
Exercise Preconditioning Blunts Early Atrogenes Expression and Atrophy in Gastrocnemius Muscle of Hindlimb Unloaded Mice

 and
and
Abstract
:1. Introduction
2. Results
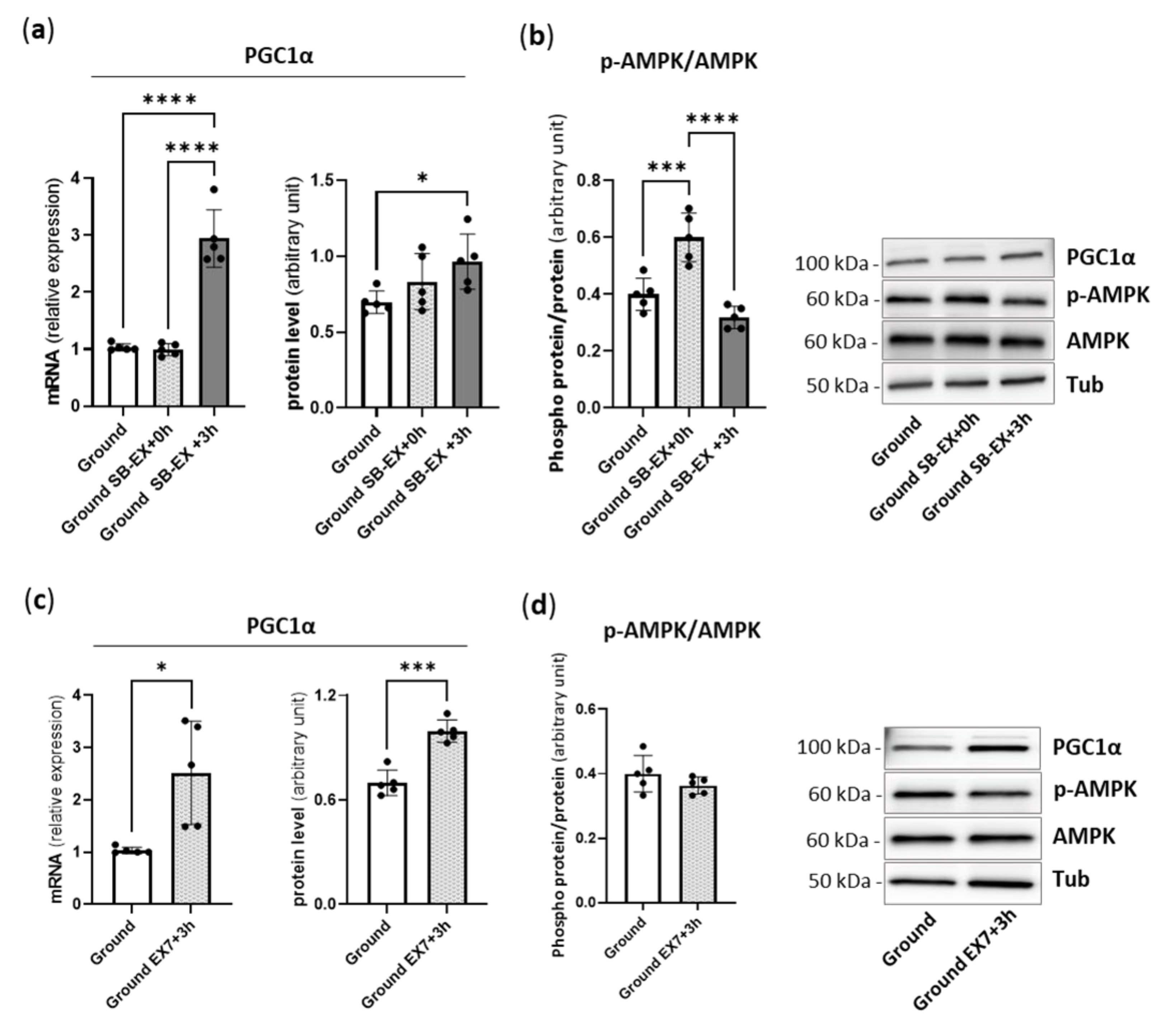
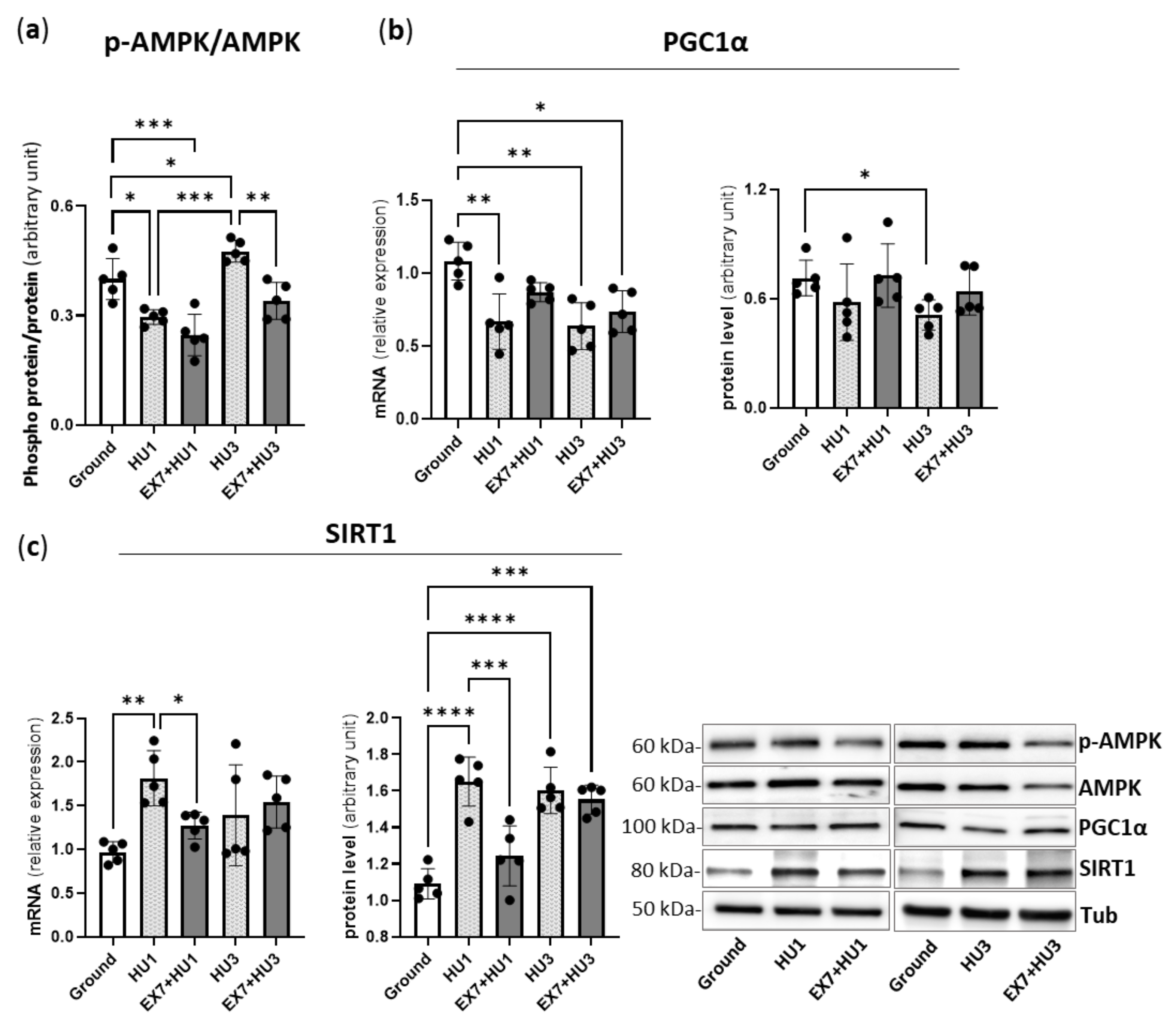
2.1. Physical Preconditioning, PGC1α and AMPK
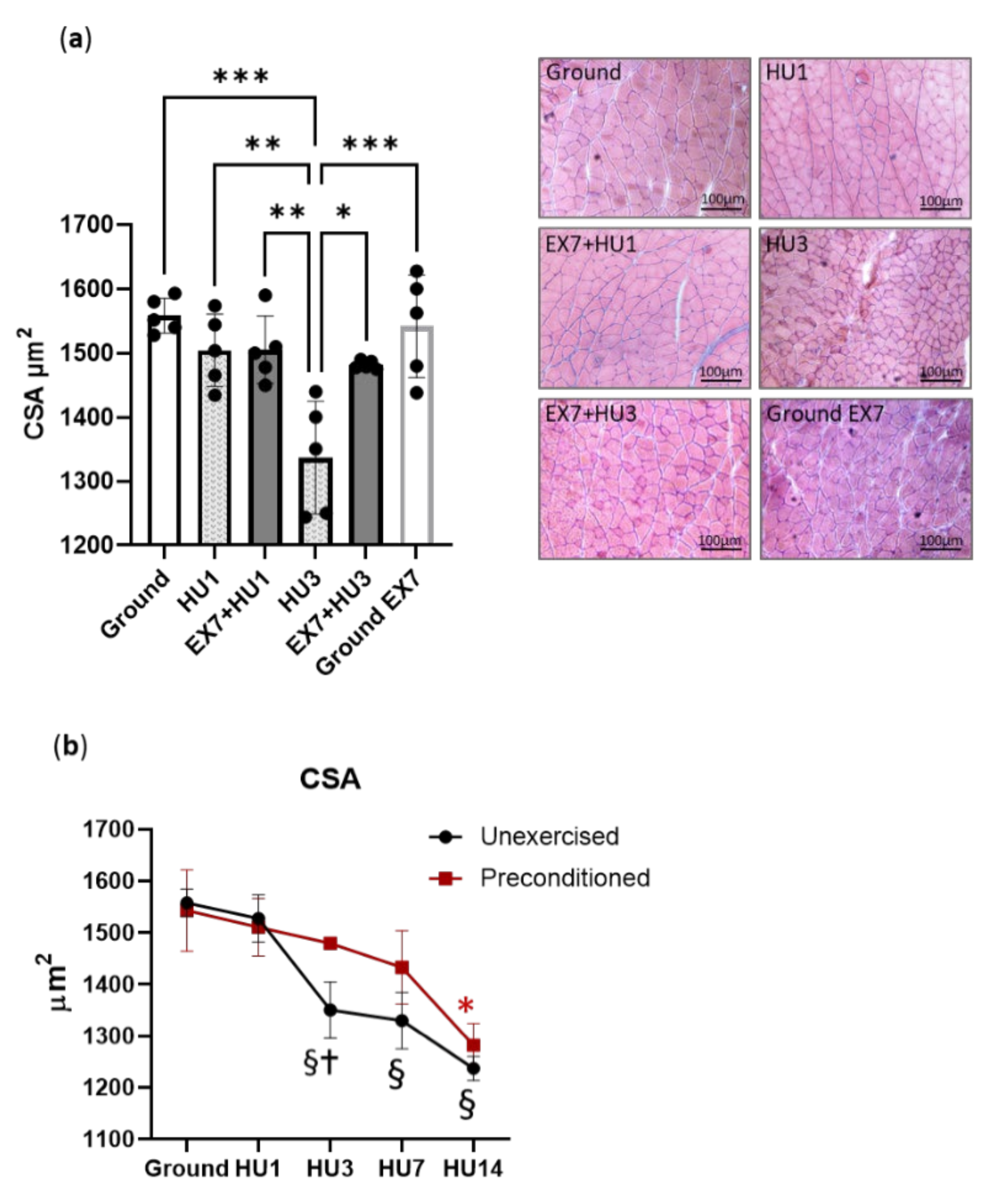
2.2. Muscle Fibers Cross Sectional Areas (CSA)
2.3. Atrophy-Related Genes
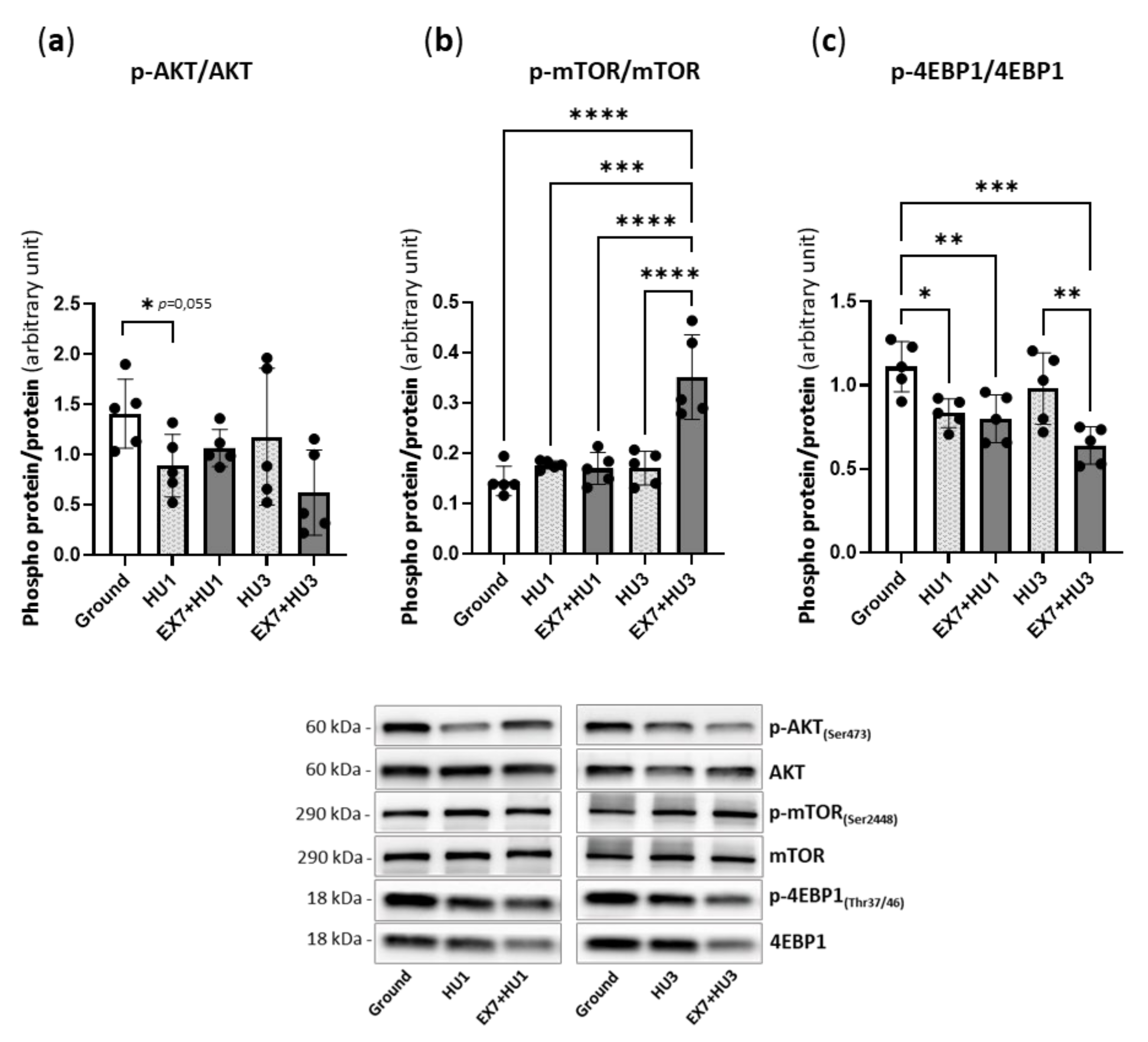
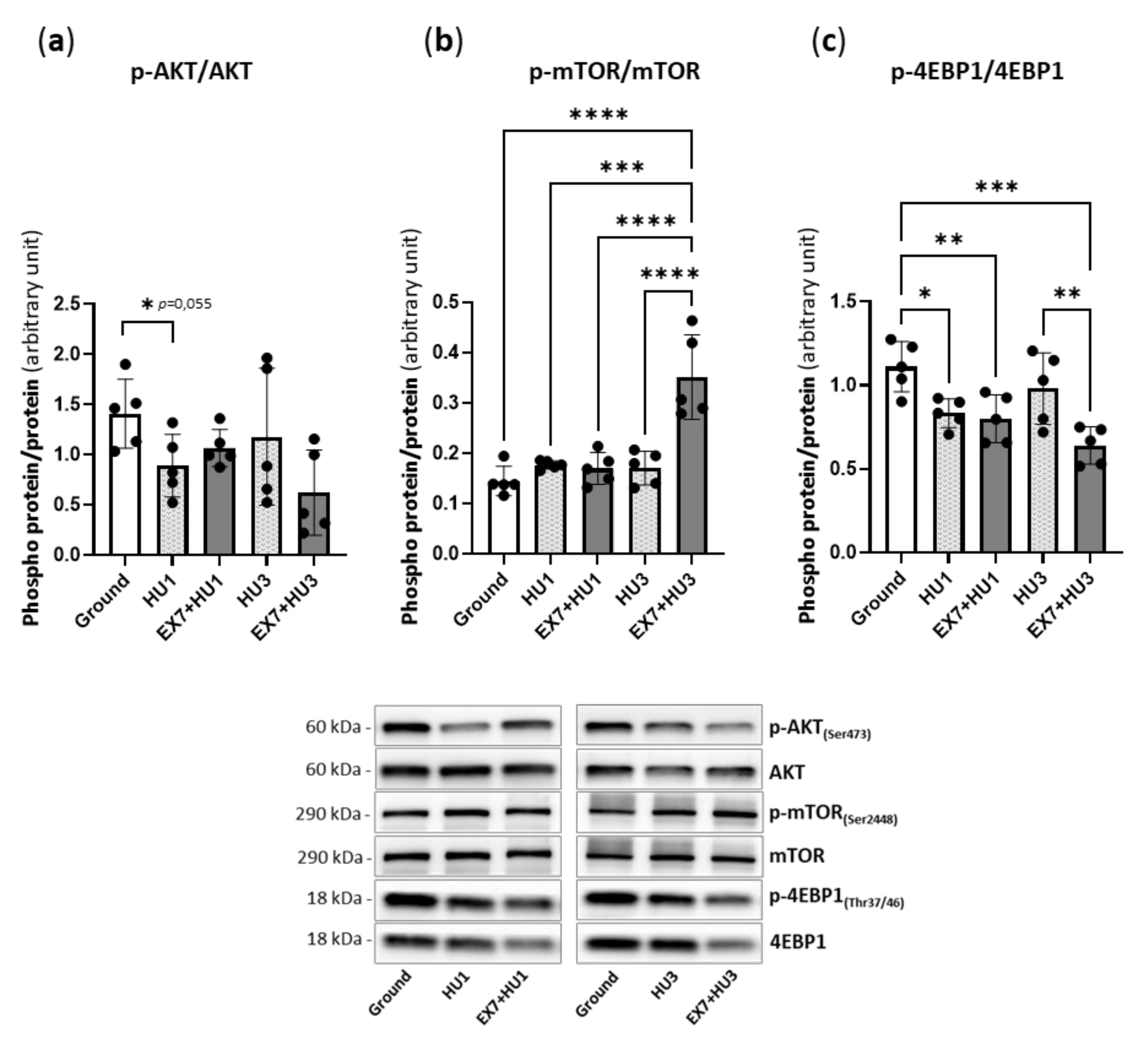
2.4. Akt/mTOR Pathway
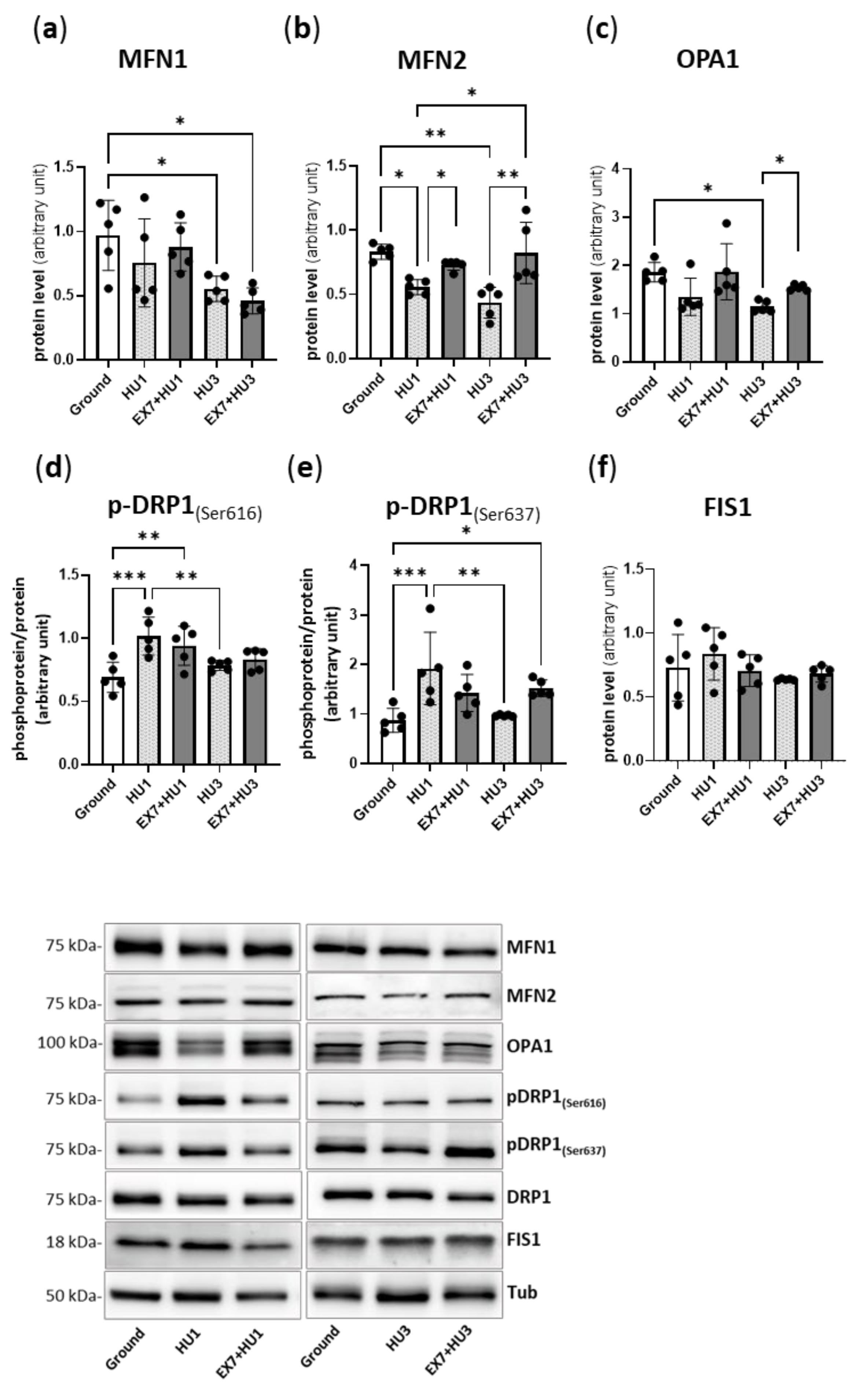
2.5. Markers of Mitochondrial Dynamics
2.6. AMPK, PGC1α, and SIRT1
3. Discussion
4. Materials and Methods
4.1. Exercise Training
4.2. Cross-Sectional Area (CSA) Analysis
4.3. Western Blot Analysis
4.4. Antibodies
4.5. Gene Expression Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mazzucco, S.; Agostini, F.; Biolo, G. Inactivity-mediated insulin resistance is associated with upregulated pro-inflammatory fatty acids in human cell membranes. Clin. Nutr. 2010, 29, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2012, 2, 1143–1211. [Google Scholar] [PubMed] [Green Version]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K.; Saltin, B. Exercise as medicine-evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sports 2015, 25 (Suppl. 3), 1–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [Green Version]
