
Checkpoints That Regulate Balanced Biosynthesis of Lipopolysaccharide and Its Essentiality in Escherichia coli




Abstract
1. Introduction
2. Regulation of the Balance between LPS and Phospholipid Biosynthesis Flux of the Common Metabolic Precursor (R)-3-hydroxymyristate in Two Pathways
3. FtsH-Mediated Control of LpxC Turnover
4. LapB Functions to Regulate FtsH-Mediated Proteolysis of LpxC and Determine the Major Checkpoint of Regulating LPS Biosynthesis in Concert with FtsH
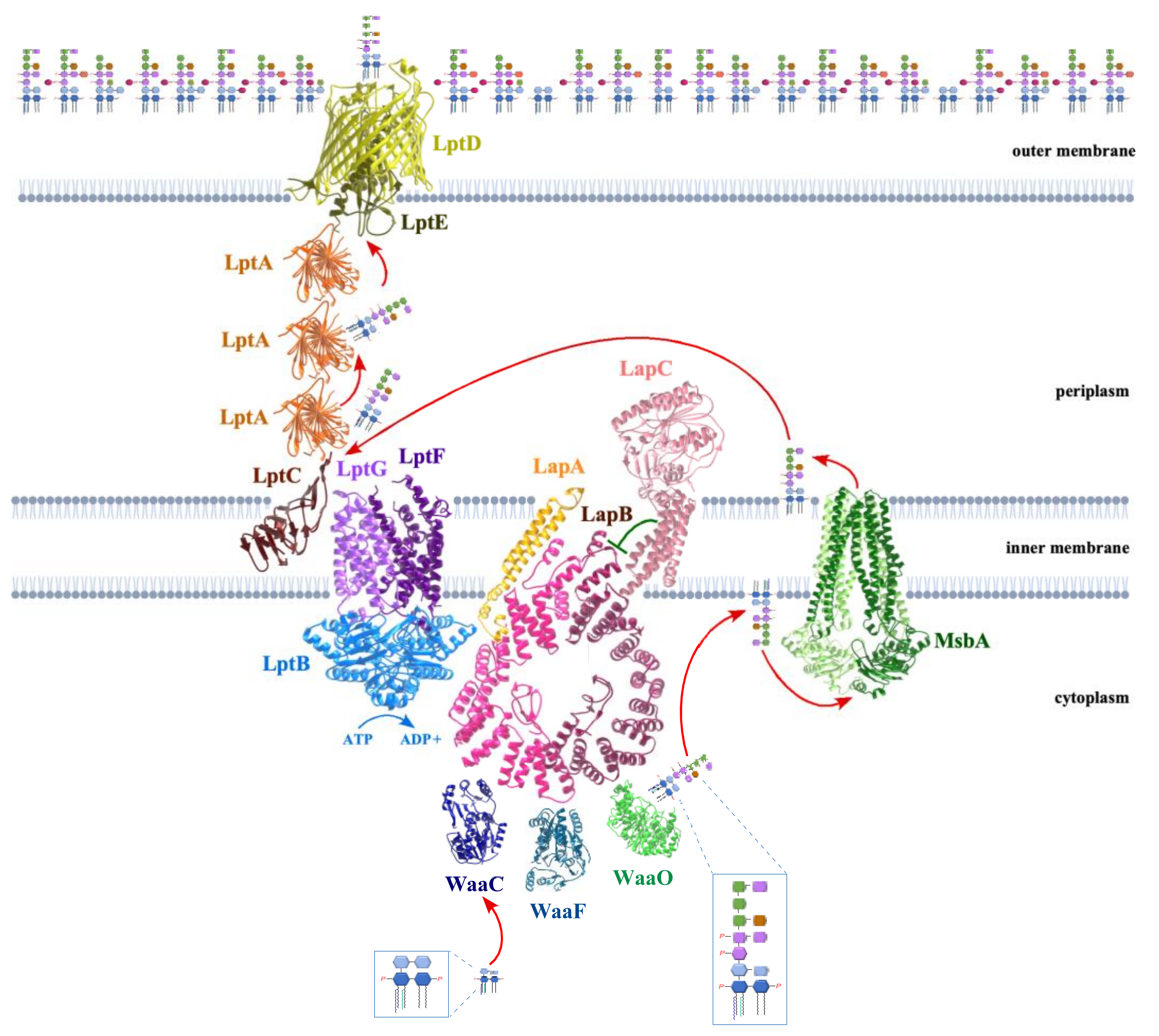
5. LapB Coordinates the LPS Synthesis and Translocation
6. Regulation of LapB/FtsH Proteolytic Control of LpxC by LapC
7. Mutations That Simultaneously Confer Sensitivity to CHIR090 and Induce Transcription from LPS Defects-Responsive rpoEP3 Promoter Identify the lapC Gene
8. How Demand for the LPS Synthesis Is Sensed by LapC and Impacts Its Interaction with LapB
9. Structural Studies of LapC and LapB Reveal LapC Recognition of Lipid A and Essential Domains of LapB
10. LpxC Alternative Proteolysis-Regulated Turnover by Heat Shock Induced HslVU Protease Complex
11. Negative Regulation of LpxC by the GcvB sRNA-Potential Transcriptional Control
12. The Cross-Talk between Various Intricate Pathways Linking Lipid and LPS Synthesis That Controls LpxC Levels
13. MsbA Flippase Acts as a Major Checkpoint to Prevent Translocation of Underacylated LPS
14. Cardiolipins Are Required for the Viability of Strains Synthesizing Underacylated LPS, Thus Providing Another Checkpoint
15. Checkpoints That Regulate Relative Abundance of LPS Glycoforms and Transcription of the Main waaQ Operon
16. Critical Role of RpoH, RpoE and Two-Component Systems in Regulating LPS Biosynthesis
17. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Raina, S. Regulated assembly of LPS, its structural alterations and cellular response to LPS defects. Int. J. Mol. Sci. 2019, 20, 356. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Raina, S. Regulated control of the assembly and diversity of LPS by noncoding sRNAs. BioMed. Res. Int. 2015, 2015, 153561. [Google Scholar] [CrossRef]
- Szczesny, M.; Beloin, C.; Ghigo, J.M. Increased osmolarity in biofilm triggers RcsB-dependent lipid A palmitoylation in Escherichia coli. mBio 2018, 9, e01415-18. [Google Scholar] [CrossRef] [PubMed]
- Dartigalongue, C.; Loferer, H.; Raina, S. EcfE, a new essential inner membrane protease: Its role in the regulation of heat shock response in Escherichia coli. EMBO J. 2001, 20, 5908–5918. [Google Scholar] [CrossRef]
- Hagiwara, D.; Yamashino, T.; Mizuno, T. A genome-wide view of the Escherichia coli BasS-BasR two-component system implicated in iron-responses. Biosci. Biotechnol. Biochem. 2004, 68, 1758–1767. [Google Scholar] [CrossRef]
- Klein, G.; Lindner, B.; Brade, H.; Raina, S. Molecular basis of lipopolysaccharide heterogeneity in Escherichia coli: Envelope stress-responsive regulators control the incorporation of glycoforms with a third 3-deoxy-α-D-manno-oct-2-ulosonic acid and rhamnose. J. Biol. Chem. 2011, 286, 42787–42807. [Google Scholar] [CrossRef]
- Gogol, E.B.; Rhodius, V.A.; Papenfort, K.; Vogel, J.; Gross, C.A. Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc. Natl. Acad. Sci. USA 2011, 108, 12875–12880. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Müller-Loennies, S.; Lindner, B.; Kobylak, N.; Brade, H.; Raina, S. Molecular and structural basis of inner core lipopolysaccharide alterations in Escherichia coli: Incorporation of glucuronic acid and phosphoethanolamine in the heptose region. J. Biol. Chem. 2013, 288, 8111–8127. [Google Scholar] [CrossRef] [PubMed]
- Gadishaw-Lue, C.; Banaag, A.; Birstonas, S.; Francis, A.S.; Barnett Foster, D. Bile salts differentially enhance resistance of enterohemorrhagic Escherichia coli O157:H7 to host defense peptides. Infect. Immun. 2021, 89, e00719-20. [Google Scholar] [CrossRef]
