes Obes. Metab. 2007, 9, 46–55. [Google Scholar] [CrossRef]
- Drain, P.; Geng, X.; Li, L. Concerted gating mechanism underlying K-ATP channel inhibition by ATP. Biophys. J. 2004, 86, 2101–2112. [Google Scholar] [CrossRef] [Green Version]
- De Zorzi, R.; Nicholson, W.V.; Guigner, J.-M.; Erne-Brand, F.; Vénien-Bryan, C. Growth of large and highly ordered 2D crystals of a K+ channel, structural role of lipidic environment. Biophys. J. 2013, 105, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Weber, P.; Pissis, C.; Navaza, R.; Mechaly, A.E.; Saul, F.; Alzari, P.M.; Haouz, A. High-Throughput Crystallization Pipeline at the Crystallography Core Facility of the Institut Pasteur. Molecules 2019, 24, 4451. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Blanc, E.; Roversi, P.; Vonrhein, C.; Flensburg, C.; Lea, S.; Bricognea, G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. Sect. D Struct Biol. 2004, 60, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B.I.I.I.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Section D Struct. Biol. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Marshall, A. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 1998, 9, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.; Bruccoleri, R.; Olafson, B.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM—A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, M.H.M.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pK(a) Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Software news and updates—CHARNIM-GUI: A web-based g raphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouawad, L.; Perahia, D. Diagonalization in a mixed basis: A method to compute low-frequency normal modes for large macromolecules. Biopolymers 1993, 33, 599–611. [Google Scholar] [CrossRef]
- Piovesan, D.; Minervini, G.; Tosatto, S.C.E. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model 1996, 14, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |
| Space group | P21212 |
| Cell dimensions (Å) | 106.77, 113.98, 89.18 |
| Cell angles | 90° 90° 90° |
| Resolution (Å) | 48.34–2.80 |
| Completeness (%) | 99.6 |
| I/σ (I) | 19.67 (1.31 in the high-resolution shell) |
| Clashscore | 8 |
| Ramachandran outliers | 1.7% |
| Side-chain outliers | 9.3% |
| Refinement with REFMAC 5.8.0258 | |
| R, R free | 0.222, 0.287 |
| R free test set | 1372 reflections (5%) |
| Wilson B-factors (Å2) | 89.3 |
| Total number of atoms | 4341 |
| Average B-factors, all atoms (Å2) | 97.464 |
| RMS of Z score of bond lengths | 0.70 |
| RMS of Z score of bond angles | 0.89 |
| Channel States | Kirbac3.1 WT (%) (16) | KirBac3.1 W46R (%) |
|---|---|---|
| Fully open | 6.8 | 7.4 |
| Fully closed | 50.2 | 49.3 |
| Half open 1 (124 open, 132 closed) | 28.8 | 32.5 |
| Half open 2 (132 open, 124 closed) | 14.2 | 10.8 |
| Current recordings of KirBac3.1 in planar lipid bilayers | 9.9 (±1.3, n = 1803) | 7.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagnen, C.; Bannwarth, L.; Oubella, I.; Zuniga, D.; Haouz, A.; Forest, E.; Scala, R.; Bendahhou, S.; De Zorzi, R.; Perahia, D.; et al. Integrative Study of the Structural and Dynamical Properties of a KirBac3.1 Mutant: Functional Implication of a Highly Conserved Tryptophan in the Transmembrane Domain. Int. J. Mol. Sci. 2022, 23, 335. https://doi.org/10.3390/ijms23010335
Fagnen C, Bannwarth L, Oubella I, Zuniga D, Haouz A, Forest E, Scala R, Bendahhou S, De Zorzi R, Perahia D, et al. Integrative Study of the Structural and Dynamical Properties of a KirBac3.1 Mutant: Functional Implication of a Highly Conserved Tryptophan in the Transmembrane Domain. International Journal of Molecular Sciences. 2022; 23(1):335. https://doi.org/10.3390/ijms23010335
Chicago/Turabian StyleFagnen, Charline, Ludovic Bannwarth, Iman Oubella, Dania Zuniga, Ahmed Haouz, Eric Forest, Rosa Scala, Saïd Bendahhou, Rita De Zorzi, David Perahia, and et al. 2022. "Integrative Study of the Structural and Dynamical Properties of a KirBac3.1 Mutant: Functional Implication of a Highly Conserved Tryptophan in the Transmembrane Domain" International Journal of Molecular Sciences 23, no. 1: 335. https://doi.org/10.3390/ijms23010335
APA StyleFagnen, C., Bannwarth, L., Oubella, I., Zuniga, D., Haouz, A., Forest, E., Scala, R., Bendahhou, S., De Zorzi, R., Perahia, D., & Vénien-Bryan, C. (2022). Integrative Study of the Structural and Dynamical Properties of a KirBac3.1 Mutant: Functional Implication of a Highly Conserved Tryptophan in the Transmembrane Domain. International Journal of Molecular Sciences, 23(1), 335. https://doi.org/10.3390/ijms23010335