
Characterization of Mutations Causing CYP21A2 Deficiency in Brazilian and Portuguese Populations

 , , ,
, , , 
Abstract
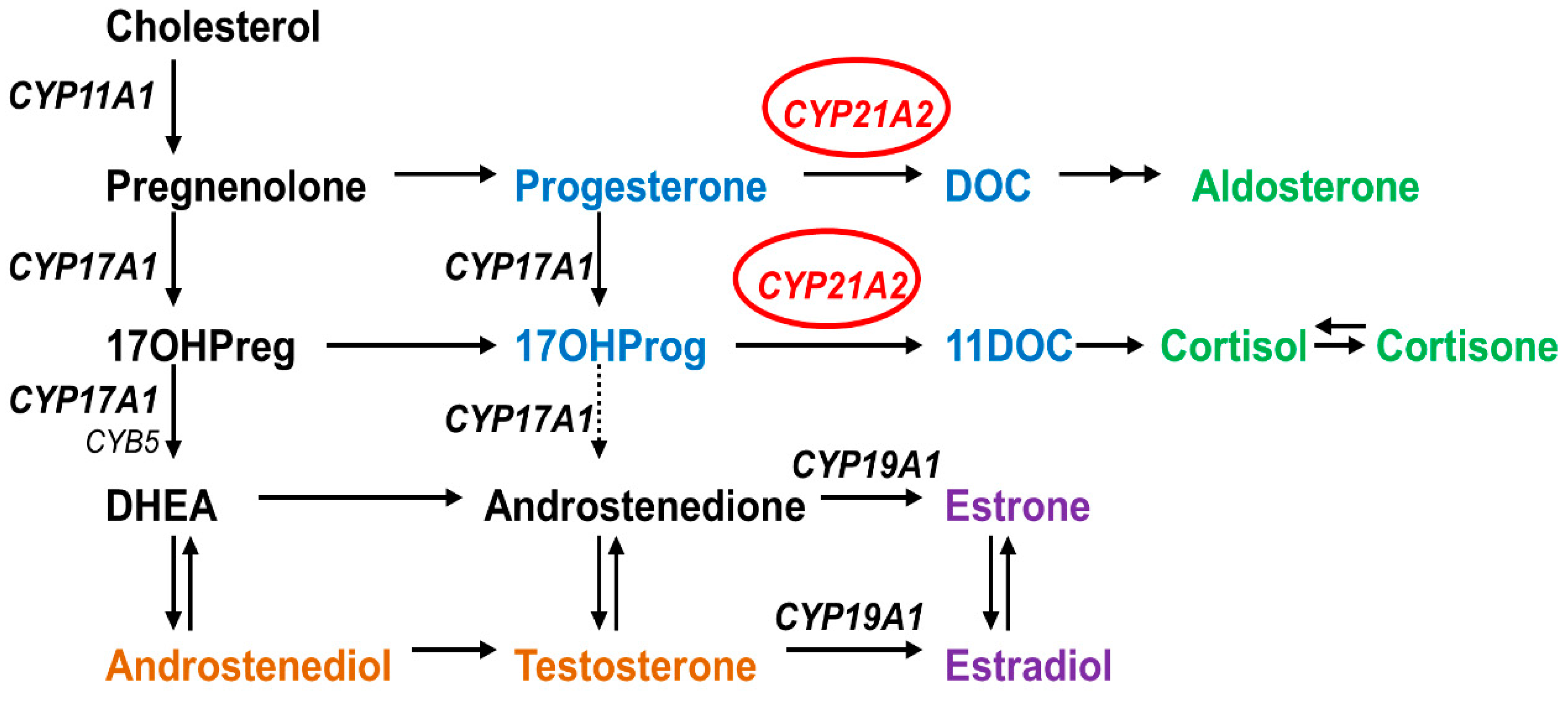
1. Introduction
2. Results
2.1. Variant Collection and Description
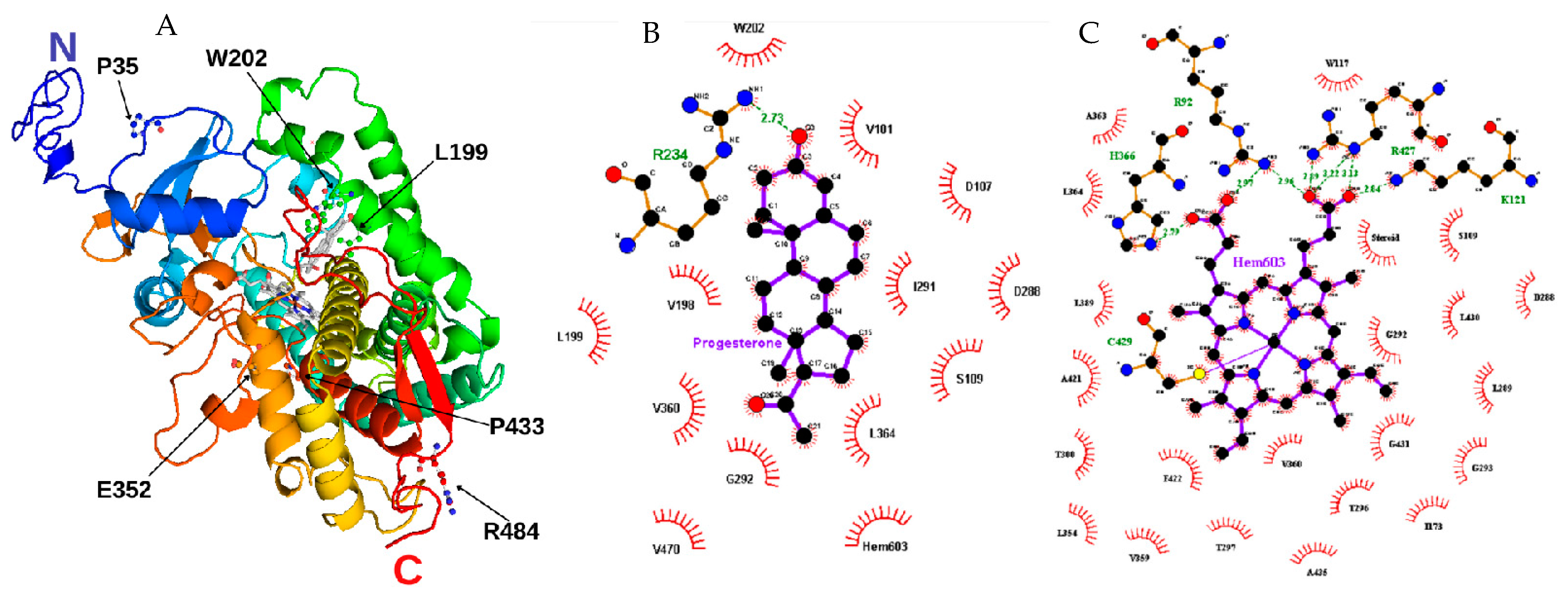
2.2. Computational Characterization Indicated the Structural Impact of the SNVs
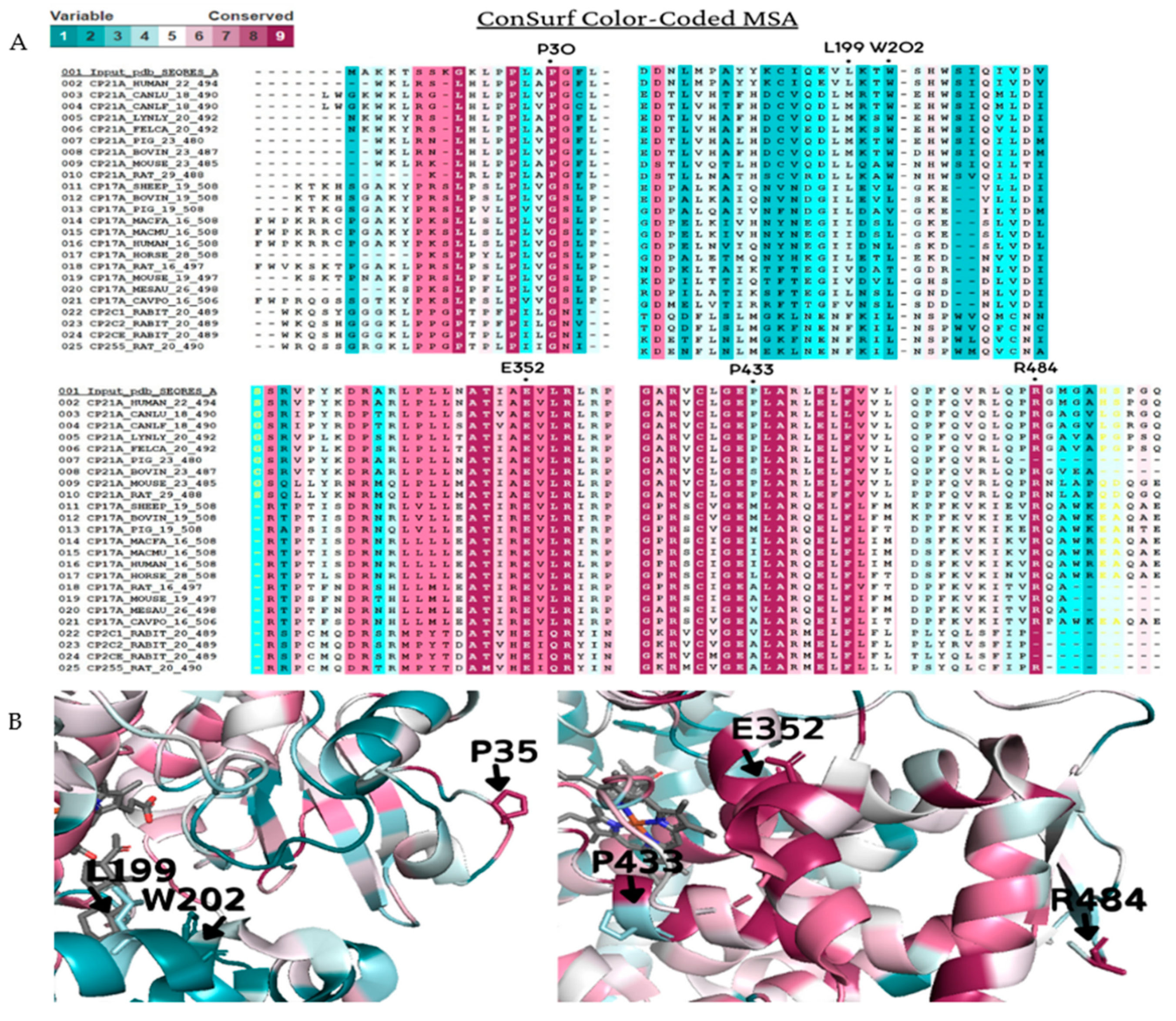
2.3. Amino Acid Chemical Proprieties, Conservation, and Structural Damage
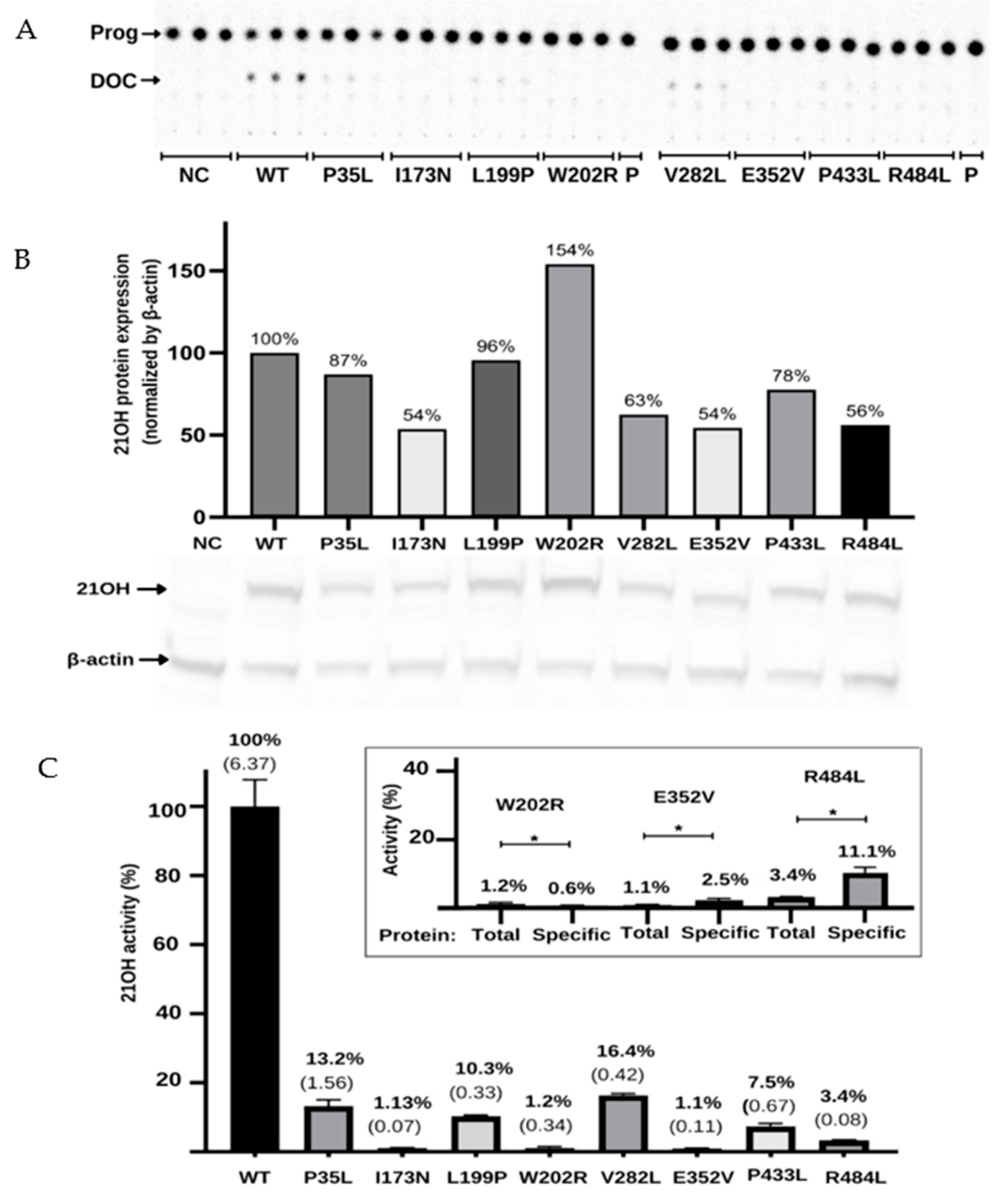
2.4. Functional Testing for the Validation of In Silico Results
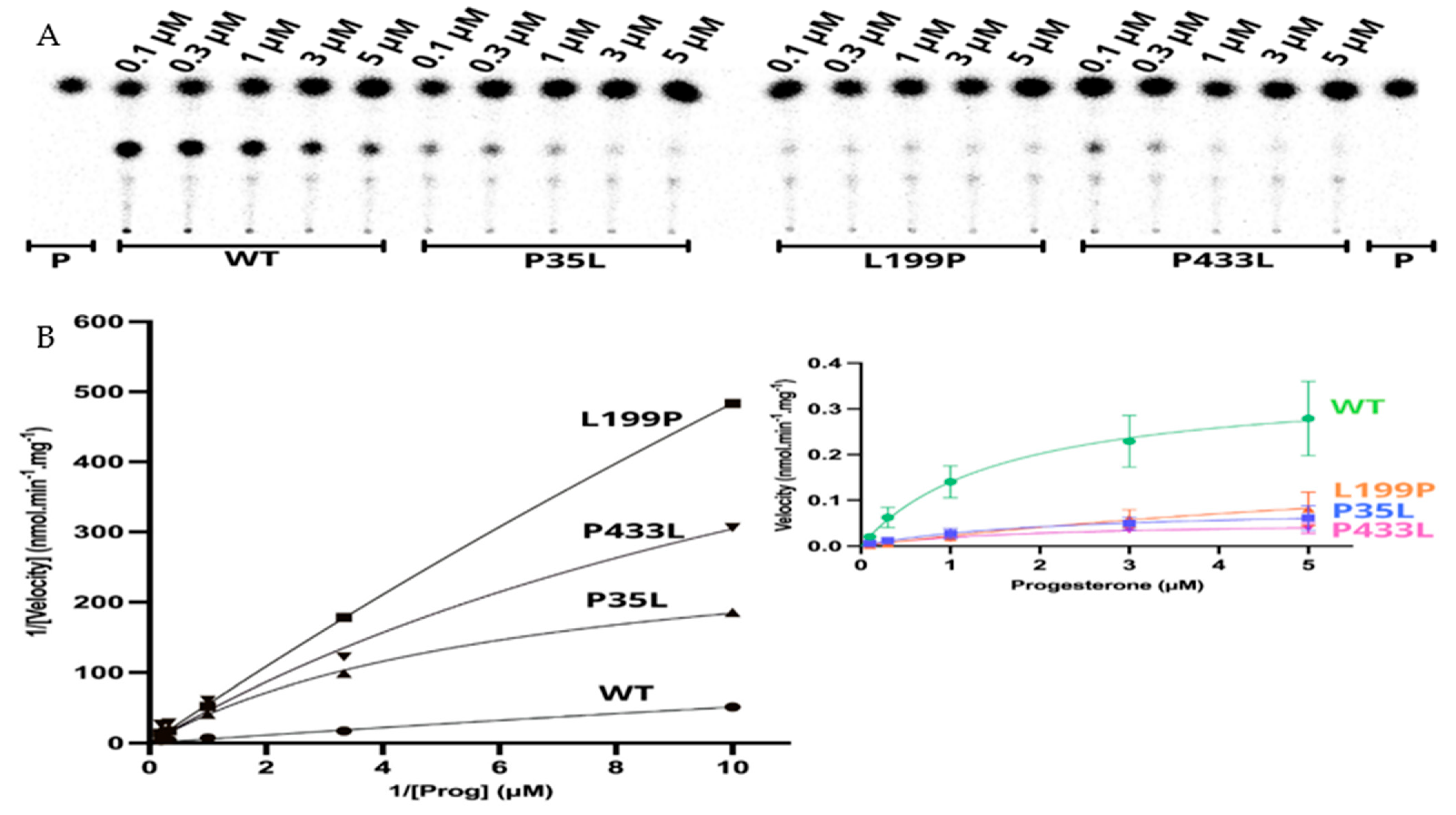
2.5. Kinetic Analysis of CYP21A2 Variants
3. Discussion
4. Materials and Methods
4.1. Search and Selection of Variants in CYP21A2
4.2. Conservation Analysis
4.3. Construction of Plasmid and Site-Directed Mutagenesis
4.4. Cell Transfection and Enzymatic Activity Assay
4.5. Western Blot
4.6. Enzyme Kinetics Assay
4.7. Statistics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef]
- Pallan, P.S.; Wang, C.; Lei, L.; Yoshimoto, F.K.; Auchus, R.J.; Waterman, M.R.; Guengerich, F.P.; Egli, M. Human Cytochrome P450 21A2, the Major Steroid 21-Hydroxylase: Structure of the enzyme·progesterone substrate complex and rate-limiting c–h bond cleavage. J. Biol. Chem. 2015, 290, 13128–13143. [Google Scholar] [CrossRef]
- New, M.I.; Abraham, M.; Gonzalez, B.; Dumic, M.; Razzaghy-Azar, M.; Chitayat, D.; Sun, L.; Zaidi, M.; Wilson, R.C.; Yuen, T. Genotype-phenotype correlation in 1507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 2013, 110, 2611–2616. [Google Scholar] [CrossRef]
- Tardy, V.; Menassa, R.; Sulmont, V.; Lienhardt-Roussie, A.; Lecointre, C.; Brauner, R.; David, M.; Morel, Y. Phenotype-Genotype Correlations of 13 Rare CYP21A2 Mutations Detected in 46 Patients Affected with 21-Hydroxylase Deficiency and in One Carrier. J. Clin. Endocrinol. Metab. 2010, 95, 1288–1300. [Google Scholar] [CrossRef]
- Janner, M.; Pandey, A.V.; Mullis, P.E.; Flück, C.E. Clinical and biochemical description of a novel CYP21A2 gene mutation 962_963insA using a new 3D model for the P450c21 protein. Eur. J. Endocrinol. 2006, 155, 143–151. [Google Scholar] [CrossRef]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.R.; Bryce, J.; Haghpanahan, H.; Lewsey, J.D.; Tan, L.E.; Atapattu, N.; Birkebaek, N.H.; Blankenstein, O.; Neumann, U.; Balsamo, A.; et al. Real-World Estimates of Adrenal Insufficiency–Related Adverse Events in Children With Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2021, 106, e192–e203. [Google Scholar] [CrossRef] [PubMed]
