
Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies


Abstract
1. Introduction
2. Inflammation in Ischemia/Reperfusion Injuries
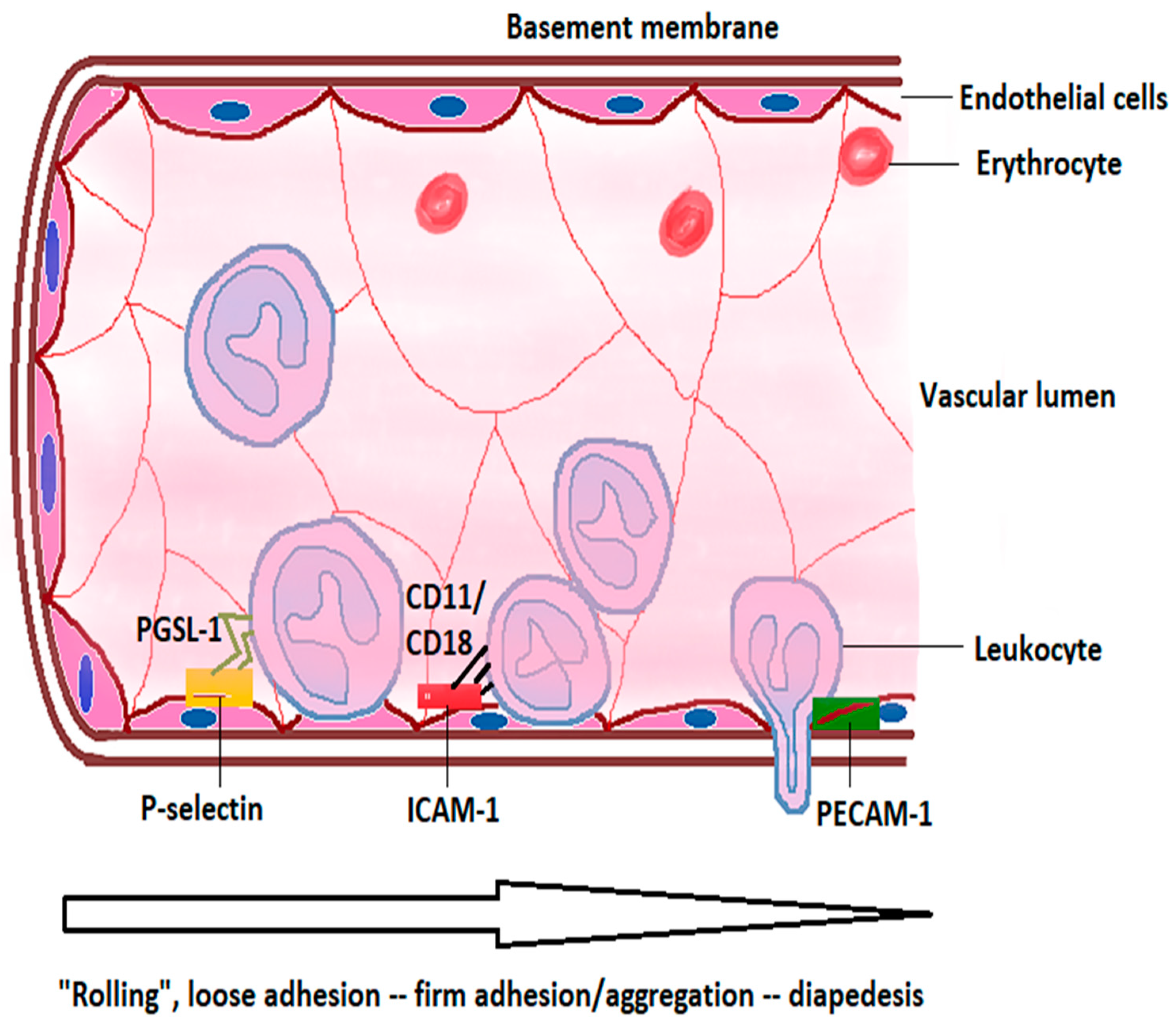
2.1. Intravascular Initiation of the Inflammatory Cascade
2.2. Parenchymal Inflammation in Cerebral I/R Injuries
2.2.1. Microglia in Neuroinflammation after Acute Ischemic Stroke
2.2.2. Astrocytes in Post-Stroke Neuroinflammation
2.2.3. Leukocytes in Neuroinflammation after Acute Ischemic Stroke
2.2.4. Platelets in Cerebral Ischemia/Reperfusion Injury
2.3. Inflammatory Mediators
2.3.1. Cytokines
Pro-Inflammatory Response
Anti-Inflammatory Cytokines
2.3.2. Chemokines
- -
- CC—two cysteine residues, immediately adjacent,
- -
- CXC—two cysteine residues separated by one amino acid,
- -
- CX3C—two cysteine residues separated by three amino acids.
2.3.3. Matrix Metalloproteinases

{kind=link}
{kind=link}
| Inflammatory Mediators | Beneficial Effects | Detrimental Effects | References |
|---|---|---|---|
| TNF-α | Stimulates the expression of antioxidants and anti-apoptotic factors, involved in ischemic preconditioning, increases expression of neurotrophic factors, modulates neuronal plasticity | Increases infarct volume, promotes leukocyte adherence to endothelium, contributes to BBB disruption, edema formation, increases apoptosis of endothelial cells | [15,28,91,92,152,160,161,203] |
| IL-6 | Enhances post-stroke angiogenesis | Contributes to leukocyte recruitment and promotes neuroinflammation, increases stroke severity | [28,164] |
| IL-8 | Augments neuroinflammation | [28,166,167] | |
| IL-10 | Diminishes cytokine release, promotes neuronal survival and neurogenesis, promotes M2 polarization of microglia/macrophages | [28,40,52,117] | |
| IL-1β | Disruption of BBB, edema formation, leukocyte recruitment | [28,59,151] | |
| MMPs | Promote vascular remodeling | BBB disruption, vasogenic edema, hemorrhagic transformation | [28,196,201] |
| Interferon-β | Downregulates ICAM-1 expression, attenuates BBB disruption | [176,177] | |
| MCP-1 | Increases BBB permeability, enhances leukocyte infiltration | [28,184,185] |

2.3.4. Neurotrophic Factors and Neuropeptides
3. Therapeutic Approaches Focusing on Modulation of Neuroinflammation
3.1. Therapies Targeting Microglia
3.2. Strategies for Blocking Neutrophil Infiltration
3.3. Targeting Lymphocytes in Acute Ischemic Stroke
3.4. Other Drugs Which Modulate Neuroinflammation
3.5. Reasons for Discrepancies
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Katan, M.; Luft, A. Global burden of stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Scott, C.A.; Rothwell, P.M.; on behalf of the Oxford Vascular Study. Trends in stroke incidence in high-income countries in the 21st century. Population-based study and systematic review. Stroke 2020, 51, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J.; Lin, J.K.; Farzadfar, F.; Khang, Y.H.; Stevens, G.A.; et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980s: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011, 378, 31–40. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef]
- Smith, W.S.; Sung, G.; Starkman, S.; Saver, J.L.; Kidwell, C.S.; Gobin, Y.P.; Lutsep, H.L.; Nesbit, G.M.; Grobelny, T.; Rymer, M.M.; et al. Safety and efficacy of mechanical embolectomy in acute ischemic stroke: Results of the MERCI trial. Stroke 2005, 36, 1432–1438. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 h after stroke with a mismatch between deficit and infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, M.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, D.M.; Hoh, B.; et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar] [PubMed]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion injury in the brain: Mechanisms and potential therapeutic strategies. Biochem. Pharmacol. 2016, 5, 213. [Google Scholar]
- Wong, C.H.Y.; Crack, P.J. Modulation of neuroinflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr. Med. Chem. 2008, 15, 1–14. [Google Scholar] [PubMed]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Refocusing neuroprotection in cerebral reperfusion era: New challenges and strategies. Front. Neurol. 2018, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front. Mol. Neurosci. 2020, 13, 28. [Google Scholar] [CrossRef]
- Chamorro, A.; Hallenbeck, J. The harms and benefits of inflammatory and immune responses in vascular disease. Stroke 2006, 37, 291–293. [Google Scholar] [CrossRef]
- Enzmann, G.; Kargaran, S.; Engelhardt, B. Ischemia-reperfusion injury in stroke: Impact of the brain barriers and brain immune privilege on neutrophil function. Ther. Adv. Neurol. Disord. 2018, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and stroke: An overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef]
- Tahsili-Fahadan, P.; Farrokh, S.; Geocadin, R.G. Hypothermia and brain inflammation after cardiac arrest. Brain Circ. 2018, 4, 1–13. [Google Scholar]
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside. Ther. Adv. Neurol. Dis. 2018, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, S.F.; Denorme, F.; Langhauser, F.; Geuss, E.; Fluri, F.; Kleinschnitz, C. Thromboinflammation in stroke brain damage. Stroke 2016, 47, 1165–1172. [Google Scholar] [CrossRef]
- Palabrica, T.; Lobb, F.; Furei, B.C.; Aronovitz, M.; Benjamin, C.; Hsu, Y.M.; Sajer, S.A.; Furie, B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992, 359, 848–851. [Google Scholar] [CrossRef]
- Rezaie, A.R. Protease-activated receptor signaling by coagulation proteases in endothelial cells. Thromb. Haemost. 2014, 112, 876–882. [Google Scholar] [PubMed]
- Turovsky, E.A.; Zinchenko, V.P.; Gaidin, S.G.; Turovskaya, M.V. Calcium-binding proteins protect GABAergic neurons of the hippocampus from hypoxia and ischemia in vitro. Biochemistry 2018, 12, 74–84. [Google Scholar] [CrossRef]
- Turovsky, E.A.; Varlamova, E.G.; Plotnikov, E.Y. Mechanisms underlying the protective effect of the peroxiredoxin-6 are mediated via the protection of astrocytes during ischemia/reoxygenation. Int. J. Mol. Sci. 2021, 22, 8805. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthius, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Simion, A.; Jurcau, A. The role of antioxidant treatment in acute ischemic stroke: Past present and future. Neurol. Res. Surg. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jala, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derived from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell. Neurosci. 2013, 7, 44. [Google Scholar] [CrossRef]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef]
- Sunnemark, D.; Eltayeb, S.; Nilsson, M.; Wallstrom, E.; Lassmann, H.; Olsson, T.; Berg, A.-L.; Ericsson-Dahlstrand, A. CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: Kinetics and cellular origin. J. Neuroinflam. 2005, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Guruswamy, R.; ElAli, A. Complex roles of microglial cells in ischemic stroke pathobiology: New insights and future directions. Int. J. Mol. Sci. 2017, 18, 496. [Google Scholar] [CrossRef]
- ElAli, A.; Rivest, S. Microglia ontology and signaling. Front. Cell Dev. Biol. 2016, 4, 72. [Google Scholar] [CrossRef]
- Ito, D.; Tanaka, K.; Suzuki, S.; Dembo, T.; Fukuuchi, Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 2001, 32, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical role of TLR4 response in the activation of microglia by ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A. Insights into the pathogenesis of neurodegenerative diseases: Focus on mitochondrial dysfunction and oxidative stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Koerner, I.P.; Möller, T. Microglia in ischemic brain injury. Future Neurol. 2011, 5, 227–246. [Google Scholar] [CrossRef]
- Ridder, D.A.; Schwaninger, M. NF-kappaB signaling in cerebral ischemia. Neuroscience 2009, 158, 995–1006. [Google Scholar] [CrossRef]