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2009, 106, 20405–20410. [Google Scholar] [CrossRef] [Green Version]
- Geng, T.; Li, P.; Yin, X.; Yan, Z. PGC-1α promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am. J. Pathol. 2011, 178, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Cannavino, J.; Brocca, L.; Sandri, M.; Bottinelli, R.; Pellegrino, M.A. PGC1-alpha over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J. Physiol. 2014, 592 Pt 20, 4575–4589. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Cannavino, J.; Brocca, L.; Sandri, M.; Grassi, B.; Bottinelli, R.; Pellegrino, M.A. The role of alterations in mitochondrial dynamics and PGC-1alpha over-expression in fast muscle atrophy following hindlimb unloading. J. Physiol. 2015, 593, 1981–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safdar, A.; Abadi, A.; Akhtar, M.; Hettinga, B.P.; Tarnopolsky, M.A. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PLoS ONE 2009, 4, e5610. [Google Scholar] [CrossRef] [PubMed]
- Norrbom, J.; Sundberg, C.J.; Ameln, H.; Kraus, W.E.; Jansson, E.; Gustafsson, T. PGC-1alpha mRNA expression is influenced by metabolic perturbation in exercising human skeletal muscle. J. Appl. Physiol. 2004, 96, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.P.; et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 2003, 52, 2874–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, S.K.; Bomkamp, M.; Ozdemir, M.; Hyatt, H. Mechanisms of exercise-induced preconditioning in skeletal muscles. Redox. Biol. 2020, 35, 101462. [Google Scholar] [CrossRef]
- Theilen, N.T.; Jeremic, N.; Weber, G.J.; Tyagi, S.C. Exercise preconditioning diminishes skeletal muscle atrophy after hindlimb suspension in mice. J. Appl. Physiol 2018, 125, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Ishihara, A.; Murakami, S.; Yasuhara, T.; Kondo, H.; Mohri, S.; Takeda, I.; Roy, R.R. Protective effects of exercise preconditioning on hindlimb unloading-induced atrophy of rat soleus muscle. Acta Physiol. 2009, 197, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Tsuzuki, T.; Chang, S.W.; Kakigi, R.; Sugiura, T.; Naito, H. Exercise preconditioning attenuates hind limb unloading-induced gastrocnemius muscle atrophy possibly via the HDAC4/Gadd45 axis in old rats. Exp. Gerontol. 2019, 122, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Signaling in muscle atrophy and hypertrophy. Physiology 2008, 23, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, E.I.; Yasuda, N.; Tarnopolsky, M.A.; Abadi, A.; Phillips, S.M. Little change in markers of protein breakdown and oxidative stress in humans in immobilization-induced skeletal muscle atrophy. Appl. Physiol. Nutr. Metab. 