- Carmina, E.; Dewailly, D.; Escobar-Morreale, H.F.; Kelestimur, F.; Moran, C.; Oberfield, S.; Witchel, S.F.; Azziz, R. Non-Classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: An update with a special focus on adolescent and adult women. Hum. Reprod. Update 2017, 23, 580–599. [Google Scholar] [CrossRef] [PubMed]
- Krone, N.; Riepe, F.G.; Grötzinger, J.; Partsch, C.J.; Sippell, W.G. Functional characterization of two novel point mutations in the CYP21 gene causing simple virilizing forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2005, 90, 445–454. [Google Scholar] [CrossRef]
- El-Maouche, D.; Arlt, W.; Merke, D.P. Congenital adrenal hyperplasia. Lancet 2017, 390, 2194–2210. [Google Scholar] [CrossRef]
- Held, P.K.; Bird, I.M.; Heather, N.L. Newborn Screening for Congenital Adrenal Hyperplasia: Review of Factors Affecting Screening Accuracy. Int. J. Neonatal Screen. 2020, 6, 67. [Google Scholar] [CrossRef]
- Flück, C.E.; Tajima, T.; Pandey, A.V.; Arlt, W.; Okuhara, K.; Verge, C.F.; Jabs, E.W.; Mendonca, B.B.; Fujieda, K.; Miller, W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004, 36, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E.; Mallet, D.; Hofer, G.; Samara-Boustani, D.; Leger, J.; Polak, M.; Morel, Y.; Pandey, A.V. Deletion of P399_E401 in NADPH cytochrome P450 oxidoreductase results in partial mixed oxidase deficiency. Biochem. Biophys. Res. Commun. 2011, 412, 572–577. [Google Scholar] [CrossRef]
- Parween, S.; Roucher-Boulez, F.; Flück, C.E.; Lienhardt-Roussie, A.; Mallet, D.; Morel, Y.; Pandey, A.V. P450 Oxidoreductase Deficiency: Loss of Activity Caused by Protein Instability From a Novel L374H Mutation. J. Clin. Endocrinol. Metab. 2016, 101, 4789–4798. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cancio, M.; Camats, N.; Fluck, C.E.; Zalewski, A.; Dick, B.; Frey, B.M.; Monne, R.; Toran, N.; Audi, L.; Pandey, A.V. Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency. Pharmaceuticals 2018, 11, 37. [Google Scholar] [CrossRef]