- Mohan, S.; Kelly, T.M.; Eveland, S.S.; Raetz, C.R.H.; Anderson, M.S. An Escherichia coli gene (fabZ) encoding (3R)-hydroxymyristoyl acyl carrier protein dehydrase. Relation to fabA and suppression of mutations in lipid A biosynthesis. J. Biol. Chem. 1994, 269, 32896–32903. [Google Scholar] [CrossRef]
- Ogura, T.; Inoue, K.; Tatsuta, T.; Suzaki, T.; Karata, K.; Young, K.; Su, L.H.; Fierke, C.A.; Jackman, J.E.; Raetz, C.R.; et al. Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli. Mol. Microbiol. 1999, 31, 833–844. [Google Scholar] [CrossRef]
- Klein, G.; Kobylak, N.; Lindner, B.; Stupak, A.; Raina, S. Assembly of lipopolysaccharide in Escherichia coli requires the essential LapB heat shock protein. J. Biol. Chem. 2014, 289, 14829–14853. [Google Scholar] [CrossRef]
- Anderson, M.S.; Bulawa, C.E.; Raetz, C.R.H. The biosynthesis of gram-negative endotoxin. Formation of lipid A precursors from UDP-GlcNAc in extracts of Escherichia coli. J. Biol. Chem. 1985, 260, 15536–15541. [Google Scholar] [CrossRef]
- Sperandeo, P.; Lau, F.K.; Carpentieri, A.; De Castro, C.; Molinaro, A.; Dehò, G.; Silhavy, T.J.; Polissi, A. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J. Bacteriol. 2008, 190, 4460–4469. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Sherman, D.J.; Silhavy, T.J.; Ruiz, N.; Kahne, D. Lipopolysaccharide transport and assembly at the outer membrane: The PEZ model. Nat. Rev. Microbiol. 2016, 14, 337–345. [Google Scholar] [CrossRef]
- Zhou, Z.; White, K.A.; Polissi, A.; Georgopoulos, C.; Raetz, C.R. Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 1998, 273, 12466–12475. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Lindner, B.; Brabetz, W.; Brade, H.; Raina, S. Escherichia coli K-12 suppressor-free mutants lacking early glycosyltransferases and late acetyltransferases. Minimal lipopolysaccharide structure and induction of envelope stress response. J. Biol. Chem. 2009, 284, 15369–15389. [Google Scholar] [CrossRef] [PubMed]
- Gorzelak, P.; Klein, G.; Raina, S. Molecular basis of essentiality of early critical steps in the lipopolysaccharide biogenesis in Escherichia coli K-12: Requirement of MsbA, cardiolipin, LpxL, LpxM and GcvB. Int. J. Mol. Sci. 2021, 22, 5099. [Google Scholar] [CrossRef] [PubMed]
- Douglass, M.V.; Cléon, F.; Trent, M.S. Cardiolipin aids in lipopolysaccharide transport to the gram-negative outer membrane. Proc. Natl. Acad. Sci. USA 2021, 118, e2018329118. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Stupak, A.; Biernacka, D.; Wojtkiewicz, P.; Lindner, B.; Raina, S. Multiple transcriptional factors regulate transcription of the rpoE gene in Escherichia coli under different growth conditions and when the lipopolysaccharide biosynthesis is defective. J. Biol. Chem. 2016, 291, 22999–23019. [Google Scholar] [CrossRef] [PubMed]
- Artsimovitch, I.; Landick, R. The transcriptional regulator RfaH stimulates RNA chain synthesis after recruitment to elongation complexes by the exposed nontemplate DNA strand. Cell 2002, 109, 193–203. [Google Scholar] [CrossRef]
- Galloway, S.M.; Raetz, C.R. A mutant of Escherichia coli defective in the first step of endotoxin biosynthesis. J. Biol. Chem. 1990, 265, 6394–6402. [Google Scholar] [CrossRef]
- Bohl, T.E.; Aihara, H. Current progress in the structural and biochemical characterization of proteins involved in the assembly of lipopolysaccharide. Int. J. Microbiol. 2018, 2018, 5319146. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Bull, H.G.; Galloway, S.M.; Kelly, T.M.; Mohan, S.; Radika, K.; Raetz, C.R. UDP-N-acetylglucosamine acyltransferase of Escherichia coli. The first step of endotoxin biosynthesis is thermodynamically unfavourable. J. Biol. Chem. 1993, 268, 19858–19865. [Google Scholar] [CrossRef]
- Anderson, M.S.; Raetz, C.R. Biosynthesis of lipid A precursors in Escherichia coli. A cytoplasmic acyltransferase that converts UDP-N-acetylglucosamine to UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine. J. Biol. Chem. 1987, 262, 5159–5169. [Google Scholar] [CrossRef]