2010, 35, 125–133. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [Green Version]
- Brocca, L.; Toniolo, L.; Reggiani, C.; Bottinelli, R.; Sandri, M.; Pellegrino, M.A. FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J. Physiol. 2017, 595, 1143–1158. [Google Scholar] [CrossRef]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- An, B.S.; Tavera-Mendoza, L.E.; Dimitrov, V.; Wang, X.; Calderon, M.R.; Wang, H.J.; White, J.H. Stimulation of Sirt1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol. Cell Biol. 2010, 30, 4890–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocca, L.; Pellegrino, M.A.; Desaphy, J.F.; Pierno, S.; Camerino, D.C.; Bottinelli, R. Is oxidative stress a cause or consequence of disuse muscle atrophy in mice? A proteomic approach in hindlimb-unloaded mice. Exp. Physiol. 2010, 95, 331–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor. coactivator-1 alpha mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 2010, 588, 1779–1790. [Google Scholar] [CrossRef]
- Desaphy, J.F.; Pierno, S.; Liantonio, A.; Giannuzzi, V.; Digennaro, C.; Dinardo, M.M.; Camerino, G.M.; Ricciuti, P.; Brocca, L.; Pellegrino, M.A.; et al. Antioxidant treatment of hindlimb-unloaded mouse counteracts fiber type transition but not atrophy of disused muscles. Pharm. Res. 2010, 61, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.M.; Harrison, B.C.; Young, M.H.; Stodieck, L.S.; Ferguson, V.L. Longitudinal characterization of functional, morphologic, and biochemical adaptations in mouse skeletal muscle with hindlimb suspension. Muscle Nerve 2013, 48, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Hyatt, J.P.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.A.; Al-Khalaf, M.; Megeney, L.A. The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet Muscle 2016, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, C.L.; Fealy, C.E.; Mulya, A.; Kirwan, J.P. Exercise training remodels human skeletal muscle mitochondrial fission and fusion machinery towards a pro-elongation phenotype. Acta Physiol. 2019, 225, e13216. [Google Scholar] [CrossRef]
- Iqbal, S.; Ostojic, O.; Singh, K.; Joseph, A.M.; Hood, D.A. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 2013, 48, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Bertaggia, E.; Coletto, L.; Sandri, M. Posttranslational modifications control FoxO3 activity during denervation. Am. J. Physiol. Cell Physiol. 