- Xu, H.; Qin, W.; Hu, X.; Mu, S.; Zhu, J.; Lu, W.; Luo, Y. Lentivirus-mediated overexpression of OTULIN ameliorates microglia activation and neuroinflammation by depressing the activation of the NF-κB signaling pathway in cerebral ischemia/reperfusion rats. J. Neuroinflamm. 2018, 15, 83. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrovski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Anttila, J.E.; Whitaker, K.W.; Wires, E.S.; Harvey, B.K.; Airavaara, M. Role of microglia in ischemic focal stroke and recovery: Focus on toll-like receptors. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 3–14. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Almolda, B.; de Labra, C.; Barrera, I.; Gruart, A.; Delgado-Garcia, J.M.; Villacampa, N.; Vilella, A.; Hofer, M.J.; Hidalgo, J.; Campbell, I.L.; et al. Alterations in microglial phenotype and hippocampal neuronal function in transgenic mice with astrocyte-targeted production of interleukin-10. Brain Behav. Immun. 2015, 45, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1and M2 macrophages: Oracles of health and disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 2010, 184, 4810–4818. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Fumagalli, S.; De Simoni, M.-G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signaling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Todd, K.G. Microglia in cerebral ischemia: Molecular actions and interactions. Can. J. Physiol. Pharmacol. 2006, 84, 49–59. [Google Scholar] [CrossRef]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef]
- Lee, E.J.; Moon, P.G.; Baek, M.C.; Kim, H.S. Comparison of the effects of matrix metalloproteinase inhibitors on TNF-α release from activated microglia and TNF-α converting enzyme activity. Biomol. Ther. 2014, 22, 414–419. [Google Scholar] [CrossRef]
- Lisi, L.; Stigliano, E.; Lauriola, L.; Navarra, P.; Dello Russo, C. Proinflammatory-activated glioma cells induce a switch in microglial polarization and activation status, from a predominant M2b phenotype to a mixture of M1 and M2a/B polarized cells. ASN Neuro 2014, 6, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 2013, 91, 1143–1151. [Google Scholar] [CrossRef]
- Locatelli, G.; Theodorou, D.; Kendirli, A.; Jordao, M.J.C.; Stazewski, O.; Phulphagar, K.; Cantuti-Castelvetri, L.; Dagkalis, A.; Bessis, A.; Simons, M.; et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat. Neurosci. 2018, 21, 1196–1208. [Google Scholar] [CrossRef]
- Fang, M.; Zhong, L.; Jin, X.; Cui, R.; Yang, W.; Gao, S.; Lv, J.; Li, B.; Liu, T. Effect of inflammation on the process of stroke rehabilitation and poststroke depression. Front. Psychiatry 2019, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.-U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Emmrich, J.V.; Ejaz, S.; Neher, J.J.; Williamson, D.J.; Baron, J.C. Regional distribution of selective neuronal loss and microglial activation across the MCA territory after transient focal ischemia: Quantitative versus semiquantitative systematic immunohistochemical assessment. J. Cereb. Blood Flow Metab. 2015, 35, 20–27. [Google Scholar] [CrossRef]
- Denes, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating resident microglia after focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1941–1953. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, C.M.; Yang, W.L.; Wang, P. Microglial CD14 activated by iNOS contributes to neuroinflammation in cerebral ischemia. Brain Res. 2013, 1506, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Facci, L.; Barbierato, M.; Marinelli, C.; Argentini, C.; Skaper, S.D.; Giusti, P. Toll-like receptors 2, -3 and -4 prime microglia but not astrocytes across the central nervous system regions for ATP-dependent interleukin-1β release. Sci. Rep. 2014, 4, 6824. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Ma, J.; Ha, T.; Xia, Y.; Kelley, J.; Williams, D.L.; Kao, R.L.; Browder, I.W.; Schweitzer, J.B.; Kalbfleisch, J.H.; et al. Activation of toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J. Neuroimmunol. 2007, 190, 101–111. [Google Scholar] [CrossRef]
- Sun, M.; Deng, B.; Zhao, X.; Gao, C.; Yang, L.; Zhao, H.; Yu, D.; Zhang, F.; Xu, L.; Chen, L.; et al. Isoflurane preconditioning provides neuroprotection against stroke by regulating the expression of the TLR4 signaling pathway to alleviate microglial activation. Sci. Rep. 2015, 5, 1445. [Google Scholar]
- Lalancette-Hebert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef]
- Kitamura, Y.; Takata, K.; Inden, M.; Tsuchiya, D.; Yanagisawa, D.; Nakata, J.; Taniguchi, T. Intracerebroventricular injection of microglia protects against focal brain ischemia. J. Pharmacol. Sci. 2004, 94, 203–206. [Google Scholar] [CrossRef]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S. Immunocytochemical studies of cellular reaction in human ischemic brain stroke. MAB anti-CD68 stains macrophages, astrocytes and microglial cells in infarcted area. Folia Neuropathol. 1996, 34, 17–24. [Google Scholar]
- Price, C.J.; Wang, D.; Menon, D.K.; Guadagno, J.V.; Cleij, M.; Fryer, T.; Aigbirhio, F.; Baron, J.-C.; Warburton, E.A. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke 2006, 37, 1749–1753. [Google Scholar] [CrossRef]
- Pappata, S.; Levasseur, M.; Gunn, R.N.; Myers, R.; Crouzel, C.; Syrota, A.; Jones, T.; Kreutzberg, G.W.; Banati, R.B. Thalamic microglial activation in ischemic stroke detected in vivo by PET and 11c 1195. Neurology 2000, 55, 1052–1054. [Google Scholar] [CrossRef]
- Dugue, R.; Dugue, A.; Barone, F.C. Roles of pro- and anti-inflammatory cytokines in traumatic brain injury and acute ischemic stroke. In Mechanisms of Neuroinflammation; Abreu, G.E.A., Ed.; IntechOpen: Rijeka, Croatia, 2017; pp. 211–261. [Google Scholar]
- Singhal, G.; Baune, B.T. Microglia: An interface between the loss of neuroplasticity and depression. Front. Cell. Neurosci. 2017, 11, 270. [Google Scholar] [CrossRef]
- Bylicky, M.; Mueller, G.P.; Day, R.M. Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef] [PubMed]
- Ketheeswaranathan, P.; Turner, N.A.; Spary, E.J.; Batten, T.F.; McColl, B.W.; Saha, S. Changes in glutamate transporter expression in mouse forebrain areas following focal ischemia. Brain Res. 2011, 1418, 93–103. [Google Scholar] [CrossRef]
- Maida, C.D.; Norrito, R.L.; Daidone, M.; Tuttolomomdo, A.; Pinto, A. Neuroinflammatory mechanisms in ischemic stroke: Focus on cardioembolic stroke, background, and therapeutic approaches. Int. J. Mol. Sci. 2020, 21, 6454. [Google Scholar] [CrossRef]
- Wang, H.; Song, G.; Chuang, H.; Chiu, C.; Abdelmaksoud, A.; Ye, Y.; Zhao, L. Portrait of glial scar in neurological diseases. Int. J. Immunopathol. Pharmacol. 2018, 31, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. Br. J. Pharmacol. 2016, 36, 1481–1507. [Google Scholar] [CrossRef]
- Nowicka, D.; Rogozinska, K.; Aleksy, M.; Witte, O.W.; Skangiel-Kramska, J. Spatiotemporal dynamics of astroglial and microglial responses after photothrombotic stroke in the rat brain. Acta Neurobiol. Exp. 2008, 68, 155. [Google Scholar]
- Li, M.; Li, Z.; Yao, Y.; Jin, W.-N.; Wood, K.; Liu, Q.; Shi, F.; Hao, J. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc. Natl. Acad. Sci. USA 2017, 114, 396–405. [Google Scholar] [CrossRef]
- Pawluk, H.; Woźniak, A.; Grześk, G.; Kolodjieska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. The role of pro-inflammatory cytokines in the pathogenesis of ischemic stroke. Clin. Interv. Aging 2020, 15, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Konstas, A.-A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007, 49, 93–102. [Google Scholar] [CrossRef]
- Jean, W.C.; Spellman, S.R.; Nussbaum, E.S.; Low, W.C. Reperfusion injury after focal cerebral ischemia: The role of inflammation and the therapeutic horizon. Neurosurgery 1998, 43, 1382–1396. [Google Scholar] [CrossRef]
- Nourshargh, S.; Krombach, F.; Dejana, E. The role of JAM-A and PECAM-1 in modulating leucocyte infiltration in inflamed ischemic tissues. J. Leukoc. Biol. 2006, 80, 714–718. [Google Scholar] [CrossRef]
- Nourshargh, S.; Hordjik, P.L.; Sixt, M. Breaching multiple barriers: Leukocyte motility through venular walls and the interstitium. Nat. Rev. Mol. Cell Biol. 2010, 11, 366–378. [Google Scholar] [CrossRef]
- Rodriguez, S.F.; Granger, D.N. Role of blood cells in ischemia-reperfusion-induced endothelial barrier failure. Cardiovasc. Res. 2010, 87, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Gelman, S. Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology 2001, 94, 1133–1138. [Google Scholar] [CrossRef]
- Del Zoppo, G.J. Acute anti-inflammatory approaches to ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 143–148. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Neuroinflammation in post-ischemic neurodegeneration of the brain: Friend, foe, or both? Int. J. Mol. Sci. 2021, 22, 4405. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, J.; Chang, J.Y.; Kim, S.H.; Lee, J.E. Inflammation after ischemic stroke: The role of leukocytes and glial cells. Exp. Neurobiol. 2016, 25, 241–251. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ito, M. Resolution of inflammation and repair after ischemic brain injury. Neuroimmunol. Neuroinflammation 2020, 7, 264–276. [Google Scholar] [CrossRef]
