
Abnormal Ca2+ Signals in Reactive Astrocytes as a Common Cause of Brain Diseases

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
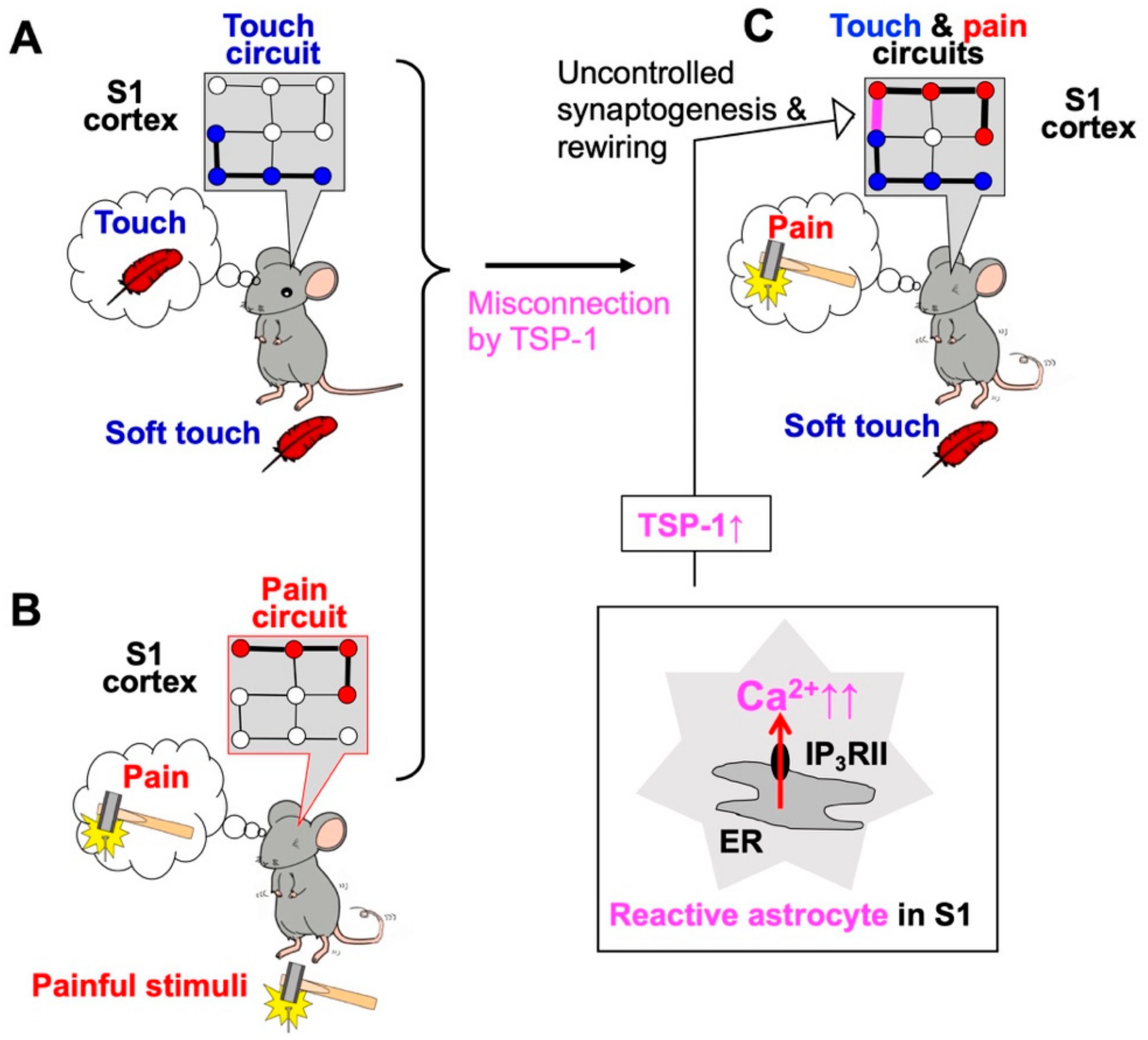
2. Neuropathic Pain and Excess Ca2+ Signals in Astrocytes of the Primary Sensory (S1) Cortex
3. Other Brain Diseases and Aberrant Astrocytic Ca2+-mediated Synapse Remodeling
4. Epileptogenesis and Aberrant Ca2+ Signals in Epileptogenic Astrocytes
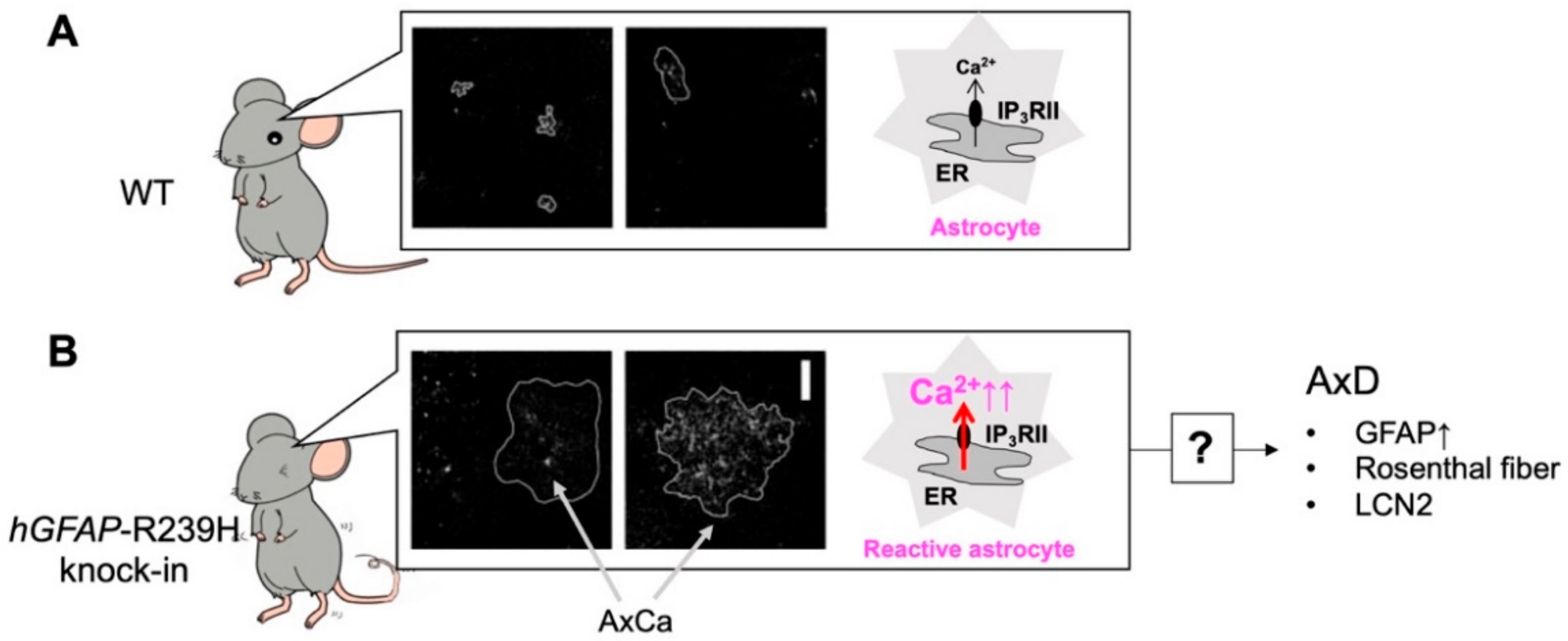
5. Alexander Disease (AxD) and Aberrant Ca2+ Signals in AxD Astrocytes
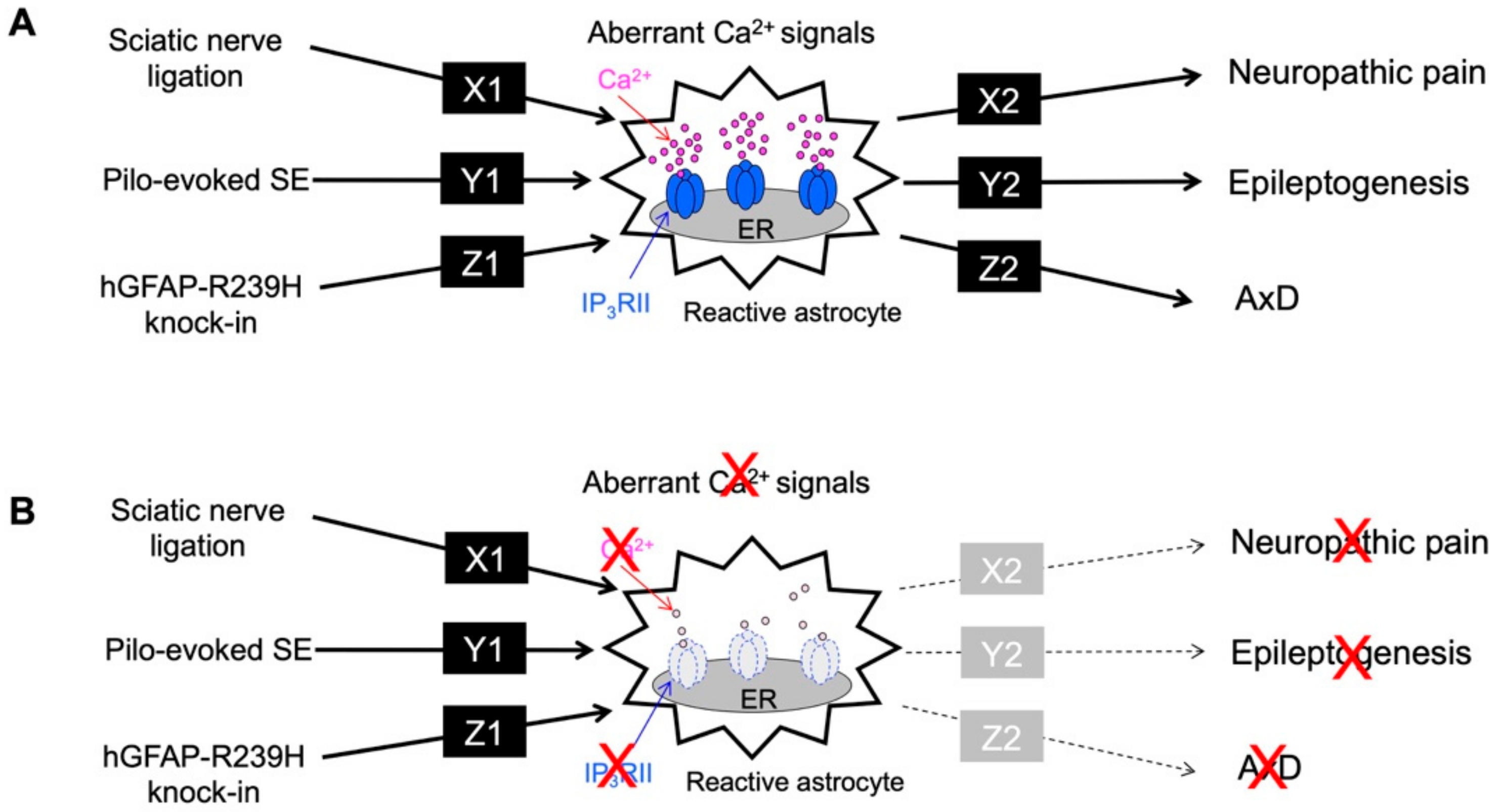
6. Complexity of Ca2+ Signals in Astrocytes
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Koizumi, S.; Fujishita, K.; Tsuda, M.; Shigemoto-Mogami, Y.; Inoue, K. Dynamic inhibition of excitatory synaptic transmission by astrocyte-derived ATP in hippocampal cultures. Proc. Natl. Acad. Sci. USA 2003, 100, 11023–11028. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.M.; Wang, H.K.; Ye, C.Q.; Ge, W.; Chen, Y.; Jiang, Z.L.; Wu, C.P.; Poo, M.M.; Duan, S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 2003, 40, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef]
- Haydon, P.G. GLIA: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef]
- Santello, M.; Bezzi, P.; Volterra, A. TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011, 69, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J. Cell Commun. Signal. 2009, 3, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Kim, S.K.; Hayashi, H.; Ishikawa, T.; Shibata, K.; Shigetomi, E.; Shinozaki, Y.; Inada, H.; Roh, S.E.; Kim, S.J.; Lee, G.; et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J. Clin. Investig. 2016, 126, 1983–1997. [Google Scholar] [CrossRef]
- Kim, S.K.; Nabekura, J.; Koizumi, S. Astrocyte-mediated synapse remodeling in the pathological brain. Glia 2017, 65, 1719–1727. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Author Correction: Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 1598. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, J.Y.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2021, 590, 612–617. [Google Scholar] [CrossRef]
- Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S. Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 996. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Eto, K.; Kim, S.K.; Wake, H.; Takeda, I.; Horiuchi, H.; Moorhouse, A.J.; Ishibashi, H.; Nabekura, J. Cortical astrocytes prime the induction of spine plasticity and mirror image pain. Pain 2018, 159, 1592–1606. [Google Scholar] [CrossRef]
- Alshelh, Z.; Di Pietro, F.; Youssef, A.M.; Reeves, J.M.; Macey, P.M.; Vickers, E.R.; Peck, C.C.; Murray, G.M.; Henderson, L.A. Chronic Neuropathic Pain: It’s about the Rhythm. J. Neurosci. 2016, 36, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Heuser, K.; Nome, C.G.; Pettersen, K.H.; Abjorsbraten, K.S.; Jensen, V.; Tang, W.; Sprengel, R.; Tauboll, E.; Nagelhus, E.A.; Enger, R. Ca2+ Signals in Astrocytes Facilitate Spread of Epileptiform Activity. Cereb. Cortex 2018, 28, 4036–4048. [Google Scholar] [CrossRef] [Green Version]
- Heuser, K.; Enger, R. Astrocytic Ca2+ Signaling in Epilepsy. Front. Cell. Neurosci. 2021, 15, 695380. [Google Scholar] [CrossRef]
- Sano, F.; Shigetomi, E.; Shinozaki, Y.; Tsuzukiyama, H.; Saito, K.; Mikoshiba, K.; Horiuchi, H.; Cheung, D.L.; Nabekura, J.; Sugita, K.; et al. Reactive astrocyte-driven epileptogenesis is induced by microglia initially activated following status epilepticus. JCI Insight 2021, 6, e135391. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Shigetomi, E.; Yasuda, R.; Sato, R.; Nakano, M.; Tashiro, K.; Tanaka, K.F.; Ikenaka, K.; Mikoshiba, K.; Mizuta, I.; et al. Aberrant astrocyte Ca2+ signals “AxCa signals” exacerbate pathological alterations in an Alexander disease model. Glia 2018, 66, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783. [Google Scholar] [CrossRef]