2012, 302, C587–C596. [Google Scholar] [CrossRef]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hou, H.; Haller, E.M.; Nicosia, S.V.; Bai, W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005, 24, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Goldberg, A.L. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beharry, A.W.; Sandesara, P.B.; Roberts, B.M.; Ferreira, L.F.; Senf, S.M.; Judge, A.R. HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J. Cell Sci. 2014, 127 Pt 7, 1441–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabi, B.; Adhihetty, P.J.; O’Leary, M.F.; Menzies, K.J.; Hood, D.A. Relationship between Sirt1 expression and mitochondrial proteins during conditions of chronic muscle use and disuse. J. Appl. Physiol. 2009, 107, 1730–1735. [Google Scholar] [CrossRef] [Green Version]
- Chacon-Cabrera, A.; Gea, J.; Barreiro, E. Short- and Long-Term Hindlimb Immobilization and Reloading: Profile of Epigenetic Events in Gastrocnemius. J. Cell Physiol. 2017, 232, 1415–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senf, S.M.; Sandesara, P.B.; Reed, S.A.; Judge, A.R. p300 Acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011, 300, C1490–C1501. [Google Scholar] [CrossRef] [Green Version]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Shefer, G.; Rauner, G.; Stuelsatz, P.; Benayahu, D.; Yablonka-Reuveni, Z. Moderate-intensity treadmill running promotes expansion of the satellite cell pool in young and old mice. FEBS J. 2013, 280, 4063–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| Ampd3 | GCGGAGAAGGTGTTTGCTA | CAGTCTTGTTGTGTTGGCATC |
| Atrogin1 | GCAAACACTGCCACATTCTCTC | CTTGAGGGGAAAGTGAGACG |
| BNIP3 | TTCCACTAGCACCTTCTGATGA | GAACACCGCATTTACAGAACAA |
| Cathepsin l | GTGGACTGTTCTCACGCTCAAG | TCCGTCCTTCGCTTCATAGG |
| FbxO31 | GTATGGCGTTTGTGAGAACC | AGCCCCAAAATGTGTCTGTA |
| FoxO3 | ACCTTCGTCTCTGAACTCCTTG | CTGTGGCTGAGTCAGTCTGAAG |
| Gadd34 | AGAGAAGACCAAGGGACGTG | CAGCAAGGAATGGACTGTG |
| GAPDH | CACCATCTTCCAGGAGCGAG | CCTTCTCCATGGTGGTGAAGAC |
| Mt1 | GCCTGCAAGAACTGCAAGTG | CCTTTGCAGACACAGCCCT |
| MUSA1 | TCGTGGAATGGTAATCTTGC | CCTCCCGTTTCTCTATCACG |
| MuRF1 | ACCTGCTGGTGGAAAACATC | ACCTGCTGGTGGAAAACATC |
| p62 | CCCAGTGTCTTGGCATTCTT | AGGGAAAGCAGAGGAAGCTC |
| PGC1α | ACCCCAGAGTCACCAAATGA | CGAAGCCTTGAAAGGGTTATC |
| Psme4 | AGGACGAGCAGAAGAACCTG | AATAGTTAGAGCCTGTGGTGGAG |
| SMART | TCAATAACCTCAAGGCGTTC | GTTTTGCACACAAGCTCCA |
| Psmd11 | GAGTTCCAGAGAGCCCAGTC | AACCCAGTTCAAGGATGCTC |
| Usp14 | CACGAGTTGCTTCGTATTCC | TTCAGGGTTCCTCCTTTCAC |
| UBC | CGTCGAGCCCAGTGTTACCACC | ACCTCCCCCATCACACCCAAGA |
