
Pathophysiology of the Different Clinical Phenotypes of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Search Strategy
2.2. Data Extraction
2.3. Qualitive Analysis and Synthesis
3. Anatomy
3.1. The Structure of Myelin Sheaths
3.2. The Structure of Ranvier’s Node and Paranodal Region
3.3. Nodal and Paranodal Associated Antibodies
4. Pathogenesis
4.1. Inflammatory Process
4.2. Demyelination Process
4.2.1. The Role of Autoantibodies against Nodal or Paranodal Proteins
4.2.2. The Role of Antibodies against the Hemi-Node-Type Region
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AnkB | the ankyrin B |
| APRIL | a proliferation-inducing ligand |
| ASPR1 | contactin-associated protein 1 |
| BL | the basal lamina |
| CAM | cell adhesion molecules |
| CCPD | combined central and peripheral demyelination |
| CIDP | chronic inflammatory demyelinating polyradiculoneuropathy |
| CISP | chronic immune sensory polyradiculopathy |
| CNTN1 | contactin-1 |
| DADS | distal acquired demyelinating symmetric neuropathy |
| EAE | experimental autoimmune encephalomyelitis |
| EFNS | European Federation of Neurological Societies |
| IL-1ra | interleukin-1 receptor antagonist |
| IVIg | intravenous immunoglobulin |
| JXP | the juxtaparanodal region |
| MADSAM | multifocal acquired demyelinating sensors and motor neuropathy |
| MAMA | multifocal acquired motor axonopathy |
| NOR | the node of Ranvier |
| PNJ | the paranodal junction |
| PNS | Peripheral Nerve Society |
References
- Burns, T.M. Chronic inflammatory demyelinating polyradiculoneuropathy. Arch. Neurol. 2004, 61, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.H. Recurrent polyneuropathies and their corticosteroid treatment with five-year observations of a placebo-controlled case treated with corticotrophin, cortisone, and prednisone. Brain 1958, 81, 157–192. [Google Scholar] [CrossRef]
- Dalakas, M.C. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat. Rev. Neurol. 2011, 7, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; O’Brien, P.C.; Oviatt, K.F.; Dinapoli, R.P.; Daube, J.R.; Bartleson, J.D.; Mokri, B.; Swift, T.; Low, P.A.; Windebank, A.J. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treatment. Ann. Neurol. 1982, 11, 136–141. [Google Scholar] [CrossRef]
- Hughes, R.A.C.; Donofrio, P.; Bril, V. Intravenous immune globulin (10% caprylate chromatography purifi ed) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): A randomized placebo-controlled trial. Lancet Neurol. 2008, 7, 136–144. [Google Scholar] [CrossRef]
- Van den Bergh, P.Y.K.; van Doorn, P.A.; Hadden, R.D.M.; Avau, B.; Vankrunkelsven, P.; Allen, J.A.; Attarian, S.; Blomkwist-Markens, P.H.; Cornblath, D.R.; Eftimov, F.; et al. European academy of neurology/peripheral nerve society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force-second revision. Eur. J. Neurol. 2021, 28, 3556–3583. [Google Scholar] [CrossRef]
