
Lipid-Induced Adaptations of the Pancreatic Beta-Cell to Glucotoxic Conditions Sustain Insulin Secretion


{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Glucose-Stimulated Insulin Secretion in a Physiological Context
1.2. Role of Fatty Acids in the Potentiation of GSIS
1.3. FFAR1 Signaling in GSIS
2. Glucose-Stimulated Insulin Secretion in a Pathophysiological Context
2.1. Pathophysiology of the β-Cell
2.2. Glucotoxicity
2.3. Lipotoxicity
2.4. Glucolipotoxicity
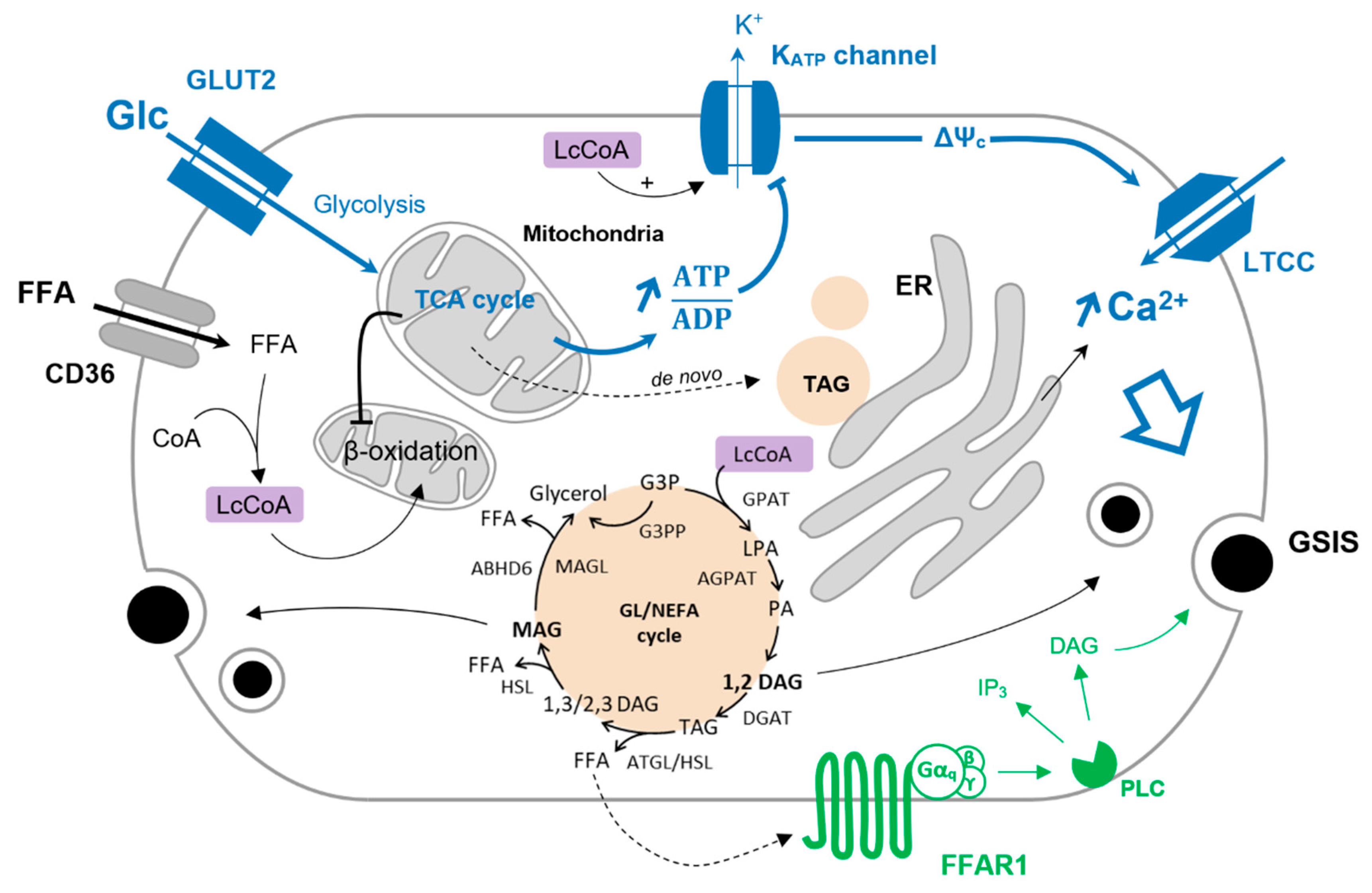
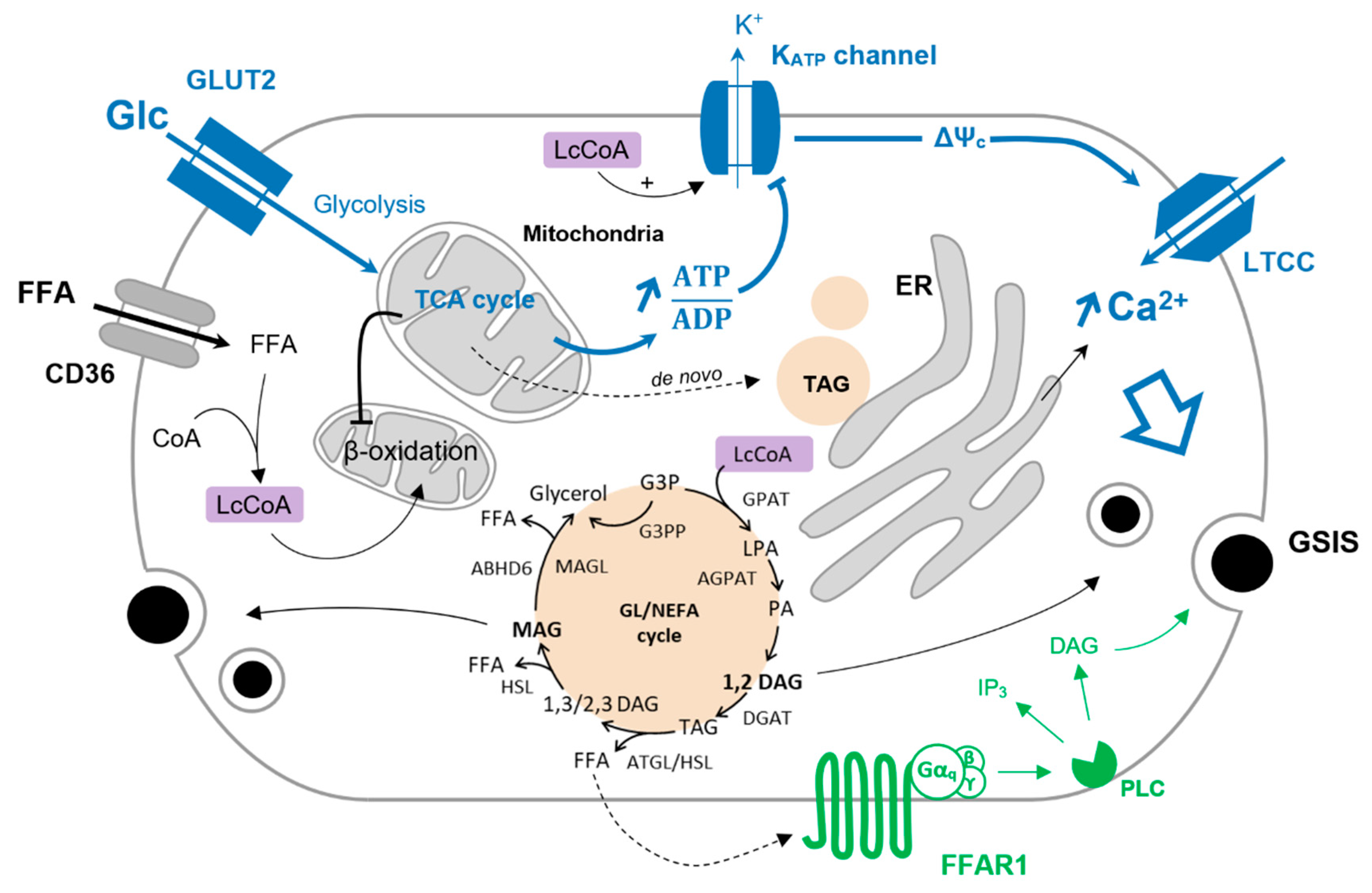
3. The Glycerolipid/NEFA Cycle
3.1. Functioning of the Glycerolipid/NEFA Cycle
3.2. The GL/NEFA Cycle in a Physiological Context
3.3. GL/NEFA Cycling in Type-2 Diabetes
4. Discussion
5. Conclusions
6. Materials and Methods
6.1. Reagents
6.2. Cell Culture and Treatments
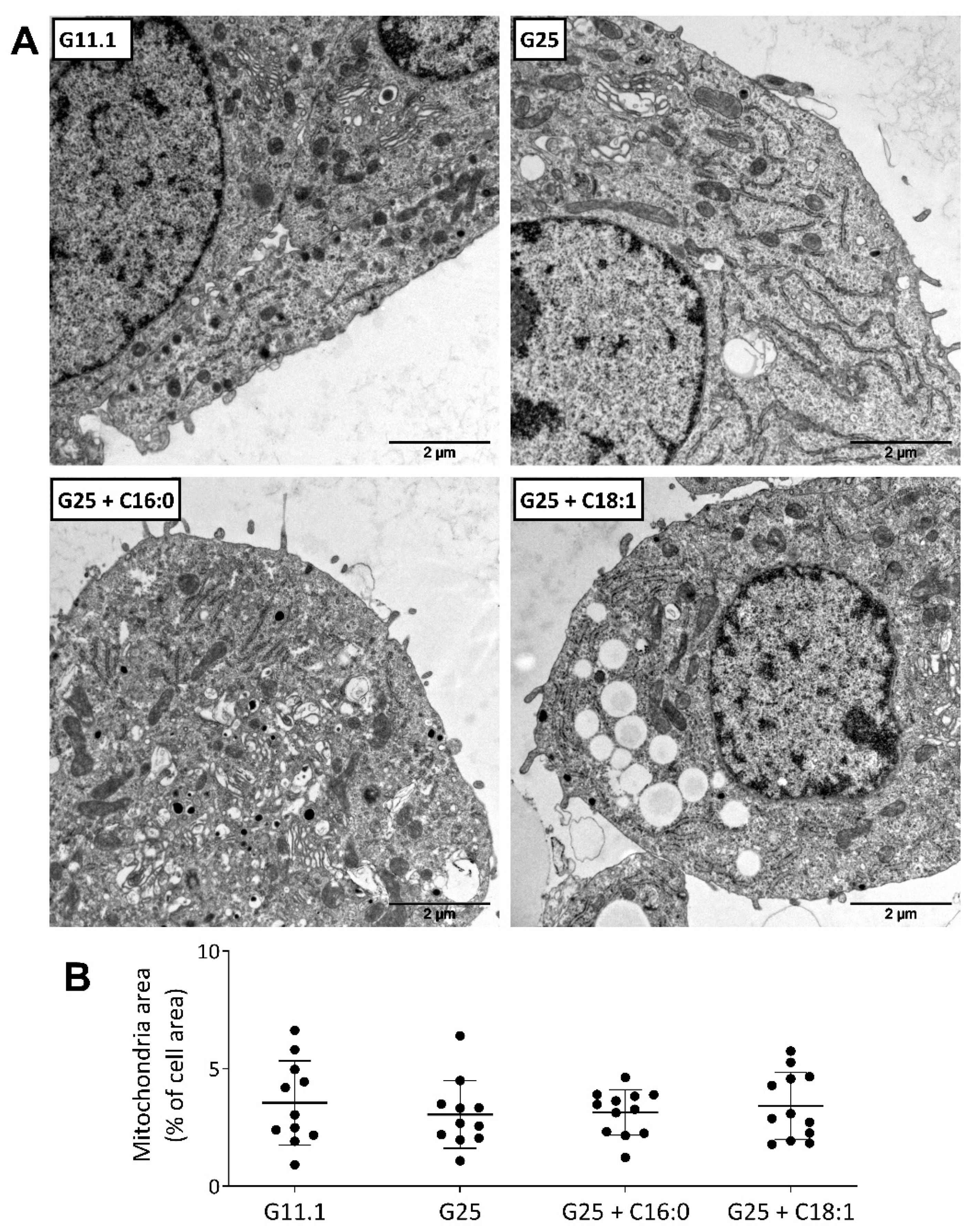
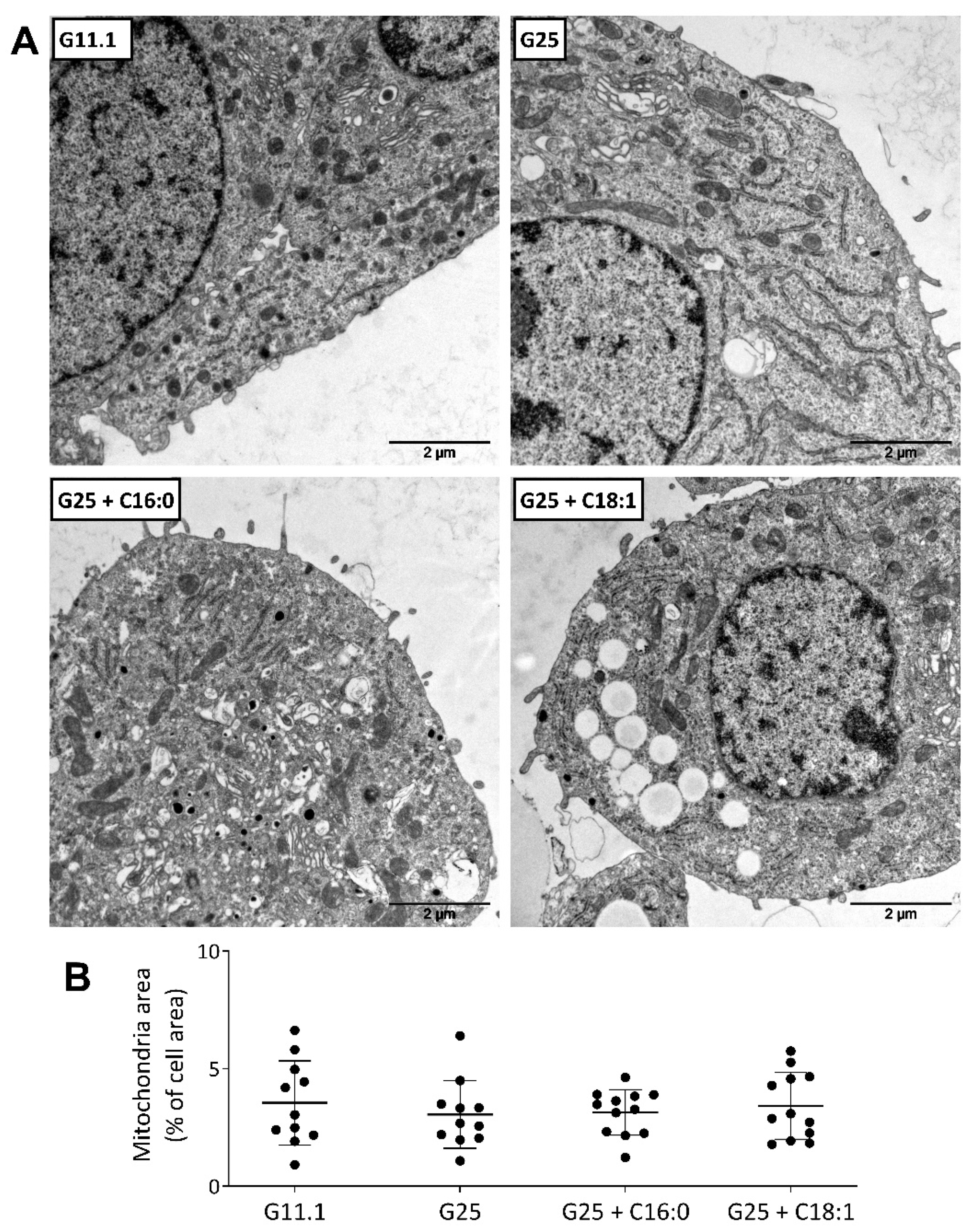
6.3. Electron Microscopy
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saltiel, A.R. Insulin Signaling in the Control of Glucose and Lipid Homeostasis. Handb. Exp. Pharmacol. 2016, 233, 51–71. [Google Scholar] [PubMed]
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorsman, P.; Renstrom, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matschinsky, F.M. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 1996, 45, 223–241. [Google Scholar] [CrossRef]
- Ashcroft, F.M. K(ATP) channels and insulin secretion: A key role in health and disease. Biochem. Soc. Trans. 2006, 34 Pt 2, 243–246. [Google Scholar] [CrossRef]
- Gembal, M.; Gilon, P.; Henquin, J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1992, 89, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- Kalwat, M.A.; Cobb, M.H. Mechanisms of the amplifying pathway of insulin secretion in the beta cell. Pharmacol. Ther. 2017, 179, 17–30. [Google Scholar] [CrossRef]
- Maechler, P. Mitochondrial function and insulin secretion. Mol. Cell. Endocrinol. 2013, 379, 12–18. [Google Scholar] [CrossRef]
- Bartley, C.; Brun, T.; Oberhauser, L.; Grimaldi, M.; Molica, F.; Kwak, B.R.; Bosco, D.; Chanson, M.; Maechler, P. Chronic fructose renders pancreatic beta-cells hyper-responsive to glucose-stimulated insulin secretion through extracellular ATP signaling. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E25–E41. [Google Scholar] [CrossRef] [Green Version]
- Ghislain, J.; Poitout, V. Targeting lipid GPCRs to treat type 2 diabetes mellitus—Progress and challenges. Nat. Rev. Endocrinol. 2021, 17, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Maechler, P. Glutamate pathways of the beta-cell and the control of insulin secretion. Diabetes Res. Clin. Pract. 2017, 131, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Egan, J.M. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol. Rev. 2008, 60, 470–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- McArthur, M.J.; Atshaves, B.P.; Frolov, A.; Foxworth, W.D.; Kier, A.B.; Schroeder, F. Cellular uptake and intracellular trafficking of long chain fatty acids. J. Lipid. Res. 1999, 40, 1371–1383. [Google Scholar] [CrossRef]
- Nolan, C.J.; Madiraju, M.S.R.; Delghingaro-Augusto, V.; Peyot, M.L.; Prentki, M. Fatty Acid Signaling in the -Cell and Insulin Secretion. Diabetes 2006, 55 (Suppl. S2), S16–S23. [Google Scholar] [CrossRef] [Green Version]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [Green Version]