| Tgif | TTTCCTCATCAGCAGCCTCT | CTTTGCCATCCTTTCTCAGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brocca, L.; Rossi, M.; Canepari, M.; Bottinelli, R.; Pellegrino, M.A. Exercise Preconditioning Blunts Early Atrogenes Expression and Atrophy in Gastrocnemius Muscle of Hindlimb Unloaded Mice. Int. J. Mol. Sci. 2022, 23, 148. https://doi.org/10.3390/ijms23010148
Brocca L, Rossi M, Canepari M, Bottinelli R, Pellegrino MA. Exercise Preconditioning Blunts Early Atrogenes Expression and Atrophy in Gastrocnemius Muscle of Hindlimb Unloaded Mice. International Journal of Molecular Sciences. 2022; 23(1):148. https://doi.org/10.3390/ijms23010148
Chicago/Turabian StyleBrocca, Lorenza, Maira Rossi, Monica Canepari, Roberto Bottinelli, and Maria Antonietta Pellegrino. 2022. "Exercise Preconditioning Blunts Early Atrogenes Expression and Atrophy in Gastrocnemius Muscle of Hindlimb Unloaded Mice" International Journal of Molecular Sciences 23, no. 1: 148. https://doi.org/10.3390/ijms23010148
APA StyleBrocca, L., Rossi, M., Canepari, M., Bottinelli, R., & Pellegrino, M. A. (2022). Exercise Preconditioning Blunts Early Atrogenes Expression and Atrophy in Gastrocnemius Muscle of Hindlimb Unloaded Mice. International Journal of Molecular Sciences, 23(1), 148. https://doi.org/10.3390/ijms23010148