- Tanaka, R.; Komine-Kobayashi, M.; Mochizuki, H.; Yamada, M.; Furuya, T.; Migita, M.; Shimada, T.; Mizuno, Y.; Urabe, T. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience 2003, 117, 531–539. [Google Scholar] [CrossRef]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translocation insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedorn, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Niewandt, B.; Windl, H.; Stoll, G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef]
- Brait, V.H.; Arumugam, T.V.; Drummond, G.R.; Sobey, C.G. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J. Cereb. Blood Flow Metab. 2012, 32, 598–611. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Wu, Q.; Yuan, Y.; Cao, W.; Zhang, X. Regulatory T cells in ischemic stroke. CNS Neurosci. Ther. 2021, 27, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Planas, A.M.; Chamorro, A. Regulatory T cells protect the brain after stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Chen, J. PRO: Regulatory T cells are protective in ischemic stroke. Stroke 2013, 44, e85–e86. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Llovera, G.; Benakis, C.; Enzmann, G.; Cai, R.; Arzberger, T.; Ghasemigharagoz, A.; Mao, X.; Malik, R.; Lazarevic, I.; Liebscher, S.; et al. The choroid plexus is a key cerebral invasion route for T cells after stroke. Acta Neuropathol. 2017, 134, 851–868. [Google Scholar] [CrossRef]
- Li, P.; Gan, Y.; Sun, B.L.; Zhang, F.; Lu, B.; Gao, Y.; Liang, W.; Thomson, A.W.; Chen, J.; Hu, X. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann. Neurol. 2013, 74, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Park, K.P.; Rosell, A.; Foerch, C.; Xing, C.; Kim, W.J.; Lee, S.; Opdenakker, G.; Furie, K.L.; Lo, E.H. Plasma and brain matrix metalloproteinase-9 after acute focal cerebral ischemia in rats. Stroke 2009, 40, 2836–2842. [Google Scholar] [CrossRef]
- Liesz, A.; Hu, X.; Kleinschnitz, C.; Offner, H. Functional role of regulatory lymphocytes in stroke: Facts and controversies. Stroke 2015, 46, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Kraft, P.; Dreykluft, A.; Hagedorn, I.; Göbel, K.; Schuhmann, M.K.; Langhauser, F.; Helluy, X.; Schwarz, T.; Bittner, S.; et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood 2013, 121, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Choudhury, G.R.; Winters, A.; Yang, S.H.; Jin, K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory response via IL-10. Eur. J. Immunol. 2015, 45, 180–191. [Google Scholar] [CrossRef]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Ruan, L.; Lau, B.W.; Wang, J.; Huang, L.; Zhuge, Q.; Wang, B.; Jin, K.; So, K.-F. Neurogenesis in neurological and psychiatric diseases and brain injury: From bench to bedside. Prog. Neurobiol. 2014, 115, 116–137. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar]
- Shaik, N.F.; Regan, R.F.; Naik, U.P. Platelets as drivers of ischemia/reperfusion injury after stroke. Blood Adv. 2021, 5, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Bladowski, M.; Gawrys, J.; Gajecki, D.; Szahidewicz-Krupska, E.; Sawicz-Bladowska, A.; Doroszko, A. Role of the platelets and nitric oxide biotransformation in ischemic stroke: A translative review from bench to bedside. Oxid. Med. Cell Longev. 2020, 2020, 2979260. [Google Scholar] [CrossRef]
- Wang, G.R.; Zhu, Y.; Halushka, P.V.; Lincoln, T.M.; Mendelsohn, M.E. Mechanism of platelet inhibition by nitric oxide: In vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 4888–4893. [Google Scholar] [CrossRef]
- Zhu, J.; Song, W.; Li, L.; Fan, X. Endothelial nitric oxide synthase: A potential therapeutic target for cerebrovascular diseases. Mol. Brain 2016, 9, 30. [Google Scholar] [CrossRef]
- Kuo, M.C.; Patschan, D.; Patschan, S.; Cohen-Gould, L.; Park, H.-C.; Ni, J.; Addabbo, F.; Goligorsky, M.S. Ischemia-induced exocytosis of Weibel-Palade bodies mobilizes stem cells. J. Am. Soc. Nephrol. 2008, 19, 2321–2330. [Google Scholar] [CrossRef]
- Zhao, B.Q.; Chauhan, A.K.; Canault, M.; Patten, I.S.; Yang, J.J.; Dockal, M.; Scheiflinger, S.; Wagner, D.D. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood 2009, 114, 3329–3334. [Google Scholar] [CrossRef]
- Fujioka, M.; Hayakawa, K.; Mishima, K.; Kunizawa, A.; Irie, K.; Higuchi, S.; Nakano, T.; Muroi, C.; Fukushima, H.; Sugimoto, M.; et al. ADAMTS13 gene deletion aggravates ischemic brain damage: A possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood 2010, 115, 1650–1653. [Google Scholar] [CrossRef]
- Page, C.; Pitchford, S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int. Immunopharmacol. 2013, 17, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.R.; Jeremy, R.W.; Weisman, H.F.; Winkelstein, J.A.; Becker, L.C. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 min of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation 2011, 123, 2579–2590. [Google Scholar]
- Ishikawa, M.; Cooper, D.; Arumugam, T.V.; Zhang, J.H.; Nanda, A.; Granger, D.N. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J. Cereb. Blood Flow Metab. 2004, 24, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Franks, Z.G.; Campbell, R.A.; Weyrich, A.S.; Rondina, M.T. Platelet-leukocyte interactions link inflammatory and thromboembolic events in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 11–17. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Invest. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [PubMed]
- Nishigaya, K.; Yoshida, Y.; Sasuga, M.; Nukui, H.; Ooneda, G. Effect of recirculation on exacerbation of ischemic vascular lesions in rat brain. Stroke 1991, 22, 635–642. [Google Scholar] [CrossRef][Green Version]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef]
- Deppermann, C.; Kraft, P.; Volz, J.; Schuhmann, M.K.; Beck, S.; Wolf, K.; Stegner, D.; Stoll, G.; Nieswandt, B. Platelet secretion is crucial to prevent bleeding in the ischemic brain but not in the inflamed skin or lung in mice. Blood 2017, 129, 1702–1706. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Pozgajova, M.; Pham, M.; Bendszus, M.; Nieswandt, B.; Stoll, G. Targeting platelets in acute experimental stroke: Impact on glycoprotein Ib, VI, and IIb/IIIb blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 2007, 115, 2323–2330. [Google Scholar] [CrossRef]
- Linke, B.; Schreiber, Y.; Picard-Willems, B.; Slattery, P.; Nüsing, R.M.; Harder, S.; Geisslinger, G.; Scholich, K. Activated platelets induce an anti-inflammatory response of monocytes/macrophages through cross-regulation of PGE2 and cytokines. Mediators Inflamm. 2017, 2017, 1463216. [Google Scholar] [CrossRef]
- Tamura, S.; Suzuki, H.; Hirowatari, Y.; Hatase, M.; Nagasawa, A.; Matsuno, K.; Kobayashi, S.; Moriyama, T. Release reaction of brain-derived neurotrophic factor (BDNF) through PAR1 activation and its two distinct pools in human platelets. Thromb. Res. 2011, 128, e55–e66. [Google Scholar] [CrossRef] [PubMed]
- Schäbitz, W.R.; Steigleder, T.; Cooper-Kuhn, C.M.; Schwab, S.; Sommer, C.; Schneider, A.; Kuhn, H.G. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke 2007, 38, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cejudo, J.; Gutiérrez-Fernández, M.; Otero-Ortega, L.; Rodríguez-Frutos, B.; Fuentes, B.; Vallejo-Cremades, M.T.; Navarro Hernanz, T.; Cerdán, S.; Díez-Tejedor, E. Brain-derived neurotrophic factor administration mediated oligodendrocyte differentiation and myelin formation in subcortical ischemic stroke. Stroke 2015, 46, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ramiro, L.; Simats, A.; Berrocoso, T.G.; Montaner, J. Inflammatory molecules might become both biomarkers and therapeutic targets for stroke management. Ther. Adv. Neurol. Disord. 2018, 11, 1–24. [Google Scholar] [CrossRef]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Rayasam, A.; Hsu, M.; Kijak, J.A.; Kissel, L.; Hernandez, G.; Sandor, M.; Fabry, Z. Immune responses in stroke: How the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology 2018, 154, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128–139. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Puleo, M.G.; Velardo, M.C.; Corpora, F.; Daidone, M.; Pinto, A. Molecular biology of atherosclerotic ischemic strokes. Int. J. Mol. Sci. 2020, 21, 9372. [Google Scholar] [CrossRef]
- Ormstad, H.; Aass, H.C.D.; Lund-Sørensen, N.; Amthor, K.-F.; Sandvik, L. Serum levels of cytokines and C-reactive protein in acute ischemic stroke patients, and their relationship to stroke lateralization, type, and infarct volume. J. Neurol. 2012, 258, 677–685. [Google Scholar] [CrossRef]
