
Akt Inhibition as Preconditioning Treatment to Protect Kidney Cells against Anoxia

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Anoxia Rapidly Inhibits Akt Phosphorylation
2.2. Inhibition of Akt Leads to Anoxia Resistance
2.3. Modulation of Glucose Metabolism Does Not Participate in Anoxia Resistance
2.4. Akt Inhibition Protects from Mitochondria-Originated ROS
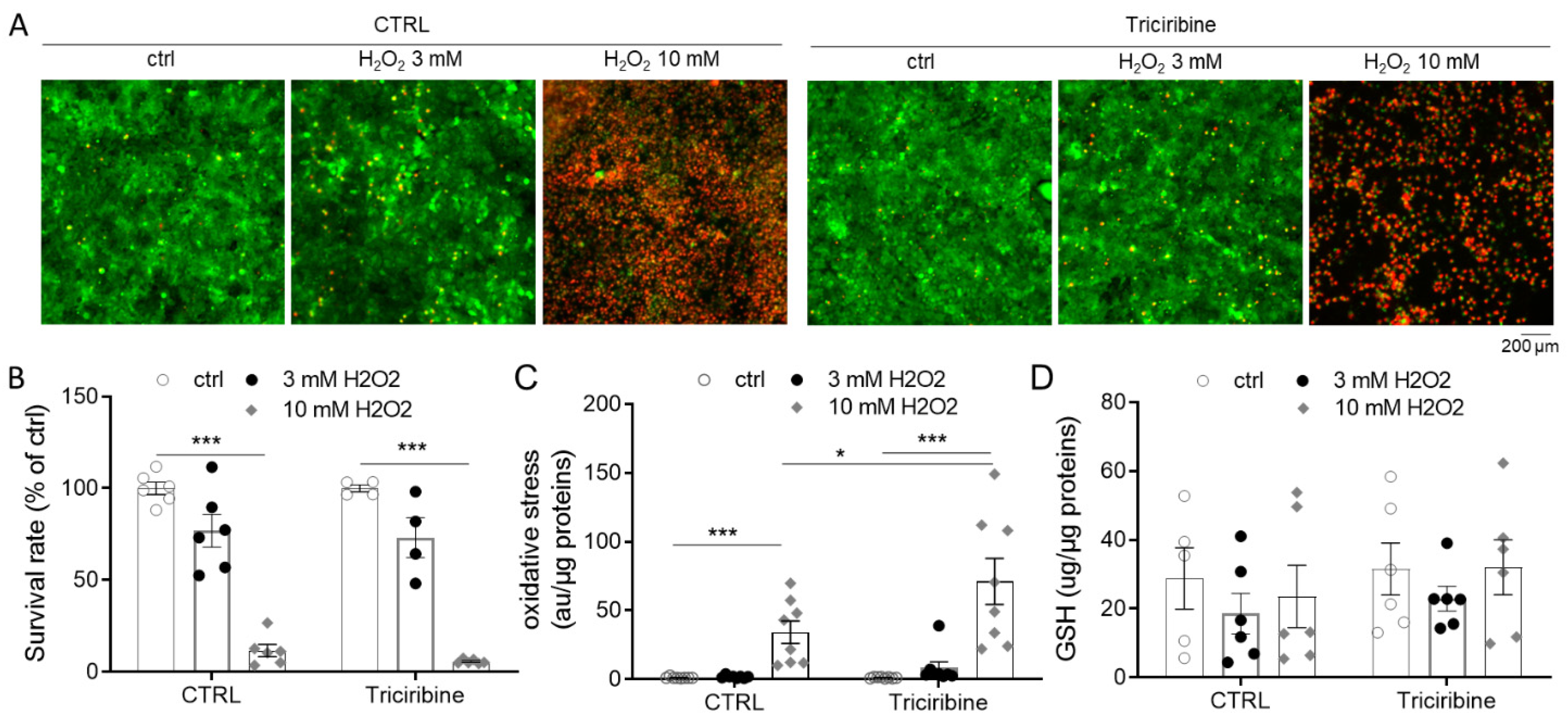
2.5. Akt Inhibition Does Not Protect from H2O2 Overproduction
2.6. Reversibility of Akt Inhibition Effect on PCT Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Metabolism Analysis
4.4. Cell Survival Analysis
4.5. Protein Analysis
4.6. Biochemical Parameter Analysis
4.7. Oxidative Stress Measurement
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Lopez-Neblina, F.; Toledo, A.H.; Toledo-Pereyra, L.H. Molecular Biology of Apoptosis in Ischemia and Reperfusion. J. Investig. Surg. 2005, 18, 335–350. [Google Scholar] [CrossRef]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2012, 40, 123–137. [Google Scholar] [CrossRef]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D.Y. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia–reperfusion. Biochim. Biophys. Acta (BBA)—Bioenergy 2011, 1813, 1382–1394. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Ricci, J.E.; Gottlieb, R.A.; Green, D.R. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.E.; Waterhouse, N.; Green, D.R. Mitochondrial functions during cell death, a complex (I–V) dilemma. Cell Death Differ. 2003, 10, 488–492. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Carcy, R.; Cougnon, M.; Poet, M.; Durandy, M.; Sicard, A.; Counillon, L.; Blondeau, N.; Hauet, T.; Tauc, M.; Pisani, D.F. Targeting oxidative stress, a crucial challenge in renal transplantation outcome. Free. Radic. Biol. Med. 2021, 169, 258–270. [Google Scholar] [CrossRef]
- Moussa, A.; Li, J. AMPK in myocardial infarction and diabetes: The yin/yang effect. Acta Pharm. Sin. B 2012, 2, 368–378. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Tsang, A.; Yellon, D.M. The Reperfusion Injury Salvage Kinase Pathway: A Common Target for Both Ischemic Preconditioning and Postconditioning. Trends Cardiovasc. Med. 2005, 15, 69–75. [Google Scholar] [CrossRef]
- Bejaoui, M.; Zaouali, M.A.; Sakly, R.; Ben Abdennebi, H. Olprinone protects the liver from ischemia–reperfusion injury through oxidative stress prevention and protein kinase Akt activation. Can. J. Physiol. Pharmacol. 2018, 96, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nie, H.; Tian, L.; Tong, L.; Deng, J.; Zhang, Y.; Dong, H.; Xiong, L. Sevoflurane preconditioning-induced neuroprotection is associated with Akt activation via carboxy-terminal modulator protein inhibition. Br. J. Anaesth. 2015, 114, 327–335. [Google Scholar] [CrossRef]