- Wyckoff, T.J.; Lin, S.; Cotter, R.J.; Dotson, G.D.; Raetz, C.R. Hydrocarbon rulers in UDP-N-acetylglucosamine acyltransferases. J. Biol. Chem. 1998, 273, 32369–32372. [Google Scholar] [CrossRef]
- Wyckoff, T.J.; Raetz, C.R. The active site of Escherichia coli UDP-N-acetylglucosamine acyltransferase. Chemical modification and site-directed mutagenesis. J. Biol. Chem. 1999, 274, 27047–27055. [Google Scholar] [CrossRef]
- Anderson, M.S.; Robertson, A.D.; Macher, I.; Raetz, C.R. Biosynthesis of lipid A in Escherichia coli: Identification of UDP-3-O-[(R)-3-hydroxymyristoyl]-α-D-glucosamine as a precursor of UDP-N2,O3-bis[(R)-3-hydroxymyristoyl]-α-D-glucosamine. Biochemistry 1988, 27, 1908–1917. [Google Scholar] [CrossRef]
- Kelly, T.M.; Stachula, S.A.; Raetz, C.R.; Anderson, M.S. The firA gene of Escherichia coli encodes UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine N-acyltransferase. The third step of endotoxin biosynthesis. J. Biol. Chem. 1993, 268, 19866–19874. [Google Scholar] [CrossRef]
- Young, K.; Silver, L.L.; Bramhill, D.; Cameron, P.; Eveland, S.S.; Raetz, C.R.; Hyland, S.A.; Anderson, M.S. The envA permeability/cell division gene of Escherichia coli encodes the second enzyme of lipid A biosynthesis. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase. J. Biol. Chem. 1995, 270, 30384–30391. [Google Scholar] [CrossRef]
- Jackman, J.E.; Raetz, C.R.H.; Fierke, C.A. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry 1999, 38, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Bartling, C.M.; Raetz, C.R.H. Steady-state kinetics and mechanism of LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry 2008, 47, 5290–5302. [Google Scholar] [CrossRef] [PubMed]
- Führer, F.; Langklotz, S.; Narberhaus, F. The C-terminal end of LpxC is required for degradation by the FtsH protease. Mol. Microbiol. 2006, 59, 1025–1036. [Google Scholar] [CrossRef]
- Zeng, D.; Zhao, J.; Chung, H.S.; Guan, Z.; Raetz, C.R.H.; Zhou, P. Mutants resistant to LpxC inhibitors by rebalancing cellular homeostasis. J. Biol. Chem. 2013, 288, 5475–5486. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, D.; Gorzelak, P.; Klein, G.; Raina, S. Regulation of the first committed step in lipopolysaccharide biosynthesis catalyzed by LpxC requires the essential protein LapC (YejM) and HslVU protease. Int. J. Mol. Sci. 2020, 21, 9088. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.L.; Rutherford, S.T.; Silhavy, T.J. Border control: Regulating LPS biogenesis. Trends Microbiol. 2021, 29, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.F.; Donachie, W.D. Transcriptional organization within an Escherichia coli cell division gene cluster: Direction of transcription of the cell separation gene envA. J. Bacteriol. 1984, 160, 724–732. [Google Scholar] [CrossRef]
- Sorensen, P.G.; Lutkenhaus, J.; Young, K.; Eveland, S.S.; Anderson, M.S.; Raetz, C.R.H. Regulation of UDP-3-O-[R-3-hydroxymyristoyl]-N-acetylglucosamine deacetylase in Escherichia coli. The second enzymatic step of lipid A biosynthesis. J. Biol. Chem. 1996, 271, 25898–25905. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, K.; Jackowski, S.; Rock, C.O.; Cronan, J.E., Jr. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol. Rev. 1993, 57, 522–542. [Google Scholar] [CrossRef]
- Führer, F.; Müller, A.; Baumann, H.; Langklotz, S.; Kutscher, B.; Narberhaus, F. Sequence and length recognition of the C-terminal turnover element of LpxC, a soluble substrate of the membrane-bound FtsH protease. J. Mol. Biol. 2007, 372, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Schäkermann, M.; Langklotz, S.; Narberhaus, F. FtsH-mediated coordination of lipopolysaccharide biosynthesis in Escherichia coli correlates with the growth rate and the alarmone (p)ppGpp. J. Bacteriol. 2013, 195, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Missiakas, D.; Betton, J.M.; Raina, S. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol. Microbiol. 1996, 21, 871–884. [Google Scholar] [CrossRef]