- De Carvalho, D.F.; Miranda, M.C.; Gomes, L.G.; Madureira, G.; Marcondes, J.A.; Billerbeck, A.E.; Rodrigues, A.S.; Presti, P.F.; Kuperman, H.; Damiani, D.; et al. Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur. J. Endocrinol. 2016, 175, 107–116. [Google Scholar] [CrossRef]
- Lidaka, L.; Bekere, L.; Lazdane, G.; Dzivite-Krisane, I.; Kivite-Urtane, A.; Gailite, L. Non-Classical Congenital Adrenal Hyperplasia-Causing Alleles in Adolescent Girls with PCOS and in Risk Group for PCOS Development. Diagnostics 2021, 11, 980. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.R.; Dunham, I.; Yu, C.Y.; Carroll, M.C.; Porter, R.R.; Campbell, R.D. Molecular characterization of the HLA-Linked steroid 21-hydroxylase B gene from an individual with congenital adrenal hyperplasia. EMBO J. 1987, 6, 1653–1661. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Simonetti, L.; Bruque, C.D.; Fernández, C.S.; Benavides-Mori, B.; Delea, M.; Kolomenski, J.E.; Espeche, L.D.; Buzzalino, N.D.; Nadra, A.D.; Dain, L. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum. Mutat. 2018, 39, 5–22. [Google Scholar] [CrossRef]
- Higashi, Y.; Tanae, A.; Inoue, H.; Hiromasa, T.; Fujii-Kuriyama, Y. Aberrant splicing and missense mutations cause steroid 21-hydroxylase [P-450(C21)] deficiency in humans: Possible gene conversion products. Proc. Natl. Acad. Sci. USA 1988, 85, 7486–7490. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Gene conversions, deletions, and polymorphisms in congenital adrenal hyperplasia. Am. J. Hum. Genet. 1988, 42, 4–7. [Google Scholar]
- Tusie-Luna, M.T.; Traktman, P.; White, P.C. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J. Biol. Chem. 1990, 265, 20916–20922. [Google Scholar] [CrossRef]
- Barbaro, M.; Soardi, F.C.; Östberg, L.J.; Persson, B.; de Mello, M.P.; Wedell, A.; Lajic, S. In Vitro functional studies of rare CYP21A2 mutations and establishment of an activity gradient for nonclassic mutations improve phenotype prediction in congenital adrenal hyperplasia. Clin. Endocrinol. 2015, 82, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Islam, B.; D’Atri, V.; Sgobba, M.; Poojari, C.; Sun, L.; Yuen, T.; Zaidi, M.; New, M.I. Structure-Phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc. Natl. Acad. Sci. USA 2013, 110, 2605–2610. [Google Scholar] [CrossRef]