- Rozier, R.; Paul, R.; Hounoum, B.M.; Villa, E.; Mhaidly, R.; Chiche, J.; Verhoeyen, E.; Marchetti, S.; Vandenberghe, A.; Raucoules, M.; et al. Pharmacological preconditioning protects from ischemia/reperfusion-induced apoptosis by modulating Bcl-xL expression through a ROS-dependent mechanism. FEBS J. 2021, 288, 3547–3569. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Z.; Zhang, X.; Zhang, X.; Dong, L.; Xing, Y.; Li, Y.; Liu, Z.; Chen, L.; Qiao, H.; et al. Protection by silibinin against experimental ischemic stroke: Up-Regulated pAkt, pmTOR, HIF-1alpha and Bcl-2, down-regulated Bax, NF-kappaB expression. Neurosci. Lett. 2012, 529, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Tao, Q.; Li, G.; Xiang, L.; Zheng, X.; Zhang, T.; Wu, C.; Li, D. Fibroblast Growth Factor 2 Attenuates Renal Ischemia-Reperfusion Injury via Inhibition of Endoplasmic Reticulum Stress. Front. Cell Dev. Biol. 2020, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Tauc, M.; Melis, N.; Bourourou, M.; Giraud, S.; Hauet, T.; Blondeau, N. A new pharmacological preconditioning-based target: From drosophila to kidney transplantation. Cond. Med. 2019, 2, 69–74. [Google Scholar]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Kharbanda, R.; Nielsen, T.T.; Redington, A.N. Translation of remote ischaemic preconditioning into clinical practice. Lancet 2009, 374, 1557–1565. [Google Scholar] [CrossRef]
- Torras, J.; Herrero-Fresneda, I.; Lloberas, N.; Riera, M.; Cruzado, J.M.; Grinyó, J.M. Promising effects of ischemic preconditioning in renal transplantation. Kidney Int. 2002, 61, 2218–2227. [Google Scholar] [CrossRef][Green Version]
- Toosy, N.; McMorris, E.L.; Grace, P.A.; Mathie, R.T. Mathie Ischaemic preconditioning protects the rat kidney from reperfusion injury. BJU Int. 1999, 84, 489–494. [Google Scholar] [CrossRef]
- Chen, C.; Sun, L.; Zhang, W.; Tang, Y.; Li, X.; Jing, R.; Liu, T. Limb ischemic preconditioning ameliorates renal microcirculation through activation of PI3K/Akt/eNOS signaling pathway after acute kidney injury. Eur. J. Med. Res. 2020, 25, 10. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.M.; Bell, R.M.; Yellon, D.M. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J. Mol. Cell. Cardiol. 2002, 34, 661–668. [Google Scholar] [CrossRef]
- Tong, H.; Chen, W.; Steenbergen, C.; Murphy, E. Ischemic Preconditioning Activates Phosphatidylinositol-3-Kinase Upstream of Protein Kinase C. Circ. Res. 2000, 87, 309–315. [Google Scholar] [CrossRef]
- Uchiyama, T.; Engelman, R.M.; Maulik, N.; Das, D.K. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation 2004, 109, 3042–3049. [Google Scholar] [CrossRef]