- Brooke, J.S.; Valvano, M.A. Biosynthesis of inner core lipopolysaccharide in enteric bacteria identification and characterization of a conserved phosphoheptose isomerase. J. Biol. Chem. 1996, 271, 3608–3614. [Google Scholar] [CrossRef]
- Gronow, S.; Brabetz, W.; Brade, H. Comparative functional characterization in vitro of heptosyltransferase I (WaaC) and II (WaaF) from Escherichia coli. Eur. J. Biochem. 2000, 267, 6602–6611. [Google Scholar] [CrossRef]
- Guo, M.S.; Updegrove, T.B.; Gogol, E.B.; Shabalina, S.A.; Gross, C.A.; Storz, G. MicL, a new σE-dependent sRNA, combats envelope stress by repressing synthesis of Lpp, the major outer membrane lipoprotein. Genes Dev. 2014, 28, 1620–1634. [Google Scholar] [CrossRef] [PubMed]
- McMillan, H.M.; Kuehn, M.J. The extracellular vesicle generation paradox: A bacterial point of view. EMBO J. 2021, 40, e108174. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Kulp, A.; Kuehn, M.J. Modulation of bacterial outer membrane vesicle production by envelope structure and content. BMC Microbiol. 2014, 14, 324. [Google Scholar] [CrossRef]
- Mahalakshmi, S.; Sunayana, M.R.; SaiSree, L.; Reddy, M. yciM is an essential gene required for regulation of lipopolysaccharide synthesis in Escherichia coli. Mol. Microbiol. 2014, 91, 145–157. [Google Scholar] [CrossRef]
- Prince, C.; Jia, Z. An unexpected duo: Rubredoxin binds nine TPR motifs to form LapB, an essential regulator of lipopolysaccharide synthesis. Structure 2015, 23, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Göpel, Y.; Khan, M.A.; Görke, B. Ménage à trois: Post-transcriptional control of the key enzyme for cell envelope synthesis by a base-pairing small RNA, an RNase adaptor protein, and a small RNA mimic. RNA Biol. 2014, 11, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Hirvas, L.; Nurminen, M.; Helander, I.M.; Vuorio, R.; Vaara, M. The lipid A biosynthesis deficiency of the Escherichia coli antibiotic-supersensitive mutant LH530 is suppressed by a novel locus, ORF195. Microbiology 1997, 143, 73–81. [Google Scholar] [CrossRef] [PubMed]
- De Lay, N.R.; Cronan, J.E. Genetic interaction between the Escherichia coli AcpT phosphopantetheinyl transferase and the YejM inner membrane protein. Genetics 2008, 178, 1327–1337. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dalebroux, Z.D.; Edrozo, M.B.; Pfuetzner, R.A.; Ressl, S.; Kulasekara, B.R.; Blanc, M.P.; Miller, S.I. Delivery of cardiolipins to the Salmonella outer membrane is necessary for survival within host tissues and virulence. Cell Host Microbe 2015, 17, 441–451. [Google Scholar] [CrossRef]
- Dalebroux, Z.D.; Matamouros, S.; Whittington, D.; Bishop, R.E.; Miller, S.I. PhoPQ regulates acidic glycerophospholipid content of the Salmonella Typhimurium outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, 1963–1968. [Google Scholar] [CrossRef]
- Cian, M.B.; Giordano, N.P.; Masilamani, R.; Minor, K.E.; Dalebroux, Z.D. Salmonella enterica serovar Typhimurium uses PdgA/YejM to regulate lipopolysaccharide assembly during bacteremia. Infect. Immun. 2020, 88, e00758-19. [Google Scholar]
- Clairfeuille, T.; Buchholz, K.R.; Li, Q.; Verschueren, E.; Liu, P.; Sangaraju, D.; Park, S.; Noland, C.L.; Storek, K.M.; Nickerson, N.N.; et al. Structure of the essential inner membrane lipopolysaccharide-PbgA complex. Nature 2020, 584, 479–483. [Google Scholar] [CrossRef]
- Nguyen, D.; Kelly, K.; Qiu, N.; Misra, R. YejM controls LpxC levels by regulating protease activity of the FtsH/YciM complex of Escherichia coli. J. Bacteriol. 2020, 202, e00303–e00320. [Google Scholar] [CrossRef]