- Xu, C.; Jia, W.; Cheng, X.; Ying, H.; Chen, J.; Xu, J.; Guan, Q.; Zhou, X.; Zheng, D.; Li, G.; et al. Genotype–Phenotype correlation study and mutational and hormonal analysis in a Chinese cohort with 21-hydroxylase deficiency. Mol. Genet. Genom. Med. 2019, 7, e671. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pallan, P.S.; Zhang, W.; Lei, L.; Yoshimoto, F.K.; Waterman, M.R.; Egli, M.; Guengerich, F.P. Functional analysis of human cytochrome P450 21A2 variants involved in congenital adrenal hyperplasia. J. Biol. Chem. 2017, 292, 10767–10778. [Google Scholar] [CrossRef]
- Coeli, F.B.; Soardi, F.C.; Bernardi, R.D.; De Araújo, M.; Paulino, L.C.; Lau, I.F.; Petroli, R.J.; De Lemos-Marini, S.H.; Baptista, M.T.; Guerra-Júnior, G.; et al. Novel deletion alleles carrying CYP21A1P/A2chimeric genes in Brazilian patients with 21-hydroxylase deficiency. BMC Med. Genet. 2010, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Silveira, E.L.; Elnecave, R.H.; dos Santos, E.P.; Moura, V.; Pinto, E.M.; van der Linden Nader, I.; Mendonca, B.B.; Bachega, T.A. Molecular analysis of CYP21A2 can optimize the follow-up of positive results in newborn screening for congenital adrenal hyperplasia. Clin. Genet. 2009, 76, 503–510. [Google Scholar] [CrossRef]
- Santos-Silva, R.; Cardoso, R.; Lopes, L.; Fonseca, M.; Espada, F.; Sampaio, L.; Brandão, C.; Antunes, A.; Bragança, G.; Coelho, R. CYP21A2 Gene Pathogenic Variants: A Multicenter Study on Genotype–Phenotype Correlation from a Portuguese Pediatric Cohort. Horm. Res. Paediatr. 2019, 91, 33–45. [Google Scholar] [CrossRef]
- Carvalho, B.; Pereira, M.; Marques, C.J.; Carvalho, D.; Leão, M.; Oliveira, J.P.; Barros, A.; Carvalho, F. Comprehensive genetic analysis and structural characterization of CYP21A2 mutations in CAH patients. Exp. Clin. Endocrinol. Diabetes 2012, 120, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Loke, K.Y.; Lee, Y.S.; Lee, W.W.; Poh, L.K. Molecular analysis of CYP-21 mutations for congenital adrenal hyperplasia in Singapore. Horm. Res. 2001, 55, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Menassa, R.T.V.; Despert, F.; Bouvattier-Morel, C.; Brossier, J.P.; Cartigny, M.; Morel, Y. p.H62L, a rare mutation of the CYP21 gene identified in two forms of 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2008, 93, 1901–1908. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soardi, F.C.; Barbaro, M.; Lau, I.F.; Lemos-Marini, S.H.V.; Baptista, M.T.M.; Guerra-Junior, G.; Wedell, A.; Lajic, S.; De Mello, M.P. Inhibition of CYP21A2 Enzyme Activity Caused by Novel Missense Mutations Identified in Brazilian and Scandinavian Patients. J. Clin. Endocrinol. Metab. 2008, 93, 2416–2420. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16 (Suppl. 8), S1. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Altman, R.B.; Bromberg, Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genom. 2013, 14, S2. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Quan, L.; Lv, Q.; Zhang, Y. STRUM: Structure-Based prediction of protein stability changes upon single-point mutation. Bioinformatics 2016, 32, 2936–2946. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schäffer, A.A.; Yu, Y.K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef]
- Schäffer, A.A.; Aravind, L.; Madden, T.L.; Shavirin, S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V.; Altschul, S.F. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001, 29, 2994–3005. [Google Scholar] [CrossRef]
- Cohen, M.; Pignatti, E.; Dines, M.; Mory, A.; Ekhilevitch, N.; Kolodny, R.; Flück, C.E.; Tiosano, D. In Silico Structural and Biochemical Functional Analysis of a Novel CYP21A2 Pathogenic Variant. Int. J. Mol. Sci. 2020, 21, 5857. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotypes | Phenotype | Clinical Data | Sex | 17OHP (nmol/L) | Na+/K+ (mmol/L) | Age a | Reference | |
|---|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | |||||||
| P35L, H63L, 30-kb del | LGC | SW | vomiting, dehydration | M | 49.3 | 116/9.2 | 2.8 m | [28] Coeli et al. (2010) |
| P35L, H63L, 30-kb del | Q319X | SW | vomiting, dehydration | M | >25 b | 120/6.8 | 13 m | [28] Coeli et al. (2010) |
| P35L, H63L, 30-kb del | c.920_921insT | SW | DSD (Prader IV) | F | >6 b | 119/5.3 | 15 d | [28] Coeli et al. (2010) |
| P35L, H63L, 30-kb del | R357W | SW | vomiting, dehydration, AC | M | >200 b | 119/9.7 | 28 d | [28] Coeli et al. (2010) |
| L199P | normal | ND | DSD (Prader I) | F | 408.5 | nd | [29] Silveira et al. (2009) | |
| W202R | LGC | SW | DSD, salt wasting | F | nd | nd | <2 m | [30] Santos-Silva et al. (2019) |
| E352V | E6 cluster | SV | nd | F | nd | nd | nd | [16] De Carvalho et al. (2016) |