- Rothwell, N.J. Interleukin-1 and neuronal injury: Mechanisms, modification, and therapeutic potential. Brain Behav. Immun. 2003, 17, 152–157. [Google Scholar] [CrossRef]
- Emsley, H.C.A.; Smith, C.J.; Gavin, C.M.; Georgiou, R.F.; Vail, A.; Barberan, M.E.; Illingworth, K.; Scarth, S.; Wickramasinghe, V.; Hoadley, M.E.; et al. Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 2007, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Matsuura, N.; Shozuhara, H.; Onodera, H.; Itoyama, Y.; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–680. [Google Scholar] [CrossRef]
- Boutin, H.; LeFeuvre, R.A.; Horai, R.; Asano, M.; Iwakura, Y.; Rothwell, N.J. Role of IL-1alpha and IL-1beta in ischemic brain damage. J. Neurosci. 2001, 21, 5528–5534. [Google Scholar] [CrossRef]
- Smith, C.J.; Emsley, H.C.; Udeh, C.; Vail, A.; Hoadley, M.E.; Rothwell, N.J.; Tyrrell, P.J.; Hopkins, S.J. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine 2012, 58, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Hulme, S.; Vail, A.; Heal, C.; Parry-Jones, A.R.; Scarth, S.; Hopkins, K.; Hoadley, M.; Allan, S.M.; Rothwell, N.J.; et al. SCIL-STROKE (Subcutaneous interleukin-1 receptor antagonist in ischemic stroke): A randomized controlled phase 2 trial. Stroke 2018, 49, 1210–1216. [Google Scholar] [CrossRef]
- Pasin, L.; Cavalli, G.; Navalesi, P.; Sella, N.; Landoni, G.; Yavorovskiy, A.G.; Likhvantsev, V.V.; Zangrillo, A.; Dagna, L.; Monti, G. Anakinra for patients with COVID-19: A meta-analysis of non-randomized cohort studies. Eur. J. Intern. Med. 2021, 86, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase III trial. Nature Med. 2021, 27, 1752–1760. [Google Scholar] [CrossRef]
- Liu, T.; Clark, R.K.; McDonnell, P.C.; Young, P.R.; White, R.F.; Barone, F.C.; Feuerstein, G.Z. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke 1994, 25, 1481–1488. [Google Scholar] [CrossRef]
- Stone, M.J.; Hayward, J.A.; Huang, C.; Huma, Z.E.; Sanchez, J. Mechanisms of regulation of the chemokine-receptor network. Int. J. Mol. Sci. 2017, 18, 342. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, J.W.; Wildeboer, D.; Koller, G.; Naus, S.; Rittger, A.; Moss, M.L.; Minai, Y.; Jockusch, H. Tumor necrosis factor-α (TNF-α) regulates shedding of TNF-α receptor 1 by the metalloprotease-disintegrin ADAM8: Evidence for a protease-regulated feedback loop in neuroprotection. J. Neurosci. 2010, 30, 12210–12218. [Google Scholar] [CrossRef]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Takahashi, H.; Machida, T.; Wakigawa, T.; Harada, E.; Miyaji, H.; Koga, M.; Nishioku, T.; et al. Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J. Neuroinflamm. 2011, 8, 106. [Google Scholar] [CrossRef]
- Ginis, I.; Jaiswal, R.; Klimanis, D.; Liu, J.; Greenspon, J.; Hallenbeck, J.M. TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kappaB transactivation: The role of NF-kappaB association with p300 adaptor. J. Cereb. Blood Flow Metab. 2002, 22, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, S.; Zanda, B.; Marchetti, B.; Fois, M.L.; Arru, G.; Pes, G.M.; Salaris, F.S.; Arru, A.; Pirisi, A.; Rosati, G. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur. J. Neurol. 2006, 13, 505–513. [Google Scholar] [CrossRef]
- Bustamante, A.; Sobrino, T.; Giralt, D.; Garcia-Berrocoso, T.; Llombart, V.; Ugarizza, I.; Espadaler, M.; Rodriguez, N.; Sudlow, C.; Castellanos, M.; et al. Prognostic value of blood interleukin-6 in the prediction of functional outcome after stroke: A systematic review and meta-analysis. J. Neuroimmunol. 2014, 15, 215–224. [Google Scholar] [CrossRef]
- Beridze, M.; Sanikidze, T.; Shakarishvilil, R.; Intskirveli, N.; Bornstein, N.M. Selected acute phase CSF factors in ischemic stroke: Findings and prognostic value. BMC Neurol. 2011, 11, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Connel, B.J.; Gordon, J.R.; Saleh, T.M. ELR-CXC chemokine antagonism is neuroprotective in a rat model of ischemic stroke. Neurosci. Lett. 2015, 606, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Villa, P.; Triulzi, S.; Cavalieri, B.; Di Bitondo, R.; Bertini, R.; Barbera, S.; Bigini, P.; Mennini, T.; Gelosa, P.; Tremoli, E.; et al. The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol. Med. 2007, 13, 125–133. [Google Scholar] [CrossRef]
- Pelidou, S.H.; Kostulas, N.; Matusevicius, D.; Kivisäkk, P.; Kostulas, V.; Link, H. High levels of IL-10 secreting cells are present in blood in cerebrovascular diseases. Eur. J. Neurol. 1999, 6, 437–442. [Google Scholar] [CrossRef]
- O’Garra, A.; Vieira, P.L.; Vieira, P.; Goldfeld, A.E. IL-10–producing and naturally occurring CD4+ Tregs: Limiting collateral damage. J. Clin. Investig. 2004, 114, 1372–1378. [Google Scholar] [CrossRef]
- Vitkovic, L.; Maeda, S.; Sternberg, E. Anti-inflammatory cytokines: Expression and action in the brain. Neuroimmunomodulation 2001, 9, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Spera, P.A.; Ellison, J.A.; Feuerstein, G.Z.; Barone, F.C. IL-10 reduces rat brain injury following focal stroke. Neurosci. Lett. 1998, 251, 189–192. [Google Scholar] [CrossRef]
- Ooboshi, H.; Ibayashi, S.; Shichita, T.; Kumai, Y.; Takada, J.; Ago, T.; Arakawa, S.; Sugimori, H.; Kamouchi, M.; Kitazono, T.; et al. Post-ischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 2005, 111, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Vila, N.; Castillo, J.; Dávalos, A.; Chamorro, A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Yushchenko, M.; Mäder, M.; Elitok, E.; Bitsch, A.; Dressel, A.; Tumani, H.; Bogumil, T.; Kitze, B.; Poser, S.; Weber, F. Interferon-beta-1 b decreased matrix metalloproteinase-9 serum levels in primary progressive multiple sclerosis. J. Neurol. 2003, 250, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Frese, A.; Nassenstein, I.; Hoppen, M.; Marziniak, M.; Ringelstein, E.B.; Kim, K.S.; Schäbitz, W.-R.; Kraus, J. Serum from interferon-β-1b treated patients with early multiple sclerosis stabilizes the blood-brain barrier in vitro. Mult. Scler. J. Exp. Transl. Clin. 2012, 18, 236–239. [Google Scholar] [CrossRef]
- Defazio, G.; Livrea, P.; Giorelli, M.; Martino, D.; Roselli, F.; Ricchiuti, F.; Trojano, M. Interferon beta-1a downregulates TNFalpha-induced intercellular adhesion molecule 1 expression on brain microvascular endothelial cells through a tyrosine kinase-dependent pathway. Brain Res. 2000, 881, 227–230. [Google Scholar] [CrossRef]
- Veldhuis, W.B.; Derksen, J.W.; Floris, S.; Van Der Meide, P.H.; De Vries, H.E.; Schepers, J.; Vos, I.M.P.; Dijkstra, C.D.; Kapelle, L.J.; Nicolay, K.; et al. Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J. Cereb. Blood Flow Metab. 2003, 23, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Liberale, L.; Vecchie, A.; Casula, M.; Carbone, F.; Dallegri, F.; Montecucco, F. Update on inflammatory biomarkers and treatments in ischemic stroke. Int. J. Mol. Sci. 2016, 17, 1967. [Google Scholar] [CrossRef]
- Ma, M.; Ma, Y.; Yi, X.; Guo, R.; Zhu, W.; Fan, X.; Xu, G.; Frey, W.H., II; Liu, X. Intranasal delivery of transforming growth factor-beta 1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci. 2008, 9, 117. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Buckwalter, M.S. Astrocytes: Integrative regulators of neuroinflammation in stroke and other neurological diseases. Neurotherapeuthics 2016, 13, 685–701. [Google Scholar] [CrossRef]
- Rietdijk, C.D.; Van Wezel, R.J.A.; Garssen, J.; Kraneveld, A.D. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. Neuroimmunol. Neuroinflamm. 2016, 3, 27. [Google Scholar] [CrossRef]
- Stamatovic, S.; Shakui, P.; Keep, R.F.; Moore, B.B.; Kunkel, S.L.; Van Rooijen, N.; Andjelkovic, A.V. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2005, 25, 593–606. [Google Scholar] [CrossRef]
- Rostène, W.; Dansereau, M.A.; Godefroy, D.; Van Steenwinckel, J.; Reaux-Le Goazigo, A.; Mélik-Parsadaniantz, S.; Apartis, E.; Hunot, S.; Beaudet, N.; Sarret, P. Neurochemokines: A menage a trois providing new insights on the functions of chemokines in the central nervous system. J. Neurochem. 2011, 118, 680–694. [Google Scholar] [CrossRef]
- Weiss, J.M.; Downie, S.A.; Lyman, W.D.; Berman, J.W. Astrocyte-derived monocyte chemoattractant protein-1 directs the transmigration of leukocytes across a model of the human blood-brain barrier. J. Immunol. 1998, 161, 6896–6903. [Google Scholar]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J. Cereb. Blood Flow Metab. 2006, 26, 797–810. [Google Scholar] [CrossRef]