- Mathey, E.K.; Park, S.B.; Hughes, R.A.; Pollard, J.D.; Armati, P.J.; Barnett, M.H.; Taylor, B.V.; Dyck, P.J.; Kiernan, M.C.; Lin, C.S. Chronic inflammatory demyelinating polyradiculoneuropathy: From pathology to phenotype. J. Neurol. Neurosurg. Psychiatry 2015, 86, 973–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, H.; Katsuno, M. Pathophysiology of chronic inflammatory demyelinating polyneuropathy: Insights into classification and therapeutic strategy. Neurol. Ther. 2020, 9, 213–227. [Google Scholar] [CrossRef]
- Ruts, L.; Drenthen, J.; Jacobs, B.C.; van Doorn, P.A. Distinguishing acute-onset CIDP from fluctuating Guillain-Barre syndrome: A prospective study. Neurology 2010, 74, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Vallat, J.-M.; Sommer, C.; Magy, L. Chronic inflammatory demyelinating polyradiculoneuropathy: Diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol. 2010, 9, 402–412. [Google Scholar] [CrossRef]
- Laughlin, R.S.; Dyck, P.J.; Melton, L.J., 3rd; Leibson, C.; Ransom, J.; Dyck, P.J.B. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology 2009, 73, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Eftimov, F.; Lucke, I.M.; Querol, L.A.; Rajabally, Y.A.; Verhamme, C. Diagnostic challenges in chronic inflammatory demyelinating polyradiculoneuropathy. Brain 2020, 143, 3214–3224. [Google Scholar] [CrossRef]
- Bunschoten, C.; Jacobs, B.C.; Van den Bergh, P.; Cornblath, D.R.; van Doorn, P. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy’. Lancet Neurol. 2019, 18, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Hagen, K.M.; Ousman, S.S. The immune response and aging in chronic inflammatory demyelinating polyradiculoneuropathy. J. Neuroinflamm. 2021, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- McGonigal, R.; Rowan, E.G.; Greenshields, K.N.; Halstead, S.K.; Humphreys, P.D.; Rother, R.P.; Furukawa, K.; Willison, H.J. Anti-GD1a antibodies activate complement and calpain to injure distal motor nodes of Ranvier in mice. Brain 2010, 133, 1944–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susuki, K.; Rasband, M.N.; Tohyama, K.; Koibuchi, K.; Okamoto, S.; Funakoshi, K.; Hirata, K.; Baba, H.; Yuki, N. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J. Neurosci. 2007, 27, 3956–3967. [Google Scholar] [CrossRef]
- Vural, A.; Doppler, K.; Meinl, E. Autoantibodies against the Node of Ranvier in Seropositive Chronic Inflammatory Demyelinating Polyneuropathy: Diagnostic, Pathogenic, and Therapeutic Relevance. Front. Immunol. 2008, 9, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vabnick, I.; Novaković, S.D.; Levinson, S.R.; Schachner, M.; Shrager, P. The clustering of axonal sodium channels during development of the peripheral nervous system. J. Neurosci. 1996, 16, 4914–4922. [Google Scholar] [CrossRef] [Green Version]