- Bastian, C.; Quinn, J.; Tripathi, A.; Aquila, D.; McCray, A.; Dutta, R.; Baltan, S.; Brunet, S. CK2 inhibition confers functional protection to young and aging axons against ischemia by differentially regulating the CDK5 and AKT signaling pathways. Neurobiol. Dis. 2019, 126, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.R.; Martins, T.R.; Couto, R.; Deus, C.; Pereira, C.V.; Simões, R.F.; Rizvanov, A.A.; Silva, F.; Cunha-Oliveira, T.; Oliveira, P.J.; et al. Berberine-induced cardioprotection and Sirt3 modulation in doxorubicin-treated H9c2 cardiomyoblasts. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 2904–2923. [Google Scholar] [CrossRef]
- Zhu, Q.-W.; Yong-Guang, L. Berberine attenuates myocardial ischemia reperfusion injury by suppressing the activation of PI3K/AKT signaling. Exp. Ther. Med. 2016, 11, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Li, H.; Pi, Y.; Li, Z.; Jin, S. Cardioprotective effect of IGF-1 against myocardial ischemia/reperfusion injury through activation of PI3K/Akt pathway in rats in vivo. J. Int. Med. Res. 2019, 47, 3886–3897. [Google Scholar] [CrossRef]
- Paiva, M.S.A.; Rutter-Locher, Z.; Gonçalves, L.M.; Providência, L.A.; Davidson, S.M.; Yellon, D.; Mocanu, M.M. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am. J. Physiol. Circ. Physiol. 2011, 300, H2123–H2134. [Google Scholar] [CrossRef]
- Wang, Z.-G.; Li, H.; Huang, Y.; Li, R.; Wang, X.-F.; Yu, L.-X.; Guang, X.-Q.; Li, L.; Zhang, H.-Y.; Zhao, Y.-Z.; et al. Nerve growth factor-induced Akt/mTOR activation protects the ischemic heart via restoring autophagic flux and attenuating ubiquitinated protein accumulation. Oncotarget 2016, 8, 5400–5413. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Deng, Q.; Zhang, L.; Zhu, W. Nobiletin alleviates ischemia/reperfusion injury in the kidney by activating PI3K/AKT pathway. Mol. Med. Rep. 2020, 22, 4655–4662. [Google Scholar] [CrossRef]
- Patel, N.S.A.; Kerr-Peterson, H.L.; Brines, M.; Collino, M.; Rogazzo, M.; Fantozzi, R.; Wood, E.G.; Johnson, F.L.; Yaqoob, M.M.; Cerami, A.; et al. Delayed Administration of Pyroglutamate Helix B Surface Peptide (pHBSP), a Novel Nonerythropoietic Analog of Erythropoietin, Attenuates Acute Kidney Injury. Mol. Med. 2012, 18, 719–727. [Google Scholar] [CrossRef]
- Bourourou, M.; Gouix, E.; Melis, N.; Friard, J.; Heurteaux, C.; Tauc, M.; Blondeau, N. Inhibition of eIF5A hypusination pathway as a new pharmacological target for stroke therapy. J. Cereb. Blood Flow Metab. 2021, 41, 1080–1090. [Google Scholar] [CrossRef]
- Giraud, S.; Kerforne, T.; Zely, J.; Ameteau, V.; Couturier, P.; Tauc, M.; Hauet, T. The inhibition of eIF5A hypusination by GC7, a preconditioning protocol to prevent brain death-induced renal injuries in a preclinical porcine kidney transplantation model. Am. J. Transpl. 2020, 20, 3326–3340. [Google Scholar] [CrossRef]
- Melis, N.; Rubera, I.; Cougnon, M.; Giraud, S.; Mograbi, B.; Belaid, A.; Pisani, D.; Huber, S.M.; Lacas-Gervais, S.; Fragaki, K.; et al. Targeting eIF5A Hypusination Prevents Anoxic Cell Death through Mitochondrial Silencing and Improves Kidney Transplant Outcome. J. Am. Soc. Nephrol. 2016, 28, 811–822. [Google Scholar] [CrossRef]