- Dénes, Á.; Ferenczi, S.; Halász, J.; Környei, Z.; Kovács, K.J. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, R.; Villa, P.; Chece, G.; Lauro, C.; Paladini, A.; Micotti, E.; Perego, C.; De Simoni, M.-G.; Fredholm, B.B.; Eusebi, F.; et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 2011, 31, 16327–16335. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.; Li, Y.; Yang, G.Y. Roles of chemokine CXCL 12 and its receptors in ischemic stroke. Curr. Drug Targets. 2012, 13, 166–172. [Google Scholar] [CrossRef]
- Terao, S.; Yilmaz, G.; Stokes, K.Y.; Russel, J.; Ishikawa, M.; Kawase, T.; Granger, D.N. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke 2008, 39, 2560–2570. [Google Scholar] [CrossRef]
- Ruscher, K.; Kuric, E.; Liu, Y.; Walter, H.L.; Issazadeh-Navikas, S.; Englund, E.; Wieloch, T. Inhibition of CXCL 12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.D.; Hess, D.C.; Martin-Studdard, A.; Carothers, J.J.; Zheng, J.; Hale, D.; Maeda, M.; Fagan, S.C.; Carroll, J.E.; Conway, S.J. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: Association with bone marrow cell homing to injury. J. Neuropathol. Experiment. Neurol. 2004, 63, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.C.; Lin, S.Z.; Yen, P.S.; Su, C.Y.; Chen, D.C.; Wang, H.J.; Li, H. Stromal cell-derived factor-1 alpha promotes neuroprotection, angiogenesis, and mobilization/homing of bone marrow-derived cells in stroke rats. J. Pharmacol. Exp. Ther. 2008, 324, 834–849. [Google Scholar] [CrossRef]
- Yang, Y.; Rosenberg, G.A. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015, 1623, 30–38. [Google Scholar] [CrossRef]
- Morancho, A.; Rosell, A.; Garcia-Bonilla, L.; Montaner, J. Metalloproteinase and stroke infarct size: Role for anti-inflammatory treatment? Ann. N. Y. Acad. Sci. 2010, 207, 123–133. [Google Scholar] [CrossRef]
- Candelario-Jalil, E. Injury and repair mechanisms in ischemic stroke: Considerations for the development of novel neurotherapeutics. Curr. Opin. Investig. Drugs 2009, 10, 644–654. [Google Scholar] [PubMed]
- Rosenberg, G.A.; Yang, Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus. 2007, 22, E4. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, L.A.; Wetzel, M.; Rosenberg, G.A. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005, 50, 329–339. [Google Scholar] [CrossRef]
- Ma, F.; Rodriguez, S.; Buxo, X.; Morancho, A.; Riba-Llena, I.; Carrera, A.; Bustamante, A.; Giralt, D.; Montaner, J.; Martinez, C.; et al. Plasma matrix metalloproteinases in patients during intensive rehabilitation therapy. Arch. Phys. Med. Rehabil. 2016, 97, 1832–1840. [Google Scholar] [CrossRef]
- Zhao, B.-Q.; Wang, S.; Kim, H.-Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nature Med. 2006, 12, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell. Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Shi, L.; Rocha, M.; Leak, R.K.; Zhao, J.; Bhatia, T.N.; Mu, H.; Wei, Z.; Yu, F.; Weiner, S.L.; Ma, F.; et al. A new era for stroke therapy: Integrating neurovascular protection with optimal reperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 2073–2091. [Google Scholar] [CrossRef] [PubMed]
- Shichita, T.; Ito, M.; Yoshimura, A. Post-ischemic inflammation regulates neural damage and protection. Front. Cell. Neurosci. 2014, 8, 319. [Google Scholar] [CrossRef]
- Rajkovic, O.; Potjewyd, G.; Pinteaux, E. Regenerative medicine therapies for targeting neuroinflammation after stroke. Front. Neurol. 2018, 9, 734. [Google Scholar] [CrossRef]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.A.; Amruta, N.; Pinteaux, E.; Bix, G.J. Neurogenesis after stroke: A therapeutic perspective. Transl. Stroke Res. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Zhang, R.L.; Zhang, Z.G.; Zhang, L.; Chopp, M. Proliferation and differentiation of progenitor cells in the cortex and subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience 2001, 105, 33–41. [Google Scholar] [CrossRef]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.P.; Sailor, K.A.; Vemuganti, R.; Dempsey, R.J. Insulin-like growth factor-1 is an endogenous mediator of focal ischemia-induced neural progenitor proliferation. Eur. J. Neurosci. 2006, 24, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zacharek, A.; Zhang, C.; Jiang, H.; Li, Y.; Roberts, C.; Lu, M.; Kapke, A.; Chopp, M. Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J. Neurosci. 2005, 25, 2366–2375. [Google Scholar] [CrossRef]
- Jin, K.; Zhu, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Greenberg, D.A. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [Google Scholar] [CrossRef]
- Yan, Y.P.; Sailor, K.A.; Lang, B.T.; Park, S.W.; Vemuganti, R.; Dempsey, R.J. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2007, 27, 1213–1224. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.G.; Zhang, R.L.; Gregg, S.R.; Hozeska-Solgot, A.; LeTourneau, Y.; Wang, Y.; Chopp, M. Matrix metalloproteinase 2 (MMP2) and MMP9 secreted by erythropoietin-activated endothelial cells promote neural progenitor cell migration. J. Neurosci. 2006, 26, 5996–6003. [Google Scholar] [CrossRef] [PubMed]
- Madelaine, R.; Sloan, S.A.; Huber, N.; Notwell, J.H.; Leung, L.C.; Skariah, G.; Halluin, C.; Pasca, S.P.; Bejerano, G.; Krasnow, M.A.; et al. MicroRNA-9 couples brain neurogenesis and angiogenesis. Cell Rep. 2017, 20, 1533–1542. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Wang, R.-L.; Zhao, H.-P.; Tao, Z.; Li, J.-C.; Ju, F.; Han, Z.-P.; Ma, Q.-F.; Liu, P.; Ma, S.-B.; et al. MEPO promotes neurogenesis and angiogenesis but suppresses gliogenesis in mice with acute ischemic stroke. Eur. J. Pharamacol. 2019, 849, 1–10. [Google Scholar] [CrossRef]
- Hayashi, T.; Noshita, N.; Sugawara, T.; Chan, P.H. temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 166–180. [Google Scholar] [CrossRef]
- Islam, O.; Gong, X.; Rose-John, S.; Heese, K. Interleukin-6 and neural stem cells: More than gliogenesis. Mol. Biol. Cell 2009, 20, 188–199. [Google Scholar] [CrossRef]
- Vallières, L.; Campbell, I.L.; Gage, F.H.; Sawchenko, P.E. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J. Neurosci. 2002, 22, 486–492. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, D.; Qi, H.; Yuan, Y.; Liu, H.; Yao, S.; Yuan, S.; Zhang, J. Enriched environment promotes post-stroke neurogenesis through NF-κB-mediated secretion of IL-17A from astrocytes. Brain Res. 2018, 1687, 20–31. [Google Scholar] [CrossRef]
- Aarum, J.; Sandberg, K.; Haeberlein, S.L.; Persson, M.A. Migration and differentiation of neural precursor cells can be directed by microglia. Proc. Natl. Acad. Sci. USA 2003, 100, 15983–15988. [Google Scholar] [CrossRef]
- Guan, J.; Williams, C.; Gunning, M.; Mallard, C.; Gluckman, P. The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats. J. Cereb. Blood Flow Metab. 1993, 13, 609–616. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug delivery to the brain. J. Cereb. Blood Flow Metab. 1997, 17, 713–731. [Google Scholar] [CrossRef]
- Guan, J. Insulin-like growth factor -1 (IGF-1) derived neuropeptides, a novel strategy for the development of pharmaceuticals for managing ischemic brain injury. CNS Neurosci. Ther. 2011, 17, 250–255. [Google Scholar] [CrossRef]
- Guan, J.; Thomas, G.B.; Lin, H.; Mathai, S.; Bachelor, D.C.; Gluckman, P.D. Neuroprotective effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate (GPE) following intravenous infusion in hypoxic-ischemic adult rats. Neuropharmacology 2004, 47, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Curatolo, L.; Williams, C.E.; Gatti, S.; Benatti, L.; Peeters, C.; Guan, J.; Dragunow, M.; Post, C.; Faull, R.L.; et al. Neuroprotective effects of Gly-Pro-Glu, the N-terminal tripeptide of IGF-1, in the hippocampus in vitro. Neuroreport 1999, 10, 161–164. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Gaidin, S.G.; Turovskaya, M.V.; Gavrish, M.S.; Babaev, A.A.; Maltseva, V.N.; Blinova, E.V.; Turovsky, E.A. The selective BDNF overexpression in neurons protects neuroglial networks against OGD and glutamate-induced excitotoxicity. Int. J. Neurosci. 2020, 130, 363–383. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; O’Connor, M.; Wang, G.; Han, F. Brain-Derived Neurotrophic Factor and its potential therapeutic role in stroke comorbidities. Neural Plast. 2020, 2020, 1969482. [Google Scholar] [CrossRef]
- Mattson, M.P. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N. Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar] [CrossRef]
- Li, Y.; Jia, Y.-C.; Cui, K.; Li, N.; Zheng, Z.-Y.; Wang, Y.-Z.; Yuan, X.-B. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature 2005, 434, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Labelle, C.; Leclerc, N. Exogenous BDNF, NT-3 and NT-4 differentially regulate neurite outgrowth in cultured hippocampal neurons. Brain Res. Dev. Brain Res. 2000, 123, 1–11. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tanaka, R.; Liu, M.; Hattori, N.; Urabe, T. Cilostazol attenuates ischemic brain injury and enhances neurogenesis in the subventricular zone of adult mice after transient focal cerebral ischemia. Neuroscience 2010, 17, 1367–1376. [Google Scholar] [CrossRef]
