
Glutathione in the Brain

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