- Park, M.H. The Post-Translational Synthesis of a Polyamine-Derived Amino Acid, Hypusine, in the Eukaryotic Translation Initiation Factor 5A (eIF5A). J. Biochem. 2006, 139, 161–169. [Google Scholar] [CrossRef]
- Park, M.H.; Nishimura, K.; Zanelli, C.F.; Valentini, S.R. Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids 2010, 38, 491–500. [Google Scholar] [CrossRef]
- Vigne, P.; Frelin, C. The role of polyamines in protein-dependent hypoxic tolerance of Drosophila. BMC Physiol. 2008, 8, 22. [Google Scholar] [CrossRef]
- Cougnon, M.; Carcy, R.; Melis, N.; Rubera, I.; Duranton, C.; Dumas, K.; Tanti, J.-F.; Pons, C.; Soubeiran, N.; Shkreli, M.; et al. Inhibition of eIF5A hypusination reprogrammes metabolism and glucose handling in mouse kidney. Cell Death Dis. 2021, 12, 283. [Google Scholar] [CrossRef] [PubMed]
- Chetram, M.A.; Jones, K.J.; Coke, C.; Don-Salu-Hewage, A.; Bethea, D.A.; Hinton, C.V. Abstract 846: ROS-Mediated activation of AKT induces apoptosis via pVHL in prostate cancer cells. Mol. Cell. Biol. 2013, 376, 63–71. [Google Scholar] [CrossRef]
- Samanta, D.; Semenza, G.L. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef]
- Conde, E.; Alegre, L.; Blanco-Sanchez, I.; Sáenz-Morales, D.; Aguado-Fraile, E.; Ponte, B.; Ramos, E.; Saiz, A.; Jiménez, C.; Ordoñez, A.; et al. Hypoxia Inducible Factor 1-Alpha (HIF-1 Alpha) Is Induced during Reperfusion after Renal Ischemia and Is Critical for Proximal Tubule Cell Survival. PLoS ONE 2012, 7, e33258. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Heyman, S.N.; Rosen, S.; Shina, A.; Goldfarb, M.; Griethe, W.; Frei, U.; Reinke, P.; Bachmann, S.; Eckardt, K.-U. Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005, 67, 531–542. [Google Scholar] [CrossRef]
- Nangaku, M.; Inagi, R.; Miyata, T.; Fujita, T. Hypoxia and Hypoxia-Inducible Factor in Renal Disease. Nephron Exp. Nephrol. 2008, 110, e1–e7. [Google Scholar] [CrossRef]
- Cai, W.J.; Chen, Y.; Shi, L.X.; Cheng, H.R.; Banda, I.; Ji, Y.H.; Wang, Y.T.; Li, X.M.; Mao, Y.X.; Zhang, D.F.; et al. AKT-GSK3beta Signaling Pathway Regulates Mitochondrial Dysfunction-Associated OPA1 Cleavage Contributing to Osteoblast Apoptosis: Preventative Effects of Hydroxytyrosol. Oxid. Med. Cell. Longev. 2019, 2019, 4101738. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [PubMed]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-Mitochondria Interaction Mediated by Akt Is Required to Inhibit Apoptosis in the Presence or Absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Lieberthal, W.; Tang, M.; Abate, M.; Lusco, M.; Levine, J.S. AMPK-mediated activation of Akt protects renal tubular cells from stress-induced apoptosis in vitro and ameliorates ischemic AKI in vivo. Am. J. Physiol. Renal Physiol. 2019, 317, F1–F11. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-K.; Kim, H.-H. The functions of mTOR in ischemic diseases. BMB Rep. 2011, 44, 506–511. [Google Scholar] [CrossRef]