- Chen, S.J.; Tsai, J.C.; Lin, C.Y.; Chang, C.K.; Tseng, T.H.; Chien, C.L. Brain-derived neurotrophic factor-transfected and nontransfected 3T3 fibroblasts enhance migratory neuroblasts and functional restoration in mice with intracerebral hemorrhage. J. Neuropathol. Exp. Neurol. 2012, 71, 1123–1136. [Google Scholar] [CrossRef]
- Kurozumi, K.; Nakamura, K.; Tamiya, T.; Kawano, Y.; Kobune, M.; Hirai, S.; Uchida, H.; Sasaki, K.; Ito, Y.; Kato, K.; et al. BDNF gene-modified mesenchymal stem cells promote functional recovery and reduce infarct size in the rat middle cerebral artery occlusion model. Mol. Ther. 2004, 9, 189–197. [Google Scholar] [CrossRef]
- Alcantara, C.C.; Garcia-Salazar, L.F.; Silva-Couto, M.A.; Santos, G.L.; Reisman, D.S.; Russo, T.L. Post-stroke BDNF concentration changes following physical exercise: A systematic review. Front. Neurol. 2018, 9, 637. [Google Scholar] [CrossRef]
- Liao, G.Y.; Kinney, C.E.; An, J.J.; Xu, B. TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc. Natl. Acad. Sci. USA 2019, 116, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pardridge, W.M. Neuroprotection in transient focal brain ischemia after delayed intravenous administration of brain-derived neurotrophic factor conjugated to a blood-brain barrier drug targeting system. Stroke 2001, 32, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.M.; Ritzel, R.; Mancini, N.S.; Jian, Y.; Yi, X.; Manickam, D.S.; Banks, W.A.; Kabanov, A.V.; McCullough, L.D.; Verma, R. Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacol. Biochem. Behav. 2016, 150–151, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Wang, Y.; Zhan, L.; Zhou, R. Targeted delivery of proteins into the central nervous system mediated by rabies virus glycoprotein-derived peptide. Pharm. Res. 2012, 29, 1562–1569. [Google Scholar] [CrossRef]
- Gottlieb, M.; Matute, C. Expression of nerve growth factor in astrocytes of the hippocampal CA1 area following transient forebrain ischemia. Neuroscience 1999, 91, 1027–1034. [Google Scholar] [CrossRef]
- Kordower, J.H.; Winn, S.R.; Liu, Y.-T.; Mufson, E.J.; Sladek, J.R.; Hammang, J.P.; Baetge, E.E.; Emerich, D.F. The aged monkey basal forebrain: Rescue and sprouting of axotomized basal forebrain neurons after grafts of encapsulated cells secreting human nerve growth factor. Proc. Natl. Acad. Sci. USA 1994, 91, 10898–10902. [Google Scholar] [CrossRef]
- Petty, B.G.; Cornblath, D.R.; Adornato, B.T.; Chaudhry, V.; Flexner, C.; Wachsman, M.; Sinicropi, D.; Burton, L.E.; Peroutka, S.J. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann. Neurol. 1994, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, A.; Kokaia, Z.; Airaksinen, M.S.; Saarma, M.; Lindvall, O. Stroke induces widespread changes of gene expression for glial cell line-derived neurotrophic factor family receptors in the adult rat brain. Neuroscience 2001, 106, 27–41. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, S.Z.; Chiou, A.L.; Williams, L.R.; Hoffer, B.J. Glial cell line-derived neurotrophic factor protects against ischemia-induced injury in the cerebral cortex. J. Neurosci. 1997, 17, 4341–4348. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, A.; Kirik, D.; Lundberg, C.; Mandel, R.J.; Andsberg, G.; Kokaia, Z.; Lindvall, O. Elevated GDNF levels following viral vector-mediated gene transfer can increase neuronal death after stroke in rats. Neurobiol Dis. 2003, 14, 542–556. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ahlenius, H.; Thored, P.; Kobayashi, R.; Kokaia, Z.; Linvall, O. Intracerebral infusion of glial cell line-derived neurotrophic factor promotes striatal neurogenesis after stroke in adult rats. Stroke 2006, 37, 2361–2367. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic potential in microvascular disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef]
- Luo, Z.Z.; Gao, Y.; Sun, N.; Zhao, Y.; Wang, J.; Tian, B.; Shi, J. Enhancing the interaction between annexin-1 and formil peptide receptors regulates microglial activation to protect neurons from ischemia-like injury. J. Neuroimmunol. 2014, 276, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gao, W.; Li, L.; Hao, J.; Yang, B.; Wang, T.; Li, L.; Bai, X.; Li, F.; Ren, H.; et al. Annexin A1 protects against cerebral ischemia-reperfusion injury by modulating microglia/macrophage polarization via FPR2/ALX-dependent AMPK-mTOR pathway. J. Neuroinflamm. 2021, 18, 119. [Google Scholar] [CrossRef]
- Zhang, W.-F.; Jin, Y.-C.; Li, X.-M.; Yang, Z.; Wang, D.; Cui, J.-J. Protective effects of leptin against cerebral ischemia/reperfusion injury (review). Exp. Ther. Med. 2019, 17, 3282–3290. [Google Scholar] [CrossRef]
- Geng, H.X.; Li, R.P.; Li, Y.G.; Wang, X.Q.; Zhang, L.; Deng, J.B.; Wang, L.; Deng, J.X. 14,15-EET suppresses neuronal apoptosis in ischemia-reperfusion through the mitochondrial pathway. Neurochem. Res. 2017, 42, 2841–2849. [Google Scholar] [CrossRef]
- Amantea, D.; Tassorelli, C.; Russo, R.; Petrelli, F.; Morrone, L.A.; Bagetta, G.; Corasaniti, M.T. Neuroprotection by leptin in a rat model of permanent cerebral ischemia: Effects on STAT3 phosphorylation in discrete cells of the brain. Cell Death Dis. 2011, 2, e238. [Google Scholar] [CrossRef]
- Deng, Z.H., Jr.; Yan, G.T.; Wang, L.H.; Zhang, J.Y.; Xue, H.; Zhang, K. Leptin relieves intestinal ischemia/reperfusion injury by promoting ERK1/2 phosphorylation and the NO signaling pathway. J. Trauma Acute Care Surg. 2012, 72, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- Zhang, J.; Deng, Z.; Liao, J.; Song, C.; Liang, C.; Xue, H.; Wang, L.; Zhang, K.; Yan, G. Leptin attenuates cerebral ischemia injury through the promotion of energy metabolism via the PI3K/Akt pathway. J. Cereb. Blood Flow Metab. 2013, 33, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Cronin, C.G.; Hudobenko, J.; Venna, V.R.; McCullough, L.D.; Liang, B.T. Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain Behav. Immun. 2017, 66, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Plane, J.M.; Shen, Y.; Pleasure, D.E.; Deng, W. Prospects for minocycline neuroprotection. Arch. Neurol. 2010, 67, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, N.; Shimazawa, M.; Yamashima, T.; Nagai, H.; Hara, H. Minocycline inhibits oxidative stress and decreases in vitro and in vivo ischemic neuronal damage. Brain Res. 2005, 1044, 8–15. [Google Scholar] [CrossRef]
- Matsukawa, N.; Yasuhara, T.; Hara, K.; Xu, L.; Maki, M.; Yu, G.; Kaneko, Y.; Ojika, K.; Hess, D.C.; Borlongan, C.V. Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC Neurosci. 2009, 10, 126. [Google Scholar] [CrossRef]
- Machado, L.S.; Sazonova, I.; Kozak, A.; Wiley, D.C.; El-Remessy, A.B.; Ergul, A.; Hess, D.C.; Waller, J.L.; Fagan, S.C. Minocycline and tissue plasminogen activator for stroke: Assessment of interaction potential. Stroke 2009, 40, 3028–3033. [Google Scholar] [CrossRef]
- Lampl, Y.; Boaz, M.; Gilad, R.; Loberboym, M.; Dabby, A.; Rapoport, A.; Anca-Hershkowitz, M.; Sadeh, M. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology 2007, 69, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Padma Srivastava, M.V.; Bhasin, A.; Bhatia, R.; Garg, A.; Gaikwad, S.; Prasad, K.; Singh, M.B.; Tripathi, M. Efficacy of minocycline in acute ischemic stroke: A single-blinded, placebo-controlled trial. Neurol. India 2012, 60, 23–28. [Google Scholar] [CrossRef]
- Kohler, E.; Prentice, D.A.; Bates, T.R.; Hankey, G.J.; Claxton, A.; Van Heerden, J.; Blacker, D. Intravenous minocycline in acute stroke: A randomized, controlled pilot study and meta-analysis. Stroke 2013, 44, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Ashayeri Ahmadabad, R.; Mirzaasgari, Z.; Gorji, A.; Khaleghi Ghadiri, M. Toll-like receptor signaling pathways: Novel therapeutic targets for cerebrovascular disorders. Int. J. Mol. Sci. 2021, 22, 6153. [Google Scholar] [CrossRef]
- Abdul, Y.; Abdelsaid, M.; Li, W.; Webb, R.C.; Sullivan, J.C.; Dong, G.; Ergul, A. Inhibition of Toll-like receptor-4 (TLR-4) improves neurobehavioral outcomes after acute ischemic stroke in diabetic rats: Possible role of vascular endothelial TLR-4. Mol. Neurobiol. 2019, 56, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Homepage on the Internet. Available online: www.clinicaltrials.gov (accessed on 12 November 2021).