- Delmont, E.; Brodovitch, A.; Kouton, L.; Allou, T.; Beltran, S.; Brisset, M.; Camdessanché, J.P.; Cauquil, C.; Cirion, J.; Dubard, T.; et al. Antibodies against the node of Ranvier: A real-life evaluation of incidence, clinical features and response to treatment based on a prospective analysis of 1500 sera. J. Neurol. 2020, 267, 3664–3672. [Google Scholar] [CrossRef]
- Rasband, M.N.; Peles, E.; Trimmer, J.S.; Levinson, S.R.; Lux, S.E.; Shrager, P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J. Neurosci. 1999, 19, 7516–7528. [Google Scholar] [CrossRef] [Green Version]
- Dubessy, A.-L. Role of a Contactin multi-molecular complex secreted by oligodendrocytes in nodal protein clustering in the CNS. Glia 2019, 67, 2248–2263. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.A.; Desmazières, A.; Simonnet, J.; Gatta, M.; Pfeiffer, F.; Aigrot, M.S.; Rappeneau, Q.; Guerreiro, S.; Michel, P.P.; Yanagawa, Y.; et al. Acceleration of conduction velocity linked to clustering of nodal components precedes myelination. Proc. Natl. Acad. Sci. USA 2015, 112, E321–E328. [Google Scholar] [CrossRef] [Green Version]
- Rydmark, M.; Berthold, C.-H. Electron microscopic serial section analysis of nodes of Ranvier in lumbar spinal roots of the cat: A morphometric study of nodal compartments in fibres of different sizes. J. Neurocytol. 1983, 12, 537–565. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Duncan, A.; Berry, M. Astrocyte associations with nodes of Ranvier: Ultrastructural analysis of HRP-filled astrocytes in the mouse optic nerve. J. Neurocytol. 1994, 23, 486–499. [Google Scholar] [CrossRef]
- Butt, A.M.; Duncan, A.; Hornby, M.F.; Kirvell, S.L.; Hunter, A.; Levine, J.M.; Berry, M. Cells expressing the NG2 antigen contact nodes of Ranvier in adult CNS white matter. Glia 1999, 26, 84–91. [Google Scholar] [CrossRef]
- Rasband, M.N.; Peles, E. Mechanisms of node of Ranvier assembly. Nat. Rev. Neurosci. 2021, 22, 7–20. [Google Scholar] [CrossRef]
- Kanda, H.; Ling, J.; Tonomura, S.; Noguchi, K.; Matalon, S.; Gu, J.G. TREK-1 and TRAAK are principal K(+) channels at the nodes of ranvier for rapid action potential conduction on mammalian myelinated afferent nerves. Neuron 2019, 104, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Kao, T.; Horvath, Z.; Lemos, J.; Sul, J.Y.; Cranstoun, S.D.; Bennett, V.; Scherer, S.S.; Cooper, E.C. A common ankyrin-G-based mechanism retains KCNQ and Na V channels at electrically active domains of the axon. J. Neurosci. 2006, 26, 2599–2613. [Google Scholar] [CrossRef] [Green Version]