- Wei, Q.; Zhao, J.; Zhou, X.; Yu, L.; Liu, Z.; Chang, Y. Propofol can suppress renal ischemia-reperfusion injury through the activation of PI3K/AKT/mTOR signal pathway. Gene 2019, 708, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Jasiulionis, M.G.; Luchessi, A.D.; Moreira, A.G.; Souza, P.P.C.; Suenaga, A.P.M.; Correa, M.; Costa, C.A.D.S.; Curi, R.; Costa-Neto, C.M. Inhibition of eukaryotic translation initiation factor 5A (eIF5A) hypusination impairs melanoma growth. Cell Biochem. Funct. 2006, 25, 109–114. [Google Scholar] [CrossRef]
- Barriere, H.; Belfodil, R.; Rubera, I.; Tauc, M.; Poujeol, C.; Bidet, M.; Poujeol, P. CFTR null mutation altered cAMP-sensitive and swelling-activated Cl− currents in primary cultures of mouse nephron. Am. J. Physiol. Renal Physiol. 2003, 284, F796–F811. [Google Scholar] [CrossRef]
- Rubera, I.; Belfodil, R.; Tauc, M.; Tonnerieux, N.; Poujeol, C.; Barhanin, J.; Poujeol, P. Swelling-activated Chloride and Potassium Conductance in Primary Cultures of Mouse Proximal Tubules. Implication of KCNE1 Protein. J. Membr. Biol. 2003, 193, 153–170. [Google Scholar] [CrossRef]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J. Mol. Cell. Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Friard, J.; Corinus, A.; Cougnon, M.; Tauc, M.; Pisani, D.F.; Duranton, C.; Rubera, I. LRRC8/VRAC channels exhibit a noncanonical permeability to glutathione, which modulates epithelial-to-mesenchymal transition (EMT). Cell Death Dis. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melis, N.; Carcy, R.; Rubera, I.; Cougnon, M.; Duranton, C.; Tauc, M.; Pisani, D.F. Akt Inhibition as Preconditioning Treatment to Protect Kidney Cells against Anoxia. Int. J. Mol. Sci. 2022, 23, 152. https://doi.org/10.3390/ijms23010152
Melis N, Carcy R, Rubera I, Cougnon M, Duranton C, Tauc M, Pisani DF. Akt Inhibition as Preconditioning Treatment to Protect Kidney Cells against Anoxia. International Journal of Molecular Sciences. 2022; 23(1):152. https://doi.org/10.3390/ijms23010152
Chicago/Turabian StyleMelis, Nicolas, Romain Carcy, Isabelle Rubera, Marc Cougnon, Christophe Duranton, Michel Tauc, and Didier F. Pisani. 2022. "Akt Inhibition as Preconditioning Treatment to Protect Kidney Cells against Anoxia" International Journal of Molecular Sciences 23, no. 1: 152. https://doi.org/10.3390/ijms23010152
APA StyleMelis, N., Carcy, R., Rubera, I., Cougnon, M., Duranton, C., Tauc, M., & Pisani, D. F. (2022). Akt Inhibition as Preconditioning Treatment to Protect Kidney Cells against Anoxia. International Journal of Molecular Sciences, 23(1), 152. https://doi.org/10.3390/ijms23010152