| E352V | IVS-13A/C>G | SW | nd | M | nd | nd | nd | [16] De Carvalho et al. (2016) |
| E352V | IVS-13A/C>G | SW | nd | M | nd | nd | nd | [16] De Carvalho et al. (2016) |
| E352V | G425S | SV | nd | M | nd | nd | nd | [16] De Carvalho et al. (2016) |
| P433L | P454S | NC | nd | F | 42.7 c | nd | 17 y | [31] Carvalho et al. (2012) |
| R484L, IVS-13A/C > G | Del CYP21A2 | SW | DSD (Prader III-IV) | F | 1537.2 | 134/4.9 | <4 m | [29] Silveira et al. (2009) |
| R484L, IVS-13A/C > G | Q319X | SW | undefined | M | 1358.7 | 130/6.2 | <9 m | [29] Silveira et al. (2009) |
| SNV a | In Silico Tools | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| dbSNP rs# | PolyPhen-2 | Score | SNAP2 | Score | MutPred2 | Score | Meta-SNP | Score | PredictSNP | Enzyme Activity % of Control | |
| ● R103L | rs6474 | Benign | 0.023 | Neutral | −47 | Neutral | 0.191 | Neutral | 0.310 | Neutral | 120 |
| ● D184E | rs397515531 | Benign | 0.000 | Neutral | −85 | Neutral | 0.260 | Neutral | 0.412 | Neutral | 100 |
| ● S269T | rs6472 | Benign | 0.016 | Neutral | −93 | Neutral | 0.076 | Neutral | 0.340 | Neutral | 103 |
| ↓ I173N | rs6475 | Damage | 1.000 | Effect | 70 | Pathogenic | 0.838 | Disease | 0.811 | Deleterious | 1.1 |
| ↓ V282L | rs6471 | Benign | 0.273 | Neutral | −76 | Neutral | 0.084 | Neutral | 0.344 | Neutral | 16.4 |
| ↓ R427H | rs151344504 | Damage | 1.000 | Effect | 82 | Pathogenic | 0.821 | Disease | 0.827 | Deleterious | 0.5 |
| P35L | rs200648381 | Damage | 0.988 | Effect | 13 | Pathogenic | 0.676 | Disease | 0.624 | Neutral | 13 |
| L199P | Damage | 0.997 | Effect | 68 | Pathogenic | 0.875 | Disease | 0.711 | Deleterious | 10.3 | |
| W202R | Damage | 1.000 | Effect | 45 | Pathogenic | 0.577 | Neutral | 0.451 | Neutral | 1.2 | |
| E352V | Damage | 0.693 | Effect | 90 | Pathogenic | 0.919 | Disease | 0.937 | Deleterious | 1.1 | |
| P433L | rs751456004 | Benign | 0.402 | Effect | 15 | Pathogenic | 0.614 | Neutral | 0.372 | Neutral | 7.5 |
| R484L | Damage | 1.000 | Effect | 81 | Pathogenic | 0.690 | Disease | 0.748 | Deleterious | 3.4 | |
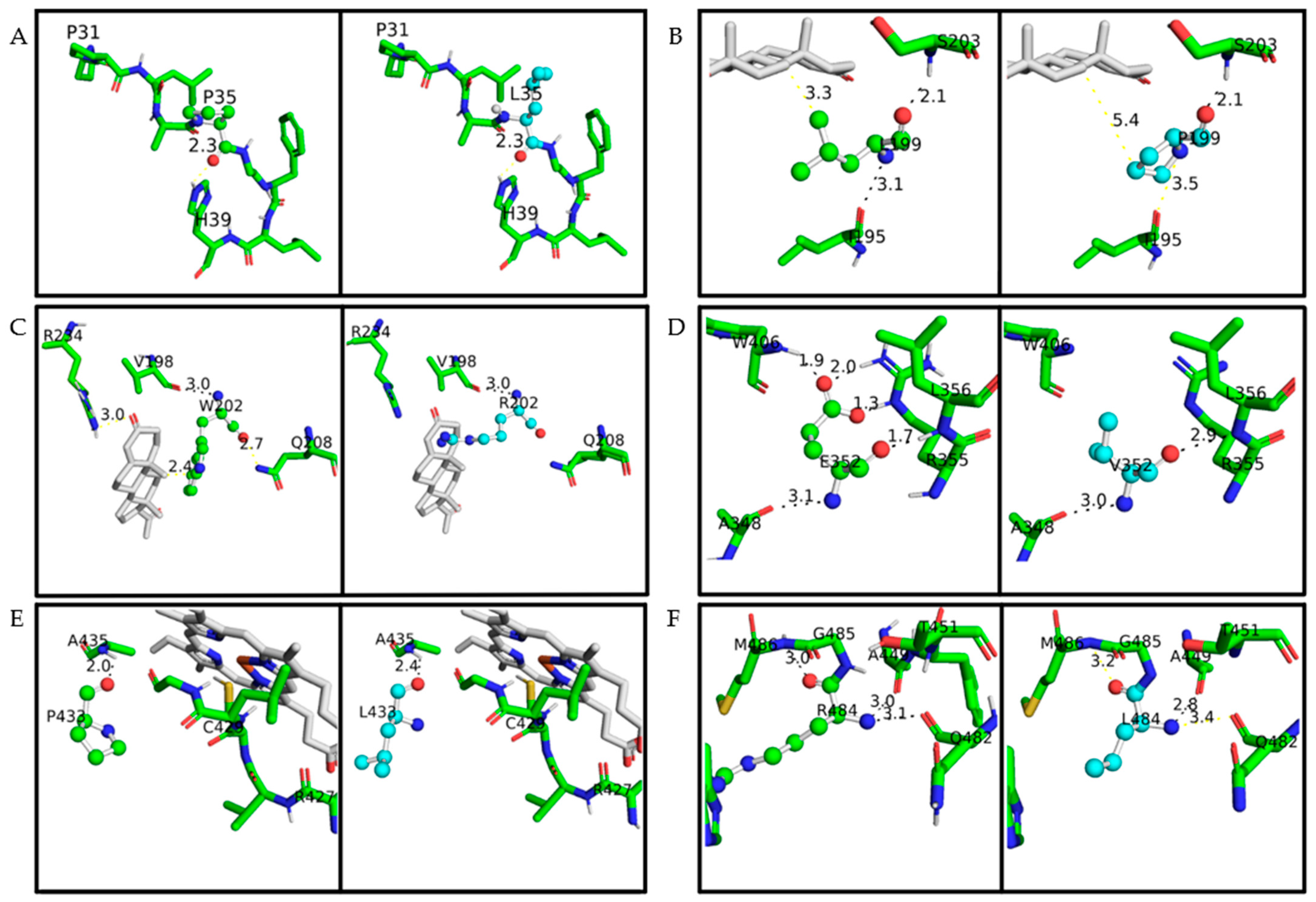
| SNV a | Structure/Protein Localization | Physicochemical Properties | ΔΔG (kcal/mol) | Enzyme Activity | The Molecular Mechanism (Hypothesis) | Mutation Group | |