- Guest, R.L.; Samé Guerra, D.; Wissler, M.; Grimm, J.; Silhavy, T.J. YejM modulates activity of the YciM/FtsH protease complex to prevent lethal accumulation of lipopolysaccharide. mBio 2020, 11, e00598-20. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Bernhardt, T.G. An essential membrane protein modulates the proteolysis of LpxC to control lipopolysaccharide synthesis in Escherichia coli. mBio 2020, 11, e00939-20. [Google Scholar] [CrossRef]
- Barb, A.W.; Zhou, P. Mechanism and inhibition of LpxC. An essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr. Pharm. Biotech. 2008, 9, 9–15. [Google Scholar]
- Zhou, P.; Hong, J. Structure- and ligand-dynamics-based design of novel antibiotics targeting lipid A enzymes LpxC and LpxH in Gram-negative bacteria. Acc. Chem. Res. 2021, 54, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Thomanek, N.; Arends, J.; Lindemann, C.; Barkovits, K.; Meyer, H.E.; Marcus, K.; Narberhaus, F. Intricate crosstalk between lipopolysaccharide, phospholipid and fatty acid metabolism in Escherichia coli modulates proteolysis of LpxC. Front. Microbiol. 2019, 9, 3285. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Park, S.H.; Lee, C.R. The inner membrane protein LapB is required for adaptation to cold stress in an LpxC-independent manner. J. Microbiol. 2021, 59, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, Z.; Tang, X.; Huang, S.; Li, H.; Peng, B.; Dong, C. Structural insights into cardiolipin transfer from the inner membrane to the outer membrane by PbgA in Gram-negative bacteria. Sci. Rep. 2016, 6, 30815. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Petersen, E.M.; Hinds, T.R.; Zheng, N.; Miller, S.I. Structure of an inner membrane protein required for PhoPQ-regulated increases in outer membrane cardiolipin. mBio 2020, 11, e03277-19. [Google Scholar] [CrossRef]
- Qiu, N.; Misra, R. Overcoming iron deficiency of an Escherichia coli tonB mutant by increasing outer membrane permeability. J. Bacteriol. 2019, 201, e00340-19. [Google Scholar] [CrossRef]
- Gabale, U.; Peña Palomino, P.A.; Kim, H.A.; Chen, W.; Ressi, S. The essential inner membrane protein YejM is a metalloenzyme. Sci. Rep. 2020, 10, 17794. [Google Scholar] [CrossRef]
- Perez-Riba, A.; Itzhaki, L.S. The tetratricopeptide-repeat motif is a versatile platform that enables diverse modes of molecular recognition. Curr. Opin. Struct. Biol. 2019, 54, 43–49. [Google Scholar] [CrossRef]
- Missiakas, D.; Schwager, F.; Betton, J.M.; Georgopoulos, C.; Raina, S. Identification and characterization of HslV HslU (ClpQ ClpY) proteins involved in overall proteolysis of misfolded proteins in Escherichia coli. EMBO J. 1996, 15, 6899–68909. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.E.; Blattner, F.R. Characterization of twenty-six new heat shock genes of Escherichia coli. J. Bacteriol. 1993, 175, 5242–5252. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.F.; Zhou, Y.N.; Gottesman, S. Redundant in vivo proteolytic activities of Escherichia coli Lon and the ClpYQ (HslUV) protease. J. Bacteriol. 1999, 181, 3681–3687. [Google Scholar] [CrossRef] [PubMed]
- Kanemori, M.; Yanagi, H.; Yura, T. The ATP-dependent HslVU/ClpQY protease participates in turnover of cell division inhibitor SulA in Escherichia coli. J. Bacteriol. 1999, 181, 3674–3680. [Google Scholar] [CrossRef]
- Baytshtok, V.; Fei, X.; Shih, T.T.; Grant, R.A.; Santos, J.C.; Baker, T.A.; Sauer, R.T. Heat activates the AAA+ HslUV protease by melting an axial autoinhibitory plug. Cell Rep. 2021, 34, 108639. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Vogel, J. Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front. Cell. Infect. Microbiol. 2014, 4, 91. [Google Scholar] [CrossRef]
- Storz, G.; Vogel, J.; Wassarman, K.M. Regulation by small RNAs in bacteria: Expanding frontiers. Mol. Cell 2011, 43, 880–891. [Google Scholar] [CrossRef]
- Fröhlich, K.S.; Papenfort, K. Regulation outside the box: New mechanisms for small RNAs. Mol. Microbiol. 2020, 114, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, V.; Papenfort, K.; Lucchini, S.; Hinton, J.C.; Vogel, J. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 2009, 16, 840–846. [Google Scholar] [CrossRef]