- Devaux, J.; Alcaraz, G.; Grinspan, J.; Bennett, V.; Joho, R.; Crest, M.; Scherer, S.S. Kv3.1b is a novel component of CNS nodes. J. Neurosci. 2003, 23, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Cooper, E.C. An ankyrin-G N-terminal gate and protein kinase CK2 dually regulate binding of voltage-gated sodium and KCNQ2/3 potassium channels. J. Biol. Chem. 2015, 290, 16619–16632. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.Q.; Lambert, S.; Bennett, V. Molecular composition of the node of Ranvier: Identification of ankyrin- binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J. Cell Biol. 1996, 135, 1355–1367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Desmazieres, A.; Zonta, B.; Melrose, S.; Campbell, G.; Mahad, D.; Li, Q.; Sherman, D.L.; Reynolds, R.; Brophy, P.J. Neurofascin 140 is an embryonic neuronal neurofascin isoform that promotes the assembly of the node of ranvier. J. Neurosci. 2015, 35, 2246–2254. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, K.; Eshed-Eisenbach, Y.; Frechter, S.; Amor, V.; Salomon, D.; Sabanay, H.; Dupree, J.L.; Grumet, M.; Brophy, P.J.; Shrager, P.; et al. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron 2010, 65, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombelli, C. Perlecan is recruited by dystroglycan to nodes of Ranvier and binds the clustering molecule gliomedin. The Journal of cell biology. Rockefeller Univ. Press 2015, 208, 313–329. [Google Scholar] [CrossRef] [Green Version]
- Goutebroze, L.; Carnaud, M.; Denisenko, N.; Boutterin, M.C.; Girault, J.A. Syndecan-3 and syndecan-4 are enriched in Schwann cell perinodal processes. BMC Neurosci. 2003, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Bang, M.L. Glial M6B stabilizes the axonal membrane at peripheral nodes of Ranvier. Glia 2018, 66, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Poliak, S.; Peles, E. The local differentiation of myelinated axons at nodes of Ranvier. Nat. Rev. Neurosci. 2003, 4, 968–980. [Google Scholar] [CrossRef]
- Boyle, M.E.; Berglund, E.O.; Murai, K.K.; Weber, L.; Peles, E.; Ranscht, B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 2001, 30, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bekku, Y.; Dzhashiashvili, Y.; Armenti, S.; Meng, X.; Sasaki, Y.; Milbrandt, J.; Salzer, J.L. Assembly and maintenance of nodes of ranvier rely on distinct sources of proteins and targeting mechanisms. Neuron 2012, 73, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, J.C.; Rubin, M.; St Martin, M.; Downey, R.T.; Einheber, S.; Rosenbluth, J.; Levinson, S.R.; Bhat, M.; Salzer, J.L. Paranodal interactions regulate expression of sodium channel subtypes and provide a diffusion barrier for the node of Ranvier. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 7001–7011. [Google Scholar] [CrossRef]
- Wang, H.; Kunkel, D.D.; Martin, T.M.; Schwartzkroin, P.A.; Tempel, B.L. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature 1993, 365, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Dupree, J.L.; Popko, B.; Karagogeos, D. The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 3016–3024. [Google Scholar] [CrossRef] [Green Version]