|---|---|---|---|---|---|---|---|
| WT | MUT | (Mean ± SD) | |||||
| P35L | N-term coil/Region of the membrane protein orientation | Polar residue with a ring at the N-term | Hydrophobic; Short side chain; Folding interactions | −0.58 | 13.2 ± 1.56% | Disturbs the protein orientation in the membrane | C |
| L199P | F-helix/Close to the active site; α-helix stabilization | Hydrophobic; Short side chain; Folding Interactions; H: I195 and S203 | Polar residue with a ring at the N-term; H: S203 | 0.79 | 10.3 ± 0.33% | Breaks the secondary structure close to the active site | C |
| W202R | Turn of the F-helix/close to the active site and steroid-bound residue (R234) | Hydrophobic; Large, rigid aromatic group; H: V198 | Hydrophobic; Long, flexible, and posit. charged side chain; Ionics-bound; H: V198 | 1.11 | 1.2 ± 0.34% | Positive charge addition causes disorder in heme binding | B |
| E352V | K-helix/ERR-triad associated with the heme group | Hydrophobic; Long, slightly flexible side chain; Strongly neg. charged; Ionics-bound; Fix metal iron; H: A348, R355 b, L356 and W406 | Hydrophobic; Short side chain; Folding interactions; H: A348 and L356 | 1.23 | 1.1 ± 0.11% | Disorder of the ERR-triad decreases heme-binding and stability | B |
| P433L | L-helix/Adjacent to essential residues for the heme bond | Polar residue with a ring at the N-term; H: A435 | Hydrophobic; Short side chain; Folding interactions; H: A435 | 0.39 | 7.5 ± 0.67% | Changes the natural orientation of heme-binding residues R427 and C429 | B |
| R484L | C-term coil/Hydrophobic cluster with ionic connection | Hydrophobic; Long, flexible, and posit. charged side chain; Ionics-bound; H: Q482, A449, and M486 | Hydrophobic; Short side chain; Folding interactions; H: A449 | −1.47 | 3.4 ± 0.8% | Loss of hydrophobic organization at the C-term region | B |
| SNV a | Conservation Score | CI | Residue Variety |
|---|---|---|---|
| P35 | 9 | 9 to 8 | L, P, G |
| L199 | 3 | 4 to 2 | T, N, L, V, F, I, A, S, G, M |
| W202 | 1 | 2 to 1 | T, E, R, W, G, C, M, L, A, I, N, Q, D, V, F, S |
| E352 | 9 | 9 to 9 | E |
| P433 | 3 | 4 to 2 | R, K, G, M, L, V, H, A, S, I, T, P, E, Q |
| R484 | 9 | 9 to 9 | R, T, L |
| Wild-Type | P35L | L199P | P433L | |
|---|---|---|---|---|
| Vmax (nmol·min−1·mg−1) | 0.360 | 0.086 | 0.252 | 0.055 |