- Lorza-Gil, E.; Kaiser, G.; Rexen Ulven, E.; Konig, G.M.; Gerst, F.; Oquendo, M.B.; Birkenfeld, A.L.; Haring, H.U.; Kostenis, E.; Ulven, T.; et al. FFA2-, but not FFA3-agonists inhibit GSIS of human pseudoislets: A comparative study with mouse islets and rat INS-1E cells. Sci. Rep. 2020, 10, 16497. [Google Scholar] [CrossRef]
- McNelis, J.C.; Lee, Y.S.; Mayoral, R.; van der Kant, R.; Johnson, A.M.; Wollam, J.; Olefsky, J.M. GPR43 Potentiates beta-Cell Function in Obesity. Diabetes 2015, 64, 3203–3217. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, M.; Villa, S.R.; Fuller, M.; Wicksteed, B.; Mackay, C.R.; Alquier, T.; Poitout, V.; Mancebo, H.; Mirmira, R.G.; Gilchrist, A.; et al. An Acetate-Specific GPCR, FFAR2, Regulates Insulin Secretion. Mol. Endocrinol. 2015, 29, 1055–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingitore, A.; Gonzalez-Abuin, N.; Ruz-Maldonado, I.; Huang, G.C.; Frost, G.; Persaud, S.J. Short chain fatty acids stimulate insulin secretion and reduce apoptosis in mouse and human islets in vitro: Role of free fatty acid receptor 2. Diabetes Obes. Metab. 2019, 21, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veprik, A.; Laufer, D.; Weiss, S.; Rubins, N.; Walker, M.D. GPR41 modulates insulin secretion and gene expression in pancreatic beta-cells and modifies metabolic homeostasis in fed and fasting states. FASEB J. 2016, 30, 3860–3869. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, M.; Layden, B.T. FFAR3 modulates insulin secretion and global gene expression in mouse islets. Islets 2015, 7, e1045182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Ahmed, K.; Gille, A.; Lu, S.; Grone, H.J.; Tunaru, S.; Offermanns, S. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015, 21, 173–177. [Google Scholar] [CrossRef]
- Shapiro, H.; Shachar, S.; Sekler, I.; Hershfinkel, M.; Walker, M.D. Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem. Biophys. Res. Commun. 2005, 335, 97–104. [Google Scholar] [CrossRef]
- Sakuma, K.; Yabuki, C.; Maruyama, M.; Abiru, A.; Komatsu, H.; Negoro, N.; Tsujihata, Y.; Takeuchi, K.; Habata, Y.; Mori, M. Fasiglifam (TAK-875) has dual potentiating mechanisms via Galphaq-GPR40/FFAR1 signaling branches on glucose-dependent insulin secretion. Pharmacol. Res. Perspect. 2016, 4, e00237. [Google Scholar] [CrossRef]
- Trexler, A.J.; Taraska, J.W. Regulation of insulin exocytosis by calcium-dependent protein kinase C in beta cells. Cell Calcium 2017, 67. [Google Scholar] [CrossRef]
- Hara, T. Ligands at Free Fatty Acid Receptor 1 (GPR40). Handb. Exp. Pharmacol. 2017, 236. [Google Scholar] [CrossRef]
- Srivastava, A.; Yano, J.; Hirozane, Y.; Kefala, G.; Gruswitz, F.; Snell, G.; Lane, W.; Ivetac, A.; Aertgeerts, K.; Nguyen, J.; et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 2014, 513, 124–127. [Google Scholar] [CrossRef]
- Lin, D.C.; Guo, Q.; Luo, J.; Zhang, J.; Nguyen, K.; Chen, M.; Tran, T.; Dransfield, P.J.; Brown, S.P.; Houze, J.; et al. Identification and pharmacological characterization of multiple allosteric binding sites on the free fatty acid 1 receptor. Mol. Pharmacol. 2012, 82, 843–859. [Google Scholar] [CrossRef] [Green Version]