- Jackson, L.; Dong, G.; Althomali, W.; Sayed, M.A.; Eldahshan, W.; Baban, B.; Johnson, M.H.; Filosa, J.; Fagan, S.C.; Ergul, A. Delayed administration of angiotensin II type 2 receptor (AT2R) agonist compound 21 prevents the development of post-stroke cognitive impairment in diabetes through the modulation of microglia polarization. Transl. Stroke Res. 2019, 11, 762–775. [Google Scholar] [CrossRef]
- Jackson-Cowan, L.; Eldahshan, W.; Dumanli, S.; Dong, G.; Jamil, S.; Abdul, Y.; Althomali, W.; Baban, B.; Fagan, S.C.; Ergul, A. Delayed administration of angiotensin receptor (AT2R) agonist C21 improves survival and preserves sensorimotor outcomes in female diabetic rats post-stroke through modulation of microglial activation. Int. J. Mol. Sci. 2021, 22, 1356. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, J.; Zhao, S.; Zhang, H.; Cai, W.; Cai, M.; Ji, X.; Leak, R.K.; Gao, Y.; Chen, J.; et al. Interleukin-4 is essential for microglial/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke 2016, 47, 498–504. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.; Zhang, H.; Ye, Q.; Wang, J.; Yang, B.; Mao, L.; Zhu, W.; Leak, R.K.; Xiao, B.; et al. ST2/IL-33-dependent microglial response limits acute ischemic brain injury. J. Neurosci. 2017, 37, 4692–4704. [Google Scholar] [CrossRef]
- Clark, W.M.; Madden, K.P.; Rothlein, R.; Zivin, J.A. Reduction of central nervous system ischemic injury by monoclonal antibody to intercellular adhesion molecule. J. Neurosurg. 1991, 75, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Bowes, M.P.; Rothlein, R.; Fagan, S.C.; Zivin, J.A. Monoclonal antibodies preventing leukocyte activation reduce experimental neurologic injury and enhance efficacy of thrombolytic therapy. Neurology 1995, 45, 815–819. [Google Scholar] [CrossRef]
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for ischemic stroke: Two decades of success and failure. NeuroRx 2004, 1, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Enlimomab Acute Stroke Trial Investigators. Use of anti-ICAM-1 therapy in ischemic stroke: Results of the Enlimomab Acute Stroke Trial. Neurology 2001, 57, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Bednar, M.M.; Gross, C.E.; Russell, S.R.; Fuller, S.P.; Ellenberger, C.L.; Schindler, E.; Klingbeil, C.; Vexler, V. Humanized anti-L-selectin monoclonal antibody DREG200 therapy in acute thromboembolic stroke. Neurol. Res. 1998, 20, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Prestigiacomo, C.J.; Kim, S.C.; Connolly, E.S.; Liao, H.; Yan, S.F.; Pinsky, D.J. CD 18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke 1999, 30, 1110–1117. [Google Scholar] [CrossRef]
- Garcia, J.H.; Liu, K.F.; Bree, M.P. Effects of CD11b/18 monoclonal antibody on rats with permanent middle cerebral artery occlusion. Am. J. Pathol. 1996, 148, 241–248. [Google Scholar]
- Krams, M.; Lees, K.R.; Hacke, W.; Grieve, A.P.; Orgogozo, J.-M.; Ford, G.A.; ASTIN Study Investigators. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): An adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke 2003, 34, 2543–2548. [Google Scholar] [CrossRef]
- Langhauser, F.; Kraft, P.; Göb, E.; Leinweber, J.; Schuhmann, M.K.; Lorenz, K.; Gelderblom, M.; Bittner, S.; Meuth, S.G.; Wiendl, H.; et al. Blocking of α4 integrin does not protect from acute ischemic stroke in mice. Stroke 2014, 45, 1799–1806. [Google Scholar] [CrossRef]
- Becker, K.; Kindrick, D.; Relton, J.; Harlan, J.; Winn, R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke 2001, 32, 206–211. [Google Scholar] [CrossRef]
- Relton, J.K.; Sloan, K.E.; Frew, E.M.; Whalley, E.T.; Adams, S.P.; Lobb, R.R. Inhibition of alpha4 integrin protects against transient focal cerebral ischemia in normotensive and hypertensive rats. Stroke 2001, 32, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Llovera, G.; Hofmann, K.; Roth, S.; Salas-Pérdomo, A.; Ferrer-Ferrer, M.; Perego, C.; Zanier, E.R.; Mamrak, U.; Rex, A.; Party, H.; et al. Results of a preclinical randomized controlled multicenter trial (pRCT): Anti-CD49d treatment for acute brain ischemia. Sci. Transl. Med. 2015, 7, 299ra121. [Google Scholar] [CrossRef]
- Elkins, J.; Veltkamp, R.; Montaner, J.; Johnston, S.C.; Singhal, A.B.; Becker, K.; Lansberg, M.G.; Tang, W.; Chang, I.; Muralidharan, K.; et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (ACTION): A randomised, placebo–controlled, double–blind phase 2 trial. Lancet Neurol. 2017, 16, 217–226. [Google Scholar] [CrossRef]
- Elkind, M.S.V.; Veltkamp, R.; Montaner, J.; Johnston, S.C.; Singhal, A.B.; Becker, K.; Lansberg, M.G.; Tang, W.; Kasliwal, R.; Elkins, J. Natalizumab in acute ischemic stroke (ACTION II): A randomized, placebo–controlled trial. Neurology 2020, 95, e1091–e1104. [Google Scholar] [CrossRef]
- Liesz, A.; Zhou, W.; Mracskó, E.; Karcher, S.; Bauer, H.; Schwarting, S.; Sun, L.; Bruder, D.; Stegeman, S.; Cerwenka, A.; et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain J. Neurol. 2011, 134, 704–720. [Google Scholar] [CrossRef]
- Kieseier, B.C. The mechanism of action of interferon-β in relapsing multiple sclerosis. CNS Drugs 2011, 25, 491–502. [Google Scholar] [CrossRef]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef]
- Liesz, A.; Zhou, W.; Na, S.-Y.; Hämmerling, G.J.; Garbi, N.; Karcher, S.; Mracsko, E.; Backs, J.; Rivest, S.; Veltkamp, R. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J. Neurosci. 2013, 33, 17350–17362. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Sun, F.; Wang, J.; Mao, X.; Xie, L.; Yang, S.-H.; Su, D.-M.; Simpkins, J.W.; Greenberg, D.A.; Jin, K. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J. Immunol. 2014, 192, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Stubbe, T.; Ebner, F.; Richter, D.; Engel, O.; Klehmet, J.; Royl, G.; Meisel, A.; Nitsch, R.; Meisel, C.; Brandt, C. Regulatory T cells accumulate and proliferate in the ischemic hemispheres for up to 30 days after MCAO. J. Cereb. Blood Flow Metab. 2013, 33, 37–47. [Google Scholar] [CrossRef]
- Na, S.-Y.; Mracsko, E.; Liesz, A.; Hünig, T.; Veltkamp, R. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke 2015, 46, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, M.K.; Kraft, P.; Stoll, G.; Lorenz, K.; Meuth, S.G.; Windl, H.; Niewandt, B.; Sparwasser, T.; Beyersdorf, N.; Kerkau, T.; et al. CD28 superagonist-mediated boost of regulatory T cells increases thrombo-inflammation and ischemic neurodegeneration during the acute phase of experimental stroke. J. Cereb. Blood Flow Metab. 2015, 35, 6–10. [Google Scholar] [CrossRef]
- Zhu, Z.; Fu, Y.; Tian, D.; Sun, N.; Han, W.; Chang, G.; Dong, Y.; Xu, X.; Liu, Q.; Huang, D.; et al. Combination of the immune modulator Fingolimod with alteplase in acute ischemic stroke: A pilot trial. Circulation 2015, 132, 1104–1112. [Google Scholar] [CrossRef]
- Kobayashi, S.; Fukuma, S.; Ikenoue, T.; Fukuhara, S.; Kobayashi, S.; on behalf of the Japan Stroke databank. Effect of edaravone on neurological symptoms in real-world patients with acute ischemic stroke. Japan Stroke Databank. Stroke 2019, 50, 1805–1811. [Google Scholar] [CrossRef]
- Cho, H.; Shukla, S. Role of edaravone as a treatment option for patients with amyotrophic lateral sclerosis. Pharmaceuticals 2020, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Licata, G.; Tuttolomondo, A.; Corrao, S.; Di Raimondi, D.; Fernandez, P.; Caruso, C.; Avellone, G.; Pinto, A. Immunoinflammatory activation during the acute phase of lacunar and non-lacunar ischemic stroke: Association with time of onset and diabetic state. Int. J. Immunopathol. Pharmacol. 2006, 19, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Maida, C.; Arnao, V.; Della Corte, V.; Simonetta, I.; Corpora, F.; Di Bona, D.; Maugeri, R.; et al. Early high-dosage atorvastatin treatment improved serum immune-inflammatory markers and functional outcome in acute ischemic strokes classified as large artery atherosclerotic stroke: A randomized trial. Medicine 2016, 95, e3186. [Google Scholar] [CrossRef]