- Nagai, J.; Rajbhandari, A.K.; Gangwani, M.R.; Hachisuka, A.; Coppola, G.; Masmanidis, S.C.; Fanselow, M.S.; Khakh, B.S. Hyperactivity with Disrupted Attention by Activation of an Astrocyte Synaptogenic Cue. Cell 2019, 177, 1280–1292. [Google Scholar] [CrossRef]
- Wang, J.; Li, K.L.; Shukla, A.; Beroun, A.; Ishikawa, M.; Huang, X.; Wang, Y.; Wang, Y.Q.; Yang, Y.; Bastola, N.D.; et al. Cocaine Triggers Astrocyte-Mediated Synaptogenesis. Biol. Psychiatry 2021, 89, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Barres, B.A. Synaptic efficacy enhanced by glial cells in vitro. Science 1997, 277, 1684–1687. [Google Scholar] [CrossRef] [Green Version]
- Ullian, E.M.; Sapperstein, S.K.; Christopherson, K.S.; Barres, B.A. Control of synapse number by glia. Science 2001, 291, 657–661. [Google Scholar] [CrossRef]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Stogsdill, J.A.; Eroglu, C. The interplay between neurons and glia in synapse development and plasticity. Curr. Opin. Neurobiol. 2017, 42, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Casati, M.E.; Murtie, J.C.; Rio, C.; Stankovic, K.; Liberman, M.C.; Corfas, G. Nonneuronal cells regulate synapse formation in the vestibular sensory epithelium via erbB-dependent BDNF expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17005–17010. [Google Scholar] [CrossRef] [Green Version]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Kucukdereli, H.; Allen, N.J.; Lee, A.T.; Feng, A.; Ozlu, M.I.; Conatser, L.M.; Chakraborty, C.; Workman, G.; Weaver, M.; Sage, E.H.; et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. USA 2011, 108, E440–E449. [Google Scholar] [CrossRef] [Green Version]
- Risher, W.C.; Patel, S.; Kim, I.H.; Uezu, A.; Bhagat, S.; Wilton, D.K.; Pilaz, L.J.; Singh Alvarado, J.; Calhan, O.Y.; Silver, D.L.; et al. Astrocytes refine cortical connectivity at dendritic spines. Elife 2014, 3, e04047. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Suarez, E.; Liu, T.F.; Kopelevich, A.; Allen, N.J. Astrocyte-Secreted Chordin-like 1 Drives Synapse Maturation and Limits Plasticity by Increasing Synaptic GluA2 AMPA Receptors. Neuron 2018, 100, 1116–1132. [Google Scholar] [CrossRef] [Green Version]