- Hauge, M.; Vestmar, M.A.; Husted, A.S.; Ekberg, J.P.; Wright, M.J.; Di Salvo, J.; Weinglass, A.B.; Engelstoft, M.S.; Madsen, A.N.; Luckmann, M.; et al. GPR40 (FFAR1)—Combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol. Metab. 2015, 4, 3–14. [Google Scholar] [CrossRef]
- Mancini, A.D.; Bertrand, G.; Vivot, K.; Carpentier, E.; Tremblay, C.; Ghislain, J.; Bouvier, M.; Poitout, V. beta-Arrestin Recruitment and Biased Agonism at Free Fatty Acid Receptor 1. J. Biol. Chem. 2015, 290, 21131–21140. [Google Scholar] [CrossRef] [Green Version]
- Zaccardi, F.; Webb, D.R.; Yates, T.; Davies, M.J. Pathophysiology of type 1 and type 2 diabetes mellitus: A 90-year perspective. Postgrad. Med. J. 2016, 92, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, E.; Storm, P.; Karajamaki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R. Impaired glucose tolerance and risk factors for progression to type 2 diabetes in youth. Pediatr. Diabetes 2007, 8 (Suppl. S9), 70–75. [Google Scholar] [CrossRef] [PubMed]
- Kloting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schon, M.R.; Kern, M.; Stumvoll, M.; Bluher, M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E506–E515. [Google Scholar] [CrossRef] [PubMed]
- Slawik, M.; Vidal-Puig, A.J. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res. Rev. 2006, 5, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Adipose tissue inflammation: A cause or consequence of obesity-related insulin resistance? Clin. Sci. 2016, 130, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.S.; Li, Y.; Lai, S.; Liu, J.; Guan, H.; Ke, W.; He, X.; Li, Y. Ectopic Fat Deposition on Insulin Sensitivity: Correlation of Hepatocellular Lipid Content and M Value. J. Diabetes Res. 2016, 2016, 3684831. [Google Scholar] [CrossRef] [Green Version]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Gastaldelli, A. Insulin resistance and reduced metabolic flexibility: Cause or consequence of NAFLD? Clin. Sci. 2017, 131, 2701–2704. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Haring, H.U. Novel phenotypes of prediabetes? Diabetologia 2016, 59, 1806–1818. [Google Scholar] [CrossRef] [Green Version]
- Unger, R.H. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995, 44, 863–870. [Google Scholar] [CrossRef]
- Prentki, M.; Corkey, B.E. Are the beta-cell signaling molecules malonyl-CoA and cystolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes 1996, 45, 273–283. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Dokken, B.B.; Saengsirisuwan, V.; Kim, J.S.; Teachey, M.K.; Henriksen, E.J. Oxidative stress-induced insulin resistance in rat skeletal muscle: Role of glycogen synthase kinase-3. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E615–E621. [Google Scholar] [CrossRef] [Green Version]
- Pistrosch, F.; Natali, A.; Hanefeld, M. Is hyperglycemia a cardiovascular risk factor? Diabetes Care 2011, 34 (Suppl. S2), S128–S131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, T.; Scarcia, P.; Li, N.; Gaudet, P.; Duhamel, D.; Palmieri, F.; Maechler, P. Changes in mitochondrial carriers exhibit stress-specific signatures in INS-1Ebeta-cells exposed to glucose versus fatty acids. PLoS ONE 2013, 8, e82364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Remedi, M.S.; Emfinger, C. Pancreatic beta-cell identity in diabetes. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Kjorholt, C.; Akerfeldt, M.C.; Biden, T.J.; Laybutt, D.R. Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of beta-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes 2005, 54, 2755–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Sanchez, C.; Brun, T.; Maechler, P. Mitochondrial Carriers Regulating Insulin Secretion Profiled in Human Islets upon Metabolic Stress. Biomolecules 2020, 10, 1543. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Jimenez-Sanchez, C.; Madsen, J.G.S.; Hadadi, N.; Duhamel, D.; Bartley, C.; Oberhauser, L.; Trajkovski, M.; Mandrup, S.; Maechler, P. AMPK Profiling in Rodent and Human Pancreatic Beta-Cells under Nutrient-Rich Metabolic Stress. Int. J. Mol. Sci. 2020, 21, 3982. [Google Scholar] [CrossRef]
- Brun, T.; Li, N.; Jourdain, A.A.; Gaudet, P.; Duhamel, D.; Meyer, J.; Bosco, D.; Maechler, P. Diabetogenic milieus induce specific changes in mitochondrial transcriptome and differentiation of human pancreatic islets. Hum. Mol. Genet. 2015, 24, 5270–5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottosson-Laakso, E.; Krus, U.; Storm, P.; Prasad, R.B.; Oskolkov, N.; Ahlqvist, E.; Fadista, J.; Hansson, O.; Groop, L.; Vikman, P. Glucose-Induced Changes in Gene Expression in Human Pancreatic Islets: Causes or Consequences of Chronic Hyperglycemia. Diabetes 2017, 66, 3013–3028. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Karaca, M.; Maechler, P. Upregulation of UCP2 in beta-cells confers partial protection against both oxidative stress and glucotoxicity. Redox Biol. 2017, 13, 541–549. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Daniel, K.W.; Liu, D.; Lyght, O.; Edelstein, D.; Brownlee, M.; Corkey, B.E.; Collins, S. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology 2009, 150, 3040–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supale, S.; Li, N.; Brun, T.; Maechler, P. Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol. Metab. 2012, 23, 477–487. [Google Scholar] [CrossRef]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Boni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in beta-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Shetty, S.; Orci, L.; Unger, R.H.; Scherer, P.E. Diabetes and apoptosis: Lipotoxicity. Apoptosis 2009, 14, 1484–1495. [Google Scholar] [CrossRef]
- Weir, G.C. Glucolipotoxicity, beta-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Ladriere, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic beta-cell dysfunction. Diabetes Obes. Metab. 2010, 12 (Suppl. S2), 76–82. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121 Pt 14, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmugi, A.; Parnis, J.; Chen, X.; Carmichael, L.; Hardy, J.; Mannan, N.; Marchetti, P.; Piemonti, L.; Bosco, D.; Johnson, P.; et al. Sorcin Links Pancreatic beta-Cell Lipotoxicity to ER Ca2+ Stores. Diabetes 2016, 65, 1009–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.C.; Green, C.D.; Olson, L.K.; Moxley, M.A.; Corbett, J.A. A role for aberrant protein palmitoylation in FFA-induced ER stress and beta-cell death. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1390–E1398. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Li, N.; Frigerio, F.; Maechler, P. The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem. Soc. Trans. 