- Furley, A.J.; Morton, S.B.; Manalo, D.; Karagogeos, D.; Dodd, J.; Jessell, T. The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell 1990, 61, 157–170. [Google Scholar] [CrossRef]
- Poliak, S.; Salomon, D.; Elhanany, H.; Sabanay, H.; Kiernan, B.; Pevny, L.; Stewart, C.L.; Xu, X.; Chiu, S.Y.; Shrager, P.; et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003, 162, 1149–1160. [Google Scholar] [CrossRef]
- Ivanovic, A.; Horresh, I.; Golan, N.; Spiegel, I.; Sabanay, H.; Frechter, S.; Ohno, S.; Terada, N.; Möbius, W.; Rosenbluth, J.; et al. The cytoskeletal adapter protein 4.1G organizes the internodes in peripheral myelinated nerves. J. Cell Biol. 2012, 196, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Hivert, B.; Marien, L.; Agbam, K.N.; Faivre-Sarrailh, C. ADAM22 and ADAM23 modulate the targeting of the Kv1 channel-associated protein LGI1 to the axon initial segment. J. Cell Sci. 2019, 132, jcs219774. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Oses-Prieto, J.; Kim, M.Y.; Horresh, I.; Peles, E.; Burlingame, A.L.; Trimmer, J.S.; Meijer, D.; Rasband, M.N. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 1038–1048. [Google Scholar] [CrossRef]
- Burnor, E.; Yang, L.; Zhou, H.; Patterson, K.R.; Quinn, C.; Reilly, M.M.; Rossor, A.M.; Scherer, S.S.; Lancaster, E. Neurofascin antibodies in autoimmune, genetic, and idiopathic neuropathies. Neurology 2018, 90, E31–E38. [Google Scholar] [CrossRef] [PubMed]
- Delmont, E.; Manso, C.; Querol, L.; Cortese, A.; Berardinelli, A.; Lozza, A.; Belghazi, M.; Malissart, P.; Labauge, P.; Taieb, G.; et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017, 140, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Devaux, J.J.; Miura, Y.; Fukami, Y.; Inoue, T.; Manso, C.; Belghazi, M.; Sekiguchi, K.; Kokubun, N.; Ichikawa, H.; Wong, A.H.; et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016, 86, 800–807. [Google Scholar] [CrossRef] [Green Version]
- Doppler, K.; Appeltshauser, L.; Wilhelmi, K.; Villmann, C.; Dib-Hajj, S.D.; Waxman, S.G.; Mäurer, M.; Weishaupt, A.; Sommer, C. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J. Neurol. Neurosurg. Psychiatry 2015, 86, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Tang, L.; Huang, Q.; Tang, X. A systematic review and meta-analysis of autoantibodies for diagnosis and prognosis in patients with chronic inflammatory demyelinating polyradiculoneuropathy. Front. Neurosci. 2021, 15, 574. [Google Scholar] [CrossRef]
- Cortese, A.; Lombardi, R.; Briani, C.; Callegari, I.; Benedetti, L.; Manganelli, F.; Luigetti, M.; Ferrari, S.; Clerici, A.M.; Marfia, G.A.; et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: Clinical relevance of IgG isotype. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L. Distinguish CIDP with autoantibody from that without autoantibody: Pathogenesis, histopathology, and clinical features. J. Neurol. 2021, 268, 2757–2768. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.W. IVIG efficacy in CIDP patients is not associated with terminal complement inhibition. J. Neuroimmunol. 2019, 330, 23–27. [Google Scholar] [CrossRef]