- Diniz, L.P.; Almeida, J.C.; Tortelli, V.; Vargas Lopes, C.; Setti-Perdigao, P.; Stipursky, J.; Kahn, S.A.; Romao, L.F.; de Miranda, J.; Alves-Leon, S.V.; et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor beta signaling through modulation of D-serine levels in cerebral cortex neurons. J. Biol. Chem. 2012, 287, 41432–41445. [Google Scholar] [CrossRef]
- Fuentes-Medel, Y.; Ashley, J.; Barria, R.; Maloney, R.; Freeman, M.; Budnik, V. Integration of a retrograde signal during synapse formation by glia-secreted TGF-beta ligand. Curr. Biol. 2012, 22, 1831–1838. [Google Scholar] [CrossRef] [Green Version]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Welsh, C.A.; Barres, B.A.; Stevens, B. Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 2015, 18, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Hama, H.; Hara, C.; Yamaguchi, K.; Miyawaki, A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron 2004, 41, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Kitaura, H.; Hiraishi, T.; Itoh, Y.; Oishi, M.; Fujii, Y.; Fukuda, M.; Kakita, A. Reactive astrocytes contribute to epileptogenesis in patients with cavernous angioma. Epilepsy Res. 2021, 176, 106732. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Kinboshi, M.; Ikeda, A.; Ohno, Y. Role of Astrocytic Inwardly Rectifying Potassium (Kir) 4.1 Channels in Epileptogenesis. Front. Neurol. 2020, 11, 626658. [Google Scholar] [CrossRef]
- Tian, G.F.; Azmi, H.; Takano, T.; Xu, Q.; Peng, W.; Lin, J.; Oberheim, N.; Lou, N.; Wang, X.; Zielke, H.R.; et al. An astrocytic basis of epilepsy. Nat. Med. 2005, 11, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Alexander, W.S. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949, 72, 373–381. [Google Scholar] [CrossRef]