2008, 36 Pt 5, 930–934. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Manukyan, L.; Ubhayasekera, S.J.; Bergquist, J.; Sargsyan, E.; Bergsten, P. Palmitate-induced impairments of beta-cell function are linked with generation of specific ceramide species via acylation of sphingosine. Endocrinology 2015, 156, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiban, J.; Caputo, L.; Colombini, M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 2008, 49, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Oberhauser, L.; Granziera, S.; Colom, A.; Goujon, A.; Lavallard, V.; Matile, S.; Roux, A.; Brun, T.; Maechler, P. Palmitate and oleate modify membrane fluidity and kinase activities of INS-1E beta-cells alongside altered metabolism-secretion coupling. Biochim. Biophys. Acta—Mol. Cell Res. 2020, 1867, 118619. [Google Scholar] [CrossRef]
- Sargsyan, E.; Artemenko, K.; Manukyan, L.; Bergquist, J.; Bergsten, P. Oleate protects beta-cells from the toxic effect of palmitate by activating pro-survival pathways of the ER stress response. Biochim. Biophys. Acta 2016, 1861, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Oberhauser, L.; Jiménez-Sánchez, C.; Madsen, J.G.S.; Duhamel, D.; Mandrup, S.; Brun, T.; Maechler, P. Glucolipotoxicity promotes the capacity of the glycerolipid/free fatty acid cycle supporting the secretory response of pancreatic beta-cells. Diabetologia 2022, in press. [Google Scholar]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J. Biol. Chem. 2005, 280, 32413–32418. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, E.; Watterson, K.R.; Stocker, C.J.; Sokol, E.; Jenkins, L.; Simon, K.; Grundmann, M.; Petersen, R.K.; Wargent, E.T.; Hudson, B.D.; et al. Activity of dietary fatty acids on FFA1 and FFA4 and characterisation of pinolenic acid as a dual FFA1/FFA4 agonist with potential effect against metabolic diseases. Br. J. Nutr. 2015, 113, 1677–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, K.; Maekawa, F.; Yada, T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: Mediation by PLC and L-type Ca2+ channel and link to insulin release. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E670–E677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steneberg, P.; Rubins, N.; Bartoov-Shifman, R.; Walker, M.D.; Edlund, H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005, 1, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Brownlie, R.; Mayers, R.M.; Pierce, J.A.; Marley, A.E.; Smith, D.M. The long-chain fatty acid receptor, GPR40, and glucolipotoxicity: Investigations using GPR40-knockout mice. Biochem. Soc. Trans. 2008, 36, 950–954. [Google Scholar] [CrossRef]
- Kebede, M.; Alquier, T.; Latour, M.G.; Semache, M.; Tremblay, C.; Poitout, V. The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes 2008, 57, 2432–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Sun, P.; Zhang, X.; Liu, H.; Jiang, H.; Zhu, W.; Wang, H. Inhibition of GPR40 protects MIN6 beta cells from palmitate-induced ER stress and apoptosis. J. Cell. Biochem. 2012, 113, 1152–1158. [Google Scholar] [CrossRef]