- De Lay, N.; Schu, D.J.; Gottesman, S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013, 288, 7996–8003. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Raina, S. Small regulatory bacterial RNAs regulating the envelope stress response. Biochem. Soc. Trans. 2017, 45, 417–425. [Google Scholar] [CrossRef]
- Lalaouna, D.; Eyraud, A.; Devinck, A.; Prévost, K.; Massé, E. GcvB small RNA uses two distinct seed regions to regulate an extensive targetome. Mol. Microbiol. 2019, 111, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.M.; Papenfort, K.; Pernitzsch, S.R.; Mollenkopf, H.J.; Hinton, J.C.; Vogel, J. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol. Microbiol. 2011, 81, 1144–1165. [Google Scholar] [CrossRef]
- Pulvermacher, S.C.; Stauffer, L.T.; Stauffer, G.V. Role of the Escherichia coli Hfq protein in GcvB regulation of oppA and dppA mRNAs. Microbiology 2009, 155, 115–123. [Google Scholar] [CrossRef]
- Coornaert, A.; Chiaruttini, C.; Springer, M.; Guillier, M. Post-transcriptional control of the Escherichia coli PhoQ-PhoP two-component system by multiple sRNAs involves a novel pairing region of GcvB. PLoS Genet. 2013, 9, e1003156. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, M.; Okayama, H.; Lejars, M.; Kanda, T.; Tanaka, Y.; Itaya, K.; Okuno, M.; Itoh, T.; Iwai, N.; Wachi, M. Mining RNA-seq data reveals the massive regulon of GcvB small RNA and its physiological significance in maintaining amino acid homeostasis in Escherichia coli. Mol. Microbiol. in press. 2021. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, M.; Chao, Y.; Vogel, J. Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. EMBO J. 2015, 34, 1478–1492. [Google Scholar] [CrossRef] [PubMed]
- Emiola, A.; Andrews, S.S.; Heller, C.; George, J. Crosstalk between the lipopolysaccharide and phospholipid pathways during outer membrane biogenesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, 3108–3113. [Google Scholar] [CrossRef] [PubMed]
- May, K.L.; Silhavy, T.J. The Escherichia coli phospholipase PldA regulates outer membrane homeostasis via lipid signaling. mBio 2018, 9, e00379-18. [Google Scholar] [CrossRef] [PubMed]
- Cronan, J.E., Jr.; Subrahmanyam, S. Fad, transcriptional co-ordination of metabolic expediency. Mol. Microbiol. 1998, 29, 937–943. [Google Scholar] [CrossRef]
- Heath, R.J.; Rock, C.O. Roles of the FabA and FabZ β-hydroxyacyl-acyl carrier protein dehydratases in Escherichia coli fatty acid biosynthesis. J. Biol. Chem. 1996, 271, 27795–27801. [Google Scholar] [CrossRef] [PubMed]
- Byers, D.M.; Gong, H. Acyl carrier protein: Structure-function relationships in a conserved multifunctional protein family. Biochem. Cell Biol. 2007, 85, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Vorachek-Warren, M.K.; Ramirez, S.; Cotter, R.J.; Raetz, C.R.H. A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. J. Biol. Chem. 2002, 277, 14194–14205. [Google Scholar] [CrossRef] [PubMed]
- Doerrler, W.T.; Raetz, C.R.H. ATPase activity of the MsbA lipid flippase of Escherichia coli. J. Biol. Chem. 2002, 277, 36697–36705. [Google Scholar] [CrossRef] [PubMed]
- Doerrler, W.T.; Gibbons, H.S.; Raetz, C.R.H. MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem. 2004, 279, 45102–45109. [Google Scholar] [CrossRef]
- Eckford, P.D.W.; Sharom, F.J. The reconstituted Escherichia coli MsbA protein displays lipid flippase activity. Biochem. J. 2010, 429, 195–203. [Google Scholar] [CrossRef][Green Version]
- Mi, W.; Li, Y.; Yoon, S.H.; Ernst, R.K.; Walz, T.; Liao, M. Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 2017, 549, 233–237. [Google Scholar] [CrossRef]
- Ho, H.; Miu, A.; Alexander, M.K.; Garcia, N.K.; Oh, A.; Zilberleyb, I.; Reichelt, M.; Austin, C.D.; Tam, C.; Shriver, S.; et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 2018, 557, 196–201. [Google Scholar] [CrossRef]