- Yoshida, T.; Nakagawa, M. Clinical aspects and pathology of Alexander disease, and morphological and functional alteration of astrocytes induced by GFAP mutation. Neuropathology 2012, 32, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.F.; Takebayashi, H.; Yamazaki, Y.; Ono, K.; Naruse, M.; Iwasato, T.; Itohara, S.; Kato, H.; Ikenaka, K. Murine model of Alexander disease: Analysis of GFAP aggregate formation and its pathological significance. Glia 2007, 55, 617–631. [Google Scholar] [CrossRef]
- Jones, J.R.; Kong, L.; Hanna, M.G.T.; Hoffman, B.; Krencik, R.; Bradley, R.; Hagemann, T.; Choi, J.; Doers, M.; Dubovis, M.; et al. Mutations in GFAP Disrupt the Distribution and Function of Organelles in Human Astrocytes. Cell Rep. 2018, 25, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigetomi, E.; Tong, X.; Kwan, K.Y.; Corey, D.P.; Khakh, B.S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 2011, 15, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koizumi, S.; Shigetomi, E.; Sano, F.; Saito, K.; Kim, S.K.; Nabekura, J. Abnormal Ca2+ Signals in Reactive Astrocytes as a Common Cause of Brain Diseases. Int. J. Mol. Sci. 2022, 23, 149. https://doi.org/10.3390/ijms23010149
Koizumi S, Shigetomi E, Sano F, Saito K, Kim SK, Nabekura J. Abnormal Ca2+ Signals in Reactive Astrocytes as a Common Cause of Brain Diseases. International Journal of Molecular Sciences. 2022; 23(1):149. https://doi.org/10.3390/ijms23010149
Chicago/Turabian StyleKoizumi, Schuichi, Eiji Shigetomi, Fumikazu Sano, Kozo Saito, Sun Kwang Kim, and Junichi Nabekura. 2022. "Abnormal Ca2+ Signals in Reactive Astrocytes as a Common Cause of Brain Diseases" International Journal of Molecular Sciences 23, no. 1: 149. https://doi.org/10.3390/ijms23010149
APA StyleKoizumi, S., Shigetomi, E., Sano, F., Saito, K., Kim, S. K., & Nabekura, J. (2022). Abnormal Ca2+ Signals in Reactive Astrocytes as a Common Cause of Brain Diseases. International Journal of Molecular Sciences, 23(1), 149. https://doi.org/10.3390/ijms23010149