- Kristinsson, H.; Smith, D.M.; Bergsten, P.; Sargsyan, E. FFAR1 is involved in both the acute and chronic effects of palmitate on insulin secretion. Endocrinology 2013, 154, 4078–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latour, M.G.; Alquier, T.; Oseid, E.; Tremblay, C.; Jetton, T.L.; Luo, J.; Lin, D.C.; Poitout, V. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 2007, 56, 1087–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, H.; Hoos, L.M.; Liu, L.; Tetzloff, G.; Hu, W.; Abbondanzo, S.J.; Vassileva, G.; Gustafson, E.L.; Hedrick, J.A.; Davis, H.R. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes 2008, 57, 2999–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croze, M.L.; Guillaume, A.; Ethier, M.; Fergusson, G.; Tremblay, C.; Campbell, S.A.; Maachi, H.; Ghislain, J.; Poitout, V. Combined Deletion of Free Fatty-Acid Receptors 1 and 4 Minimally Impacts Glucose Homeostasis in Mice. Endocrinology 2021, 162, bqab002. [Google Scholar] [CrossRef] [PubMed]
- Nagasumi, K.; Esaki, R.; Iwachidow, K.; Yasuhara, Y.; Ogi, K.; Tanaka, H.; Nakata, M.; Yano, T.; Shimakawa, K.; Taketomi, S.; et al. Overexpression of GPR40 in pancreatic beta-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes 2009, 58, 1067–1076. [Google Scholar] [CrossRef]
- Wagner, R.; Kaiser, G.; Gerst, F.; Christiansen, E.; Due-Hansen, M.E.; Grundmann, M.; Machicao, F.; Peter, A.; Kostenis, E.; Ulven, T.; et al. Reevaluation of fatty acid receptor 1 as a drug target for the stimulation of insulin secretion in humans. Diabetes 2013, 62, 2106–2111. [Google Scholar] [CrossRef] [Green Version]
- Panse, M.; Gerst, F.; Kaiser, G.; Teutsch, C.A.; Dolker, R.; Wagner, R.; Haring, H.U.; Ullrich, S. Activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) by free fatty acid receptor 1 (FFAR1/GPR40) protects from palmitate-induced beta cell death, but plays no role in insulin secretion. Cell. Physiol. Biochem. 2015, 35, 1537–1545. [Google Scholar] [CrossRef]
- Teutsch, C.A.; Panse, M.; Grundmann, M.; Kaiser, G.; Kostenis, E.; Haring, H.U.; Ullrich, S. Detection of free fatty acid receptor 1 expression: The critical role of negative and positive controls. Diabetologia 2014, 57, 776–780. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thevenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Jacqueminet, S.; Briaud, I.; Rouault, C.; Reach, G.; Poitout, V. Inhibition of insulin gene expression by long-term exposure of pancreatic beta cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metab. Clin. Exp. 2000, 49, 532–536. [Google Scholar] [CrossRef]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Roche, E.; Assimacopoulos-Jeannet, F.; Corkey, B.E.; Kim, K.H.; Prentki, M. Evidence for an anaplerotic/malonyl-CoA pathway in pancreatic beta-cell nutrient signaling. Diabetes 1996, 45, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontes, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Gjoni, E.; Brioschi, L.; Cinque, A.; Coant, N.; Islam, M.N.; Ng, C.K.; Verderio, C.; Magnan, C.; Riboni, L.; Viani, P.; et al. Glucolipotoxicity impairs ceramide flow from the endoplasmic reticulum to the Golgi apparatus in INS-1 beta-cells. PLoS ONE 2014, 9, e110875. [Google Scholar]
- Bagnati, M.; Ogunkolade, B.W.; Marshall, C.; Tucci, C.; Hanna, K.; Jones, T.A.; Bugliani, M.; Nedjai, B.; Caton, P.W.; Kieswich, J.; et al. Glucolipotoxicity initiates pancreatic beta-cell death through TNFR5/CD40-mediated STAT1 and NF-kappaB activation. Cell Death Dis. 2016, 7, e2329. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Corkey, B.E.; Glennon, M.C.; Chen, K.S.; Deeney, J.T.; Matschinsky, F.M.; Prentki, M. A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic beta-cells. J. Biol. Chem. 1989, 264, 21608–21612. [Google Scholar] [CrossRef]