| Km (µM) | 1.57 | 2.07 | 10.24 | 1.91 |
| Vmax/Km | 0.229 | 0.041 | 0.025 | 0.029 |
| SNP | Oligonucleotide Sequence (5′→3′) |
|---|---|
| P35L_Fwd | GTGCAAGAAGCCCAGGGCAAGAGGCGG |
| P35L_Rev | CCGCCTCTTGCCCTGGGCTTCTTGCAC |
| I173N_Fwd | CTCACCTGCAGCATCAACTGTTACCTCACCTTC |
| I173N_Rev | GAAGGTGAGGTAACAGTTGATGCTGCAGGTGAG |
| L199P_Fwd | CCAGTGGCTCCAGGTTTTTGGCACCTCCTGGATACATTTG |
| L199P_Rev | CAAATGTATCCAGGAGGTGCCAAAAACCTGGAGCCACTGG |
| W202R_Fwd | GGACCAGTGGCTCCTGGTTTTTAACACCTCCT |
| W202R_Rev | AGGAGGTGTTAAAAACCAGGAGCCACTGGTCC |
| V282L_Fwd | GCAGCCATGTGCAGGTGCCCTTCCAGG |
| V282L_Rev | CCTGGAAGGGCACCTGCACATGGCTGC |
| E352V_Fwd | GGCGCAGCACCACGGCGATGGTG |
| E352V_Rev | CACCATCGCCGTGGTGCTGCGCC |
| P433L_Fwd | GGCGCGCCAGCAGCTCGCCCAGG |
| P433L_Rev | CCTGGGCGAGCTGCTGGCGCGCC |
| R484L_Fwd | CCCCCATCCCCAGGGGCTGCAGC |
| R484L_Rev | GCTGCAGCCCCTGGGGATGGGGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prado, M.J.; Singh, S.; Ligabue-Braun, R.; Meneghetti, B.V.; Rispoli, T.; Kopacek, C.; Monteiro, K.; Zaha, A.; Rossetti, M.L.R.; Pandey, A.V. Characterization of Mutations Causing CYP21A2 Deficiency in Brazilian and Portuguese Populations. Int. J. Mol. Sci. 2022, 23, 296. https://doi.org/10.3390/ijms23010296
Prado MJ, Singh S, Ligabue-Braun R, Meneghetti BV, Rispoli T, Kopacek C, Monteiro K, Zaha A, Rossetti MLR, Pandey AV. Characterization of Mutations Causing CYP21A2 Deficiency in Brazilian and Portuguese Populations. International Journal of Molecular Sciences. 2022; 23(1):296. https://doi.org/10.3390/ijms23010296
Chicago/Turabian StylePrado, Mayara J., Shripriya Singh, Rodrigo Ligabue-Braun, Bruna V. Meneghetti, Thaiane Rispoli, Cristiane Kopacek, Karina Monteiro, Arnaldo Zaha, Maria L. R. Rossetti, and Amit V. Pandey. 2022. "Characterization of Mutations Causing CYP21A2 Deficiency in Brazilian and Portuguese Populations" International Journal of Molecular Sciences 23, no. 1: 296. https://doi.org/10.3390/ijms23010296
APA StylePrado, M. J., Singh, S., Ligabue-Braun, R., Meneghetti, B. V., Rispoli, T., Kopacek, C., Monteiro, K., Zaha, A., Rossetti, M. L. R., & Pandey, A. V. (2022). Characterization of Mutations Causing CYP21A2 Deficiency in Brazilian and Portuguese Populations. International Journal of Molecular Sciences, 23(1), 296. https://doi.org/10.3390/ijms23010296