- Ogata, H.; Yamasaki, R.; Hiwatashi, A.; Oka, N.; Kawamura, N.; Matsuse, D.; Kuwahara, M.; Suzuki, H.; Kusunoki, S.; Fujimoto, Y.; et al. Characterization of IgG4 anti-neurofascin 155 antibody-positive polyneuropathy. Ann. Clin. Transl. Neurol. 2015, 2, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Diaz, C.; Dubourg, O.; Irinopoulou, T.; Vigny, M.; Lachkar, S.; Decker, L.; Charnay, P.; Denisenko, N.; Maisonobe, T.; Léger, J.M.; et al. Nodes of ranvier and paranodes in chronic acquired neuropathies. PLoS ONE 2011, 6, e14533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaudineau, Y.; Devauchelle-Pensec, V.; Hanrotel, C.; Pers, J.O.; Saraux, A.; Youinou, P. Monoclonal anti-CD20 antibodies: Mechanisms of action and monitoring of biological effects. Jt. Bone Spine 2009, 76, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Querol, L.; Rojas-García, R.; Diaz-Manera, J.; Barcena, J.; Pardo, J.; Ortega-Moreno, A.; Sedano, M.J.; Seró-Ballesteros, L.; Carvajal, A.; Ortiz, N.; et al. Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e149. [Google Scholar] [CrossRef] [Green Version]
- Oaklander, A.L.; Lunn, M.P.; Hughes, R.A.; van Schaik, I.N.; Frost, C.; Chalk, C.H. Treatments for chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): An overview of systematic reviews. Cochrane Database Syst. Rev. 2017, 1, CD010369. [Google Scholar] [CrossRef]
- Carrera-García, L.; Natera-de Benito, D.; Lleixà, C.; Ortez, C.; Colomer, J.; Nascimento, A.; Saiz, A.; Dalmau, J.; Querol, L.; Armangué, T. Chronic inflammatory demyelinating polyneuropathy associated with contactin-1 antibodies in a child. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zheng, P.; Devaux, J.J.; Wang, Y.; Liu, C.; Li, J.; Guo, S.; Song, Y.; Wang, Q.; Feng, X. Chronic inflammatory demyelinating polyneuropathy with anti-NF155 IgG4 in China. J. Neuroimmunol. 2019, 337, 577074. [Google Scholar] [CrossRef] [PubMed]
- Wieske, L. Serum contactin-1 in CIDP: A cross-sectional study. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1040. [Google Scholar] [CrossRef] [PubMed]
- Pinatel, D.; Faivre-Sarrailh, C. Assembly and function of the juxtaparanodal Kv1 complex in health and disease. Life 2020, 8, 8. [Google Scholar] [CrossRef]
- Hu, J.; Sun, C.; Lu, J.; Zhao, C.; Lin, J. Efficacy of rituximab treatment in chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review and meta-analysis. J. Neurol. 2021. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M. Macrophages and autoantibodies in demyelinating diseases. Cells 2021, 10, 844. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Koike, H.; Nishi, R.; Kawagashira, Y.; Iijima, M.; Katsuno, M.; Sobue, G. Clinicopathological characteristics of subtypes of chronic inflammatory demyelinating polyradiculoneuropathy. J. Neurol. Neurosurg. Psychiatry 2019, 90, 988–996. [Google Scholar] [CrossRef]