- Brun, T.; Roche, E.; Kim, K.H.; Prentki, M. Glucose regulates acetyl-CoA carboxylase gene expression in a pancreatic beta-cell line (INS-1). J. Biol. Chem. 1993, 268, 18905–18911. [Google Scholar] [CrossRef]
- Prentki, M.; Madiraju, S.R. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol. Cell. Endocrinol. 2012, 353, 88–100. [Google Scholar] [CrossRef]
- Noel, R.J.; Antinozzi, P.A.; McGarry, J.D.; Newgard, C.B. Engineering of glycerol-stimulated insulin secretion in islet beta cells. Differential metabolic fates of glucose and glycerol provide insight into mechanisms of stimulus-secretion coupling. J. Biol. Chem. 1997, 272, 18621–18627. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, K.; Chang, B.H.; Fujimiya, M.; Chen, W.; Kulkarni, R.N.; Eguchi, Y.; Kimura, H.; Kojima, H.; Chan, L. Aquaporin 7 is a beta-cell protein and regulator of intraislet glycerol content and glycerol kinase activity, beta-cell mass, and insulin production and secretion. Mol. Cell. Biol. 2007, 27, 6026–6037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, H.; Holst, L.S.; Svensson, H.; Degerman, E.; Sundler, F.; Ahren, B.; Rorsman, P.; Holm, C. Hormone-sensitive lipase, the rate-limiting enzyme in triglyceride hydrolysis, is expressed and active in beta-cells. Diabetes 1999, 48, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.B.; Fridriksson, J.; Madsen, L.; Rishi, V.; Vinson, C.; Holmsen, H.; Berge, R.K.; Mandrup, S. Glucose-induced lipogenesis in pancreatic beta-cells is dependent on SREBP-1. Mol. Cell. Endocrinol. 2005, 240, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Fex, M.; Mulder, H. Lipases in the pancreatic beta-cell: Implications for insulin secretion. Biochem. Soc. Trans. 2008, 36 Pt 5, 885–890. [Google Scholar] [CrossRef]
- Chlouverakis, C. The action of glucose on lipolysis. Metabolism 1967, 16, 469–472. [Google Scholar] [CrossRef]
- Winzell, M.S.; Strom, K.; Holm, C.; Ahren, B. Glucose-stimulated insulin secretion correlates with beta-cell lipolysis. Nutr. Metab. Cardiovasc. Dis. 2006, 16 (Suppl. S1), S11–S16. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H.; Yang, S.; Winzell, M.S.; Holm, C.; Ahren, B. Inhibition of lipase activity and lipolysis in rat islets reduces insulin secretion. Diabetes 2004, 53, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fex, M.; Haemmerle, G.; Wierup, N.; Dekker-Nitert, M.; Rehn, M.; Ristow, M.; Zechner, R.; Sundler, F.; Holm, C.; Eliasson, L.; et al. A beta cell-specific knockout of hormone-sensitive lipase in mice results in hyperglycaemia and disruption of exocytosis. Diabetologia 2009, 52, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attane, C.; Peyot, M.L.; Lussier, R.; Poursharifi, P.; Zhao, S.; Zhang, D.; Morin, J.; Pineda, M.; Wang, S.; Dumortier, O.; et al. A beta cell ATGL-lipolysis/adipose tissue axis controls energy homeostasis and body weight via insulin secretion in mice. Diabetologia 2016, 59, 2654–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichmann, T.O.; Lass, A. DAG tales: The multiple faces of diacylglycerol—Stereochemistry, metabolism, and signaling. Cell. Mol. Life Sci. 2015, 72, 3931–3952. [Google Scholar] [CrossRef] [Green Version]