- Choi, J.C.; Lee, J.S.; Park, T.H.; Cho, Y.-J.; Park, J.-M.; Kang, K.; Lee, K.B.; Lee, S.-J.; Ko, Y.; Lee, J.; et al. Effect of pre-stroke statin use on stroke severity and early functional recovery: A retrospective cohort study. BMC Neurol. 2015, 15, 120. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Cognition, statins, and cholesterol in elderly ischemic stroke patients: A neurologist’s perspective. Medician 2021, 57, 616. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L. Anti-inflammatory effects of vinpocetine in atherosclerosis and ischemic stroke: A review of the literature. Molecules 2014, 20, 335–347. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Yan, C. An update on Vinpocetine: New discoveries and clinical implications. Eur. J. Pharmacol. 2018, 819, 30–34. [Google Scholar] [CrossRef]
- Spaccapelo, L.; Bitto, A.; Galantucci, M.; Ottani, A.; Irrera, N.; Minutoli, L.; Altavilla, D.; Novellino, E.; Grieco, P.; Zaffe, D.; et al. Melanocortin MC(4) receptor agonists counteract late inflammatory and apoptotic responses and improve neuronal functionality after cerebral ischemia. Eur. J. Pharmacol. 2011, 670, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Zendedel, A.; Habib, P.; Dang, J.; Lammerding, L.; Hoffmann, S.; Beyer, C.; Slowik, A. Omega-3 polyunsaturated fatty acids ameliorate neuroinflammation and mitigate ischemic stroke damage through interactions with astrocytes and microglia. J. Neuroimmunol. 2015, 278, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, Y.; DeMars, K.M.; Rosenberg, G.A.; Candelario-Jalil, E. Genetic deletion or pharmacological inhibition of cyclooxygenase-2 reduces blood-brain barrier damage in experimental ischemic stroke. Front. Neurol. 2020, 11, 887. [Google Scholar] [CrossRef]
- Sasaki, T.; Kitagawa, K.; Sugiura, S.; Omura-Matsuoka, E.; Tanaka, S.; Yagita, Y.; Okano, H.; Matsumoto, M.; Hori, M. Implication of cyclooxygenase-2 on enhanced proliferation of neural progenitor cells in the adult mouse hippocampus after ischemia. J. Neurosci. Res. 2003, 72, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Horváth-Puhó, E.; Christiansen, C.F.; Petersen, K.L.; Bøtker, H.E.; Sørensen, H.T. Preadmission use of nonaspirin nonsteroidal anti-inflammatory drugs and 30-day stroke mortality. Neurology 2014, 83, 2013–2022. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Wang, Y.-Y.; Chang, C.-Y.; Wu, C.-C.; Chen, W.-Y.; Liao, S.-L.; Chen, C.-J. TNF-α receptor inhibitor alleviates metabolic and inflammatory changes in a rat model of ischemic stroke. Antioxidants 2021, 10, 851. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Liu, X.; Feng, Z.; Du, L.; Huang, Y.; Ge, J.; Deng, Y.; Mei, Z. The potential role of microRNA-124 in cerebral ischemia injury. Int. J. Mol. Sci. 2020, 21, 120. [Google Scholar] [CrossRef] [PubMed]
- Volny, O.; Kasickova, L.; Coufalova, D.; Cimflova, P.; Novak, J. MicroRNAs in cerebrovascular disease. Adv. Exp. Med. Biol. 2015, 888, 155–195. [Google Scholar]
- Hamzei, T.S.; Kho, W.; Aswendt, M.; Collmann, F.M.; Green, C.; Adamczak, J.; Tennstaedt, A.; Hoehn, M. Dynamic modulation of microglia/macrophage polarization by miR-124 after focal cerebral ischemia. J. Neuroimmune Pharmacol. 2016, 11, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Talhada, D.; Rai, A.; Ferreira, R.; Ferreira, L.; Bernardino, L.; Ruscher, K. MicroRNA-124-loaded nanoparticles increase survival and neuronal differentiation of neural stem cells in vitro but do not contribute to stroke outcome in vivo. PLoS ONE 2018, 13, e0193609. [Google Scholar] [CrossRef]
- Liu, X.S.; Chopp, M.; Zhang, R.L.; Tao, T.; Wang, X.L.; Kassis, H.; Hozeska-Solgot, A.; Zhang, L.; Chen, C.; Zhang, Z.G. MicroRNA profiling in subventricular zone after stroke: MiR-124a regulates proliferation of neural progenitor cells through Notch signaling pathway. PLoS ONE 2011, 6, e23461. [Google Scholar] [CrossRef]
- Orset, C.; Haelewyn, B.; Allan, S.M.; Ansar, S.; Campos, F.; Cho, T.H.; Durand, A.; El Amki, M.; Fatar, M.; Garcia-Yébenes, I.; et al. Efficacy of alteplase in a mouse model of acute ischemic stroke: A retrospective pooled analysis. Stroke 2016, 47, 1312–1318. [Google Scholar] [CrossRef]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Janowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamm. 2019, 16, 178. [Google Scholar] [CrossRef]
- Zhang, L.; Yi, L.; Chopp, M.; Kramer, B.C.; Romanko, M.; Gosiewska, A.; Hong, K. Intravenous administration of human umbilical tissue-derived cells improves neurological function in aged rats after embolic stroke. Cell Transplant. 2013, 22, 1569–1576. [Google Scholar] [CrossRef]
- Guzman, R.; Janowski, M.; Walczak, P. Intra-arterial delivery of cell therapies for stroke. Stroke 2018, 49, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-R.; Duan, W.-M.; Reyes, M.; Keene, C.D.; Verfaillie, C.M.; Low, W.C. Human bone marrow stem cells exhibit neural phenotypes and ameliorate neurological deficits after grafting into the ischemic brain of rats. Exp. Neurol. 2002, 174, 11–20. [Google Scholar] [CrossRef]
- Yoo, S.-W.; Chang, D.-Y.; Lee, H.-S.; Kim, G.-H.; Park, J.-S.; Ryu, B.-Y.; Joe, E.-H.; Lee, Y.-D.; Kim, S.-S.; Suh-Kim, H. Immune following suppression mesenchymal stem cell transplantation in the ischemic brain is mediated by TGF-β. Neurobiol. Dis. 2013, 58, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Karlupia, N.; Manley, N.C.; Prasad, K.; Schafer, R.; Steinberg, G.K. Intraarterial transplantation of human umbilical cord mononuclear cells is more efficacious and safer compared with umbilical cord mesenchymal stromal cells in a rodent stroke model. Stem Cell Res. Ther. 2014, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chopp, M. Exosome therapy for stroke. Stroke 2018, 49, 1083–1090. [Google Scholar] [CrossRef]
- Liu, F.; McCullogh, L.D. Middle cerebral artery occlusion model in rodents: Methods and potential pitfalls. J. Biomed. Biotechnol. 2011, 2011, 464701. [Google Scholar] [CrossRef] [PubMed]
- Hug, A.; Dalpke, A.; Wieczorek, N.; Giese, T.; Lorenz, A.; Auffarth, G.; Liesz, A.; Veltkamp, R. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke 2009, 40, 3226–3232. [Google Scholar] [CrossRef] [PubMed]

| Time Course | Pathophysiological Mechanisms |
|---|---|
| Acute phase (minutes–hours) | Reduced cerebral blood flow with diminished oxygen and glucose delivery Anaerobic metabolism and lactic acidosis Failure of ATPase, cellular depolarization, increased intracellular ion influx Release of neuromediators (excitotoxicity) Increased expression of stress signaling genes |
| Subacute phase (hours–days) | Increased production of ROS Apoptosis Expression of adhesion molecules Microglial activation and leukocyte infiltration of the brain Release of pro-inflammatory mediators Increased activity of proteolytic enzymes and damage of BBB and endothelium |
| Chronic phase (days–weeks) | Release of trophic factors (BDNF, IGF, GDNF) Neurogenesis, angiogenesis, synaptogenesis Activation of stem cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 14. https://doi.org/10.3390/ijms23010014
Jurcau A, Simion A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. International Journal of Molecular Sciences. 2022; 23(1):14. https://doi.org/10.3390/ijms23010014
Chicago/Turabian StyleJurcau, Anamaria, and Aurel Simion. 2022. "Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies" International Journal of Molecular Sciences 23, no. 1: 14. https://doi.org/10.3390/ijms23010014
APA StyleJurcau, A., & Simion, A. (2022). Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. International Journal of Molecular Sciences, 23(1), 14. https://doi.org/10.3390/ijms23010014