- Padayatti, P.S.; Lee, S.C.; Stanfield, R.L.; Wen, P.C.; Tajkhorshid, E.; Wilson, I.A.; Zhang, Q. Structural insights into the lipid A transport pathway in MsbA. Structure 2019, 27, 1114–1123.e3. [Google Scholar] [CrossRef]
- Ward, A.; Reyes, C.L.; Yu, J.; Roth, C.B.; Chang, G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA 2007, 104, 19005–19010. [Google Scholar] [CrossRef]
- Thélot, F.A.; Zhang, W.; Song, K.; Xu, C.; Huang, J.; Liao, M. Distinct allosteric mechanisms of first-generation MsbA inhibitors. Science 2021, 374, 580–585. [Google Scholar] [CrossRef]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef]
- Tan, B.K.; Bogdanov, M.; Zhao, J.; Dowhan, W.; Raetz, C.R.; Guan, Z. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc. Natl. Acad. Sci. USA 2012, 109, 16504–16509. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, S.; Matsuzaki, H.; Shibuya, I. Active increase in cardiolipin synthesis in the stationary growth phase and its physiological significance in Escherichia coli. FEBS Lett. 1993, 336, 221–224. [Google Scholar] [CrossRef]
- Rowlett, V.W.; Mallampalli, V.K.P.S.; Karlstaedt, A.; Dowhan, W.; Taegtmeyer, H.; Margolin, W.; Vitrac, H. Impact of membrane phospholipid alterations in Escherichia coli on cellular function and bacterial stress adaptation. J. Bacteriol. 2017, 199, e00849-16. [Google Scholar] [CrossRef] [PubMed]
- Wojtkiewicz, P.; Biernacka, D.; Gorzelak, P.; Stupak, A.; Klein, G.; Raina, S. Multicopy suppressor analysis of strains lacking cytoplasmic peptidyl-prolyl cis/trans isomerases identifies three new PPIase activities in Escherichia coli that includes the DksA transcription factor. Int. J. Mol. Sci. 2020, 21, 5843. [Google Scholar] [CrossRef] [PubMed]
- Müller-Loennies, S.; Lindner, B.; Brade, H. Structural analysis of oligosaccharides from lipopolysaccharide (LPS) of Escherichia coli K12 strain W3100 reveals a link between inner and outer core LPS biosynthesis. J. Biol. Chem. 2003, 278, 34090–34101. [Google Scholar] [CrossRef]
- Bailey, M.J.; Hughes, C.; Koronakis, V. RfaH and the ops element, components of a novel system controlling bacterial transcription elongation. Mol. Microbiol. 1997, 26, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jensen, S.; Hallman, R.; Reeves, P.R. Expression of the O antigen gene cluster is regulated by RfaH through the JUMPstart sequence. FEMS Microbiol. Lett. 1998, 165, 201–206. [Google Scholar] [CrossRef]
- Wang, B.; Artsimovitch, I. NusG, an ancient yet rapidly evolving transcription factor. Front. Microbiol. 2021, 11, 619618. [Google Scholar] [CrossRef]
- Zuber, P.K.; Schweimer, K.; Rösch, P.; Artsimovitch, I.; Knauer, S.H. Reversible fold-switching controls the functional cycle of the antitermination factor RfaH. Nat. Commun. 2019, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Raina, S.; Georgopoulos, C. The htrM gene, whose product is essential for Escherichia coli viability only at elevated temperatures, is identical to the rfaD gene. Nucleic Acids Res. 1991, 19, 3811–3819. [Google Scholar] [CrossRef]
- Klein, G.; Wojtkiewicz, P.; Biernacka, D.; Stupak, A.; Gorzelak, P.; Raina, S. Identification of substrates of cytoplasmic peptidyl-prolyl cis/trans isomerases and their collective essentiality in Escherichia coli. Int. J. Mol. Sci. 2020, 21, 4212. [Google Scholar] [CrossRef]
- Dartigalongue, C.; Missiakas, D.; Raina, S. Characterization of the Escherichia coli σE regulon. J. Biol. Chem. 2001, 276, 20866–20875. [Google Scholar] [CrossRef]
- Raina, S.; Missiakas, D.; Georgopoulos, C. The rpoE gene encoding the σE (σ24) heat shock sigma factor of Escherichia coli. EMBO J. 1995, 14, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Rouviere, P.E.; De Las Peñas, A.; Mecsas, J.; Lu, C.Z.; Rudd, K.E.; Gross, C.A. rpoE, the gene encoding the second heat-shock sigma factor, σE, in Escherichia coli. EMBO J. 1995, 14, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Rhodius, V.A.; Suh, W.C.; Nonaka, G.; West, J.; Gross, C.A. Conserved and variable functions of the σE stress response in realated genomes. PLoS Biol. 2005, 4, e2. [Google Scholar] [CrossRef]