- Eichmann, T.O.; Kumari, M.; Haas, J.T.; Farese, R.V., Jr.; Zimmermann, R.; Lass, A.; Zechner, R. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J. Biol. Chem. 2012, 287, 41446–41457. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Mugabo, Y.; Iglesias, J.; Xie, L.; Delghingaro-Augusto, V.; Lussier, R.; Peyot, M.L.; Joly, E.; Taib, B.; Davis, M.A.; et al. alpha/beta-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab. 2014, 19, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Sheu, L.; Pasyk, E.A.; Ji, J.; Huang, X.; Gao, X.; Varoqueaux, F.; Brose, N.; Gaisano, H.Y. Regulation of insulin exocytosis by Munc13-1. J. Biol. Chem. 2003, 278, 27556–27563. [Google Scholar] [CrossRef] [Green Version]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [Green Version]
- Nolan, C.J.; Leahy, J.L.; Delghingaro-Augusto, V.; Moibi, J.; Soni, K.; Peyot, M.L.; Fortier, M.; Guay, C.; Lamontagne, J.; Barbeau, A.; et al. Beta cell compensation for insulin resistance in Zucker fatty rats: Increased lipolysis and fatty acid signalling. Diabetologia 2006, 49, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Mugabo, Y.; Zhao, S.; Seifried, A.; Gezzar, S.; Al-Mass, A.; Zhang, D.; Lamontagne, J.; Attane, C.; Poursharifi, P.; Iglesias, J.; et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc. Natl. Acad. Sci. USA 2016, 113, E430–E439. [Google Scholar] [CrossRef] [Green Version]
- Peyot, M.L.; Pepin, E.; Lamontagne, J.; Latour, M.G.; Zarrouki, B.; Lussier, R.; Pineda, M.; Jetton, T.L.; Madiraju, S.R.; Joly, E.; et al. Beta-cell failure in diet-induced obese mice stratified according to body weight gain: Secretory dysfunction and altered islet lipid metabolism without steatosis or reduced beta-cell mass. Diabetes 2010, 59, 2178–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguayo-Mazzucato, C. Functional changes in beta cells during ageing and senescence. Diabetologia 2020, 63, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Merglen, A.; Theander, S.; Rubi, B.; Chaffard, G.; Wollheim, C.B.; Maechler, P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 2004, 145, 667–678. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oberhauser, L.; Maechler, P. Lipid-Induced Adaptations of the Pancreatic Beta-Cell to Glucotoxic Conditions Sustain Insulin Secretion. Int. J. Mol. Sci. 2022, 23, 324. https://doi.org/10.3390/ijms23010324
Oberhauser L, Maechler P. Lipid-Induced Adaptations of the Pancreatic Beta-Cell to Glucotoxic Conditions Sustain Insulin Secretion. International Journal of Molecular Sciences. 2022; 23(1):324. https://doi.org/10.3390/ijms23010324
Chicago/Turabian StyleOberhauser, Lucie, and Pierre Maechler. 2022. "Lipid-Induced Adaptations of the Pancreatic Beta-Cell to Glucotoxic Conditions Sustain Insulin Secretion" International Journal of Molecular Sciences 23, no. 1: 324. https://doi.org/10.3390/ijms23010324
APA StyleOberhauser, L., & Maechler, P. (2022). Lipid-Induced Adaptations of the Pancreatic Beta-Cell to Glucotoxic Conditions Sustain Insulin Secretion. International Journal of Molecular Sciences, 23(1), 324. https://doi.org/10.3390/ijms23010324