- Koike, H.; Nishi, R.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Katsuno, M.; Sobue, G. Ultrastructural mechanisms of macrophage-induced demyelination in CIDP. Neurology 2018, 91, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Kira, J.I. Anti-neurofascin 155 antibody-positive chronic inflammatory demyelinating polyneuropathy/combined central and peripheral demyelination: Strategies for diagnosis and treatment based on the disease mechanism. Front. Neurol. 2021, 12, 665136. [Google Scholar] [CrossRef]
- Ogata, H.; Zhang, X.; Yamasaki, R.; Fujii, T.; Machida, A.; Morimoto, N.; Kaida, K.; Masuda, T.; Ando, Y.; Kuwahara, M.; et al. Intrathecal cytokine profile in neuropathy with anti-neurofascin 155 antibody. Ann. Clin. Transl. Neurol. 2019, 6, 2304–2316. [Google Scholar] [CrossRef] [Green Version]
- Culver, E.L.; Vermeulen, E.; Makuch, M.; van Leeuwen, A.; Sadler, R.; Cargill, T.; Klenerman, P.; Aalberse, R.C.; van Ham, S.M.; Barnes, E.; et al. Increased IgG4 responses to multiple food and animal antigens indicate a polyclonal expansion and differentiation of pre-existing B cells in IgG4-related disease. Ann. Rheum. Dis. 2015, 74, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Yamashita, K.; Kudo, M. IgG4-related disease and innate immunity. Curr. Top. Microbiol. Immunol. 2017, 401, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Watanabe, T.; Minaga, K.; Kamata, K.; Kudo, M. Cytokines produced by innate immune cells in IgG4-related disease. Mod. Rheumatol. 2019, 29, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Koneczny, I. Update on IgG4-mediated autoimmune diseases: New insights and new family members. Autoimmun. Rev. 2020, 19, 102646. [Google Scholar] [CrossRef]
- Kira, J.I.; Yamasaki, R.; Ogata, H. Anti-neurofascin autoantibody and demyelination. Neurochem. Int. 2019, 130, 104360. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, M.G.; Querol, L.A.; Niks, E.H.; Plomp, J.J.; van der Maarel, S.M.; Graus, F.; Dalmau, J.; Illa, I.; Verschuuren, J.J. The expanding field of IgG4-mediated neurological autoimmune disorders. Eur. J. Neurol. 2015, 22, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Faivre-Sarrailh, C.; Devaux, J.J. Neuro-glial interactions at the nodes of Ranvier: Implication in health and diseases. Front Cell Neurosci. 2013, 7, 196. [Google Scholar] [CrossRef] [Green Version]
- Ng, J.K.M. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology 2012, 79, 2241–2248. [Google Scholar] [CrossRef] [Green Version]
- Kouton, L. Electrophysiological features of chronic inflammatory demyelinating polyradiculoneuropathy associated with IgG4 antibodies targeting neurofascin 155 or contactin 1 glycoproteins. Clin. Neurophysiol. 2020, 131, 921–927. [Google Scholar] [CrossRef]