- Moon, K.; Six, D.A.; Lee, H.-J.; Raetz, C.R.H.; Gottesman, S. Complex transcriptional and post-transcriptional regulation of an enzyme for lipopolysaccharide modification. Mol. Microbiol. 2013, 89, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, A.; Lu, A.; Mandin, P.; Springer, M.; Gottesman, S.; Guillier, M. MicA sRNA links the PhoP regulon to cell envelope stress. Mol. Microbiol. 2010, 76, 467–479. [Google Scholar] [CrossRef]
- Wang, Q.P.; Kaguni, J.M. A novel sigma factor is involved in expression of the rpoH gene of Escherichia coli. J. Bacteriol. 1989, 171, 4248–4253. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.W.; Gross, C.A. Identification of the σE subunit of Escherichia coli RNA polymerase: A second alternative σ factor involved in high-temperature gene expression. Genes Dev. 1989, 3, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Garénaux, E.; Carriere, J.; Rolhion, N.; Guérardel, Y.; Barnich, N.; Bonnet, R.; Darfeuille-Michaud, A. Analysis of the σE regulon in Crohn’s disease-associated Escherichia coli revealed involvement of the waaWVL operon in biofilm formation. J. Bacteriol. 2015, 197, 1451–1465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Langklotz, S.; Schäkermann, M.; Narberhaus, F. Control of lipopolysaccharide biosynthesis by FtsH-mediated proteolysis of LpxC is conserved in enterobacteria but not in all gram-negative bacteria. J. Bacteriol. 2011, 193, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Andolina, G.; Bencze, L.C.; Zerbe, K.; Müller, M.; Steinmann, J.; Kocherla, H.; Mondal, M.; Sobek, J.; Moehle, K.; Malojčić, G.; et al. A peptidomimetic antibiotic interacts with the periplasmic domain of LptD from Pseudomonas aeruginosa. ACS Chem. Biol. 2018, 13, 666–675. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | EC Number | Product |
|---|---|---|
| lpxA | 2.3.1.129 | acyl-[acyl-carrier-protein]-UDP-N-acetyloglucosamine O-acyltransferase |
| lpxB | 2.4.1.182 | lipid A disaccharide synthase |
| lpxC | 3.5.1.108 | UDP-3-O-acyl N-acetylglucosamine deacetylase |
| lpxD | 2.3.1.191 | UDP-3-O-(3-hydroxyacyl)-glucosamine N-acyltransferase |
| lpxH | 3.6.1.54 | UDP-2,3-diacylglucosamine diphosphatase |
| lpxK | 2.7.1.130 | tetraacyldisaccharide A 4′-kinase |
| lpxL | 2.3.1.241 | Kdo2-lipid IVA acyltransferase |
| lpxM | 2.3.1.243 | acyl-Kdo2-lipid IVA acyltransferase |
| lpxP | 2.3.1.242 | Kdo2-lipid IVA palmitoleoyltransferase |
| lpxT | 2.7.4.29 | Kdo2-lipid IVA phosphotransferase |
| waaA | 2.4.99.12/ 2.4.99.13 | lipid IVA 3-deoxy-α-D-manno-octulosonic acid transferase/ (Kdo)-lipid IVA 3-deoxy-α-D-manno-octulosonic acid transferase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, G.; Wieczorek, A.; Szuster, M.; Raina, S. Checkpoints That Regulate Balanced Biosynthesis of Lipopolysaccharide and Its Essentiality in Escherichia coli. Int. J. Mol. Sci. 2022, 23, 189. https://doi.org/10.3390/ijms23010189
Klein G, Wieczorek A, Szuster M, Raina S. Checkpoints That Regulate Balanced Biosynthesis of Lipopolysaccharide and Its Essentiality in Escherichia coli. International Journal of Molecular Sciences. 2022; 23(1):189. https://doi.org/10.3390/ijms23010189
Chicago/Turabian StyleKlein, Gracjana, Alicja Wieczorek, Martyna Szuster, and Satish Raina. 2022. "Checkpoints That Regulate Balanced Biosynthesis of Lipopolysaccharide and Its Essentiality in Escherichia coli" International Journal of Molecular Sciences 23, no. 1: 189. https://doi.org/10.3390/ijms23010189
APA StyleKlein, G., Wieczorek, A., Szuster, M., & Raina, S. (2022). Checkpoints That Regulate Balanced Biosynthesis of Lipopolysaccharide and Its Essentiality in Escherichia coli. International Journal of Molecular Sciences, 23(1), 189. https://doi.org/10.3390/ijms23010189