- Duflocq, A.; Chareyre, F.; Giovannini, M.; Couraud, F.; Davenne, M. Characterization of the axon initial segment (AIS) of motor neurons and identification of a para-AIS and a juxtapara-AIS, organized by protein 4.1B. BMC Biol. 2011, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Kalafatakis, I.; Savvaki, M.; Velona, T.; Karagogeos, D. Implication of contactins in demyelinating pathologies. Life 2021, 11, 51. [Google Scholar] [CrossRef] [PubMed]

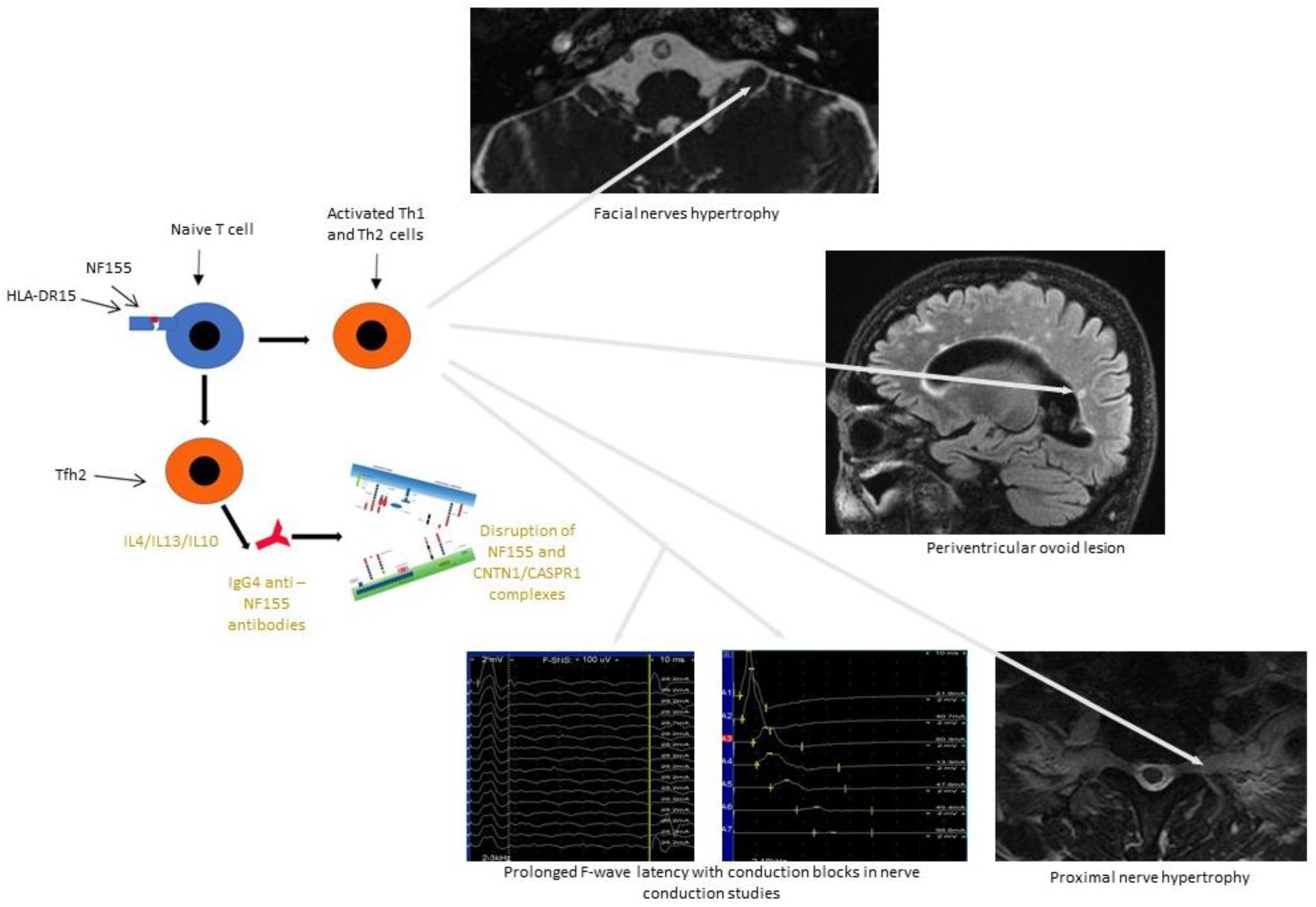

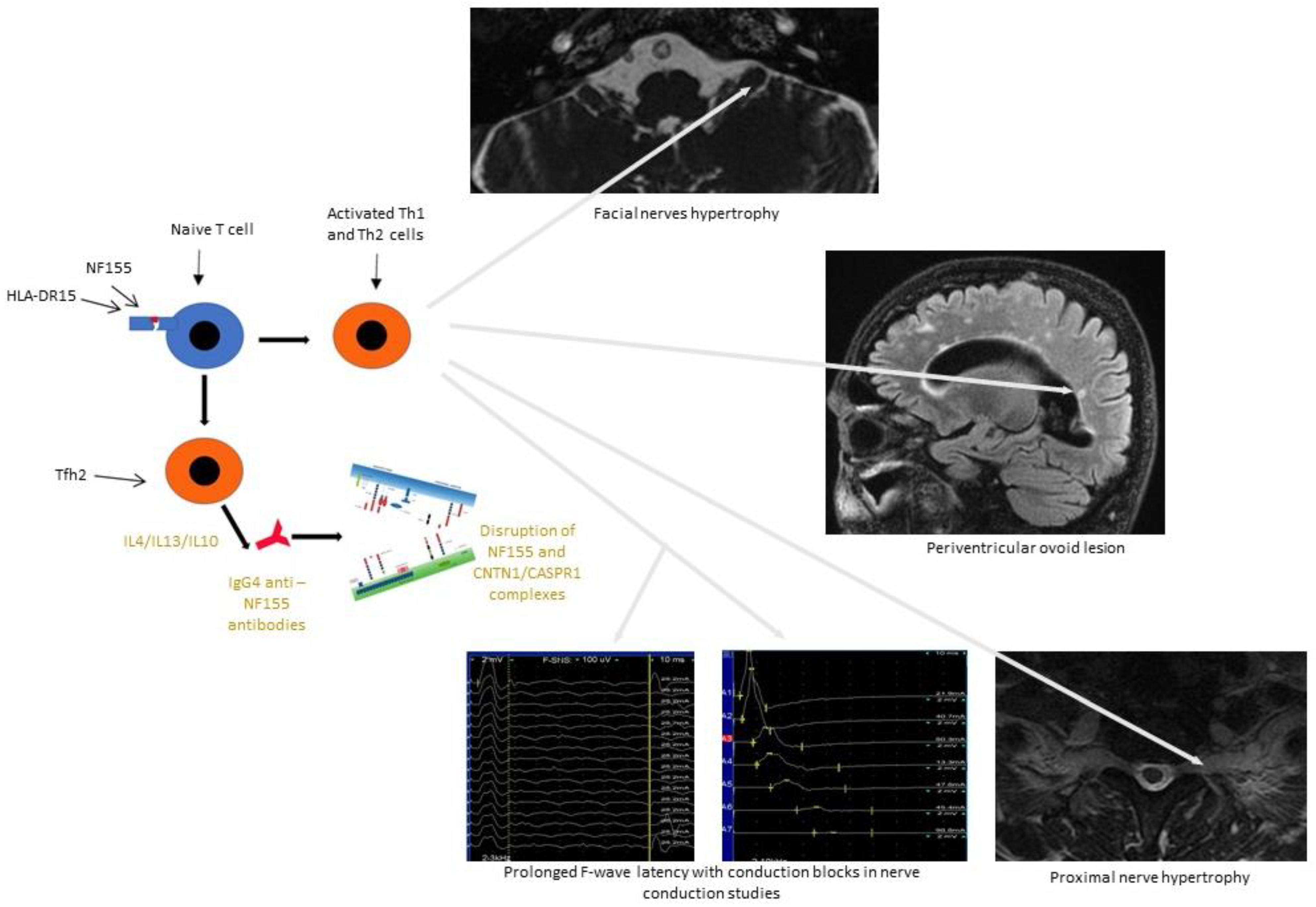
{kind=link}
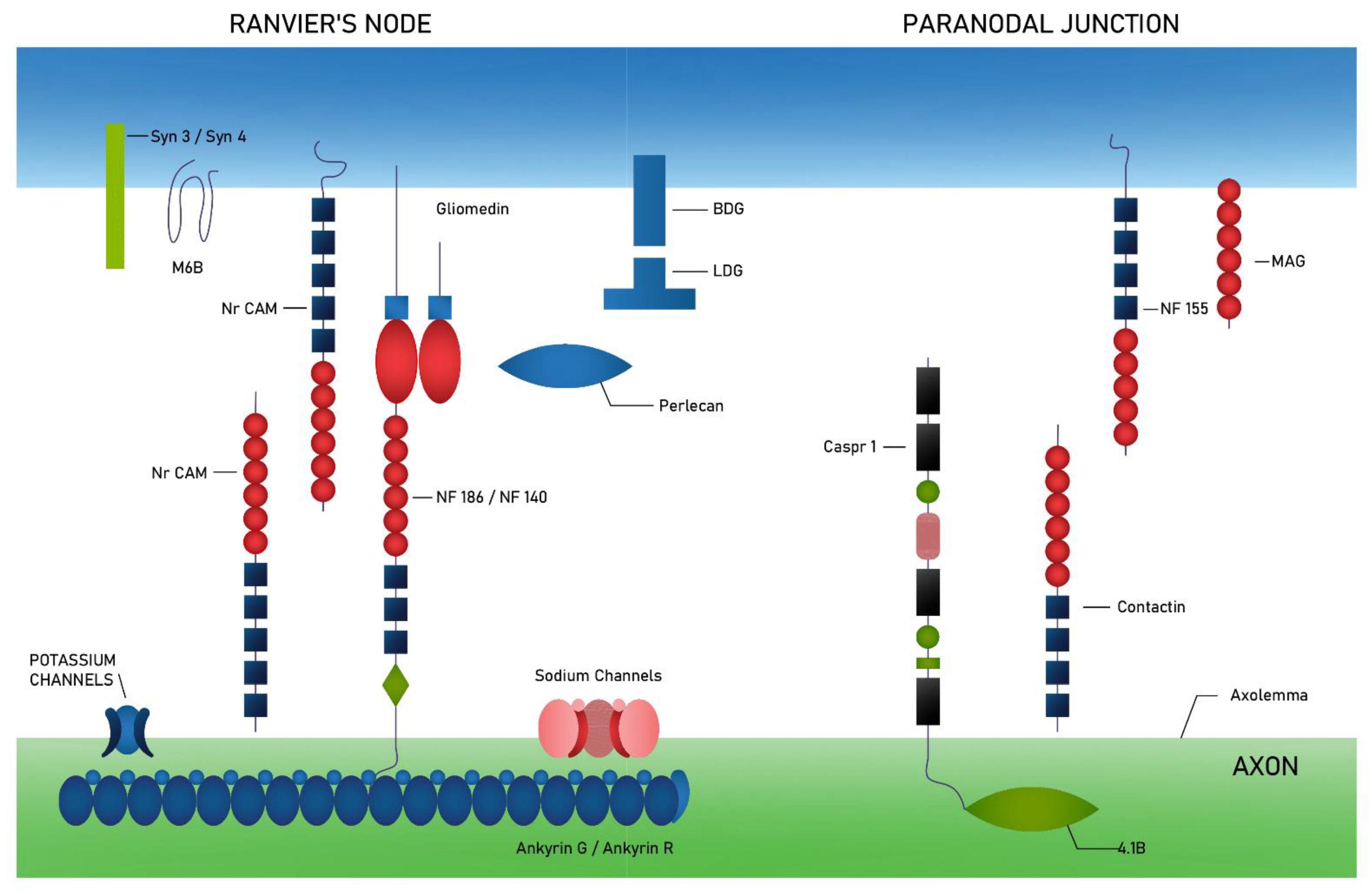
{kind=link}
| Typical CIDP | CIDP Variants |
|---|---|
All the following:
| One of the following, but otherwise as in typical CIDP (tendon reflexes may be normal in unaffected limbs):
|
| Antibodies | Frequency in CIDP Patients | Localisation | IvIg | Corticosteroids | Plasma Exchange | Rituximab |
|---|---|---|---|---|---|---|
| NF155 | 1–21% [19,62,63] | Paranodal | Poor response | Partial response | Potentially good response | Potentially good response |
| CNTN1 | 0.7–8% [19,52,62] | Paranodal | Poor response | Partial response | Partial response | Potentially good response |
| NF140/NF186 | 2–5% [48,49] | Nodal | Partial response | Partial response | Potentially good response | Potentially good response |
| Caspr1 | 0.2–3% [19,64] | Paranodal | Poor response | Partial response | Partial response | Potentially good response |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziadkowiak, E.; Waliszewska-Prosół, M.; Nowakowska-Kotas, M.; Budrewicz, S.; Koszewicz, Z.; Koszewicz, M. Pathophysiology of the Different Clinical Phenotypes of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP). Int. J. Mol. Sci. 2022, 23, 179. https://doi.org/10.3390/ijms23010179
Dziadkowiak E, Waliszewska-Prosół M, Nowakowska-Kotas M, Budrewicz S, Koszewicz Z, Koszewicz M. Pathophysiology of the Different Clinical Phenotypes of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP). International Journal of Molecular Sciences. 2022; 23(1):179. https://doi.org/10.3390/ijms23010179
Chicago/Turabian StyleDziadkowiak, Edyta, Marta Waliszewska-Prosół, Marta Nowakowska-Kotas, Sławomir Budrewicz, Zofia Koszewicz, and Magdalena Koszewicz. 2022. "Pathophysiology of the Different Clinical Phenotypes of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)" International Journal of Molecular Sciences 23, no. 1: 179. https://doi.org/10.3390/ijms23010179
APA StyleDziadkowiak, E., Waliszewska-Prosół, M., Nowakowska-Kotas, M., Budrewicz, S., Koszewicz, Z., & Koszewicz, M. (2022). Pathophysiology of the Different Clinical Phenotypes of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP). International Journal of Molecular Sciences, 23(1), 179. https://doi.org/10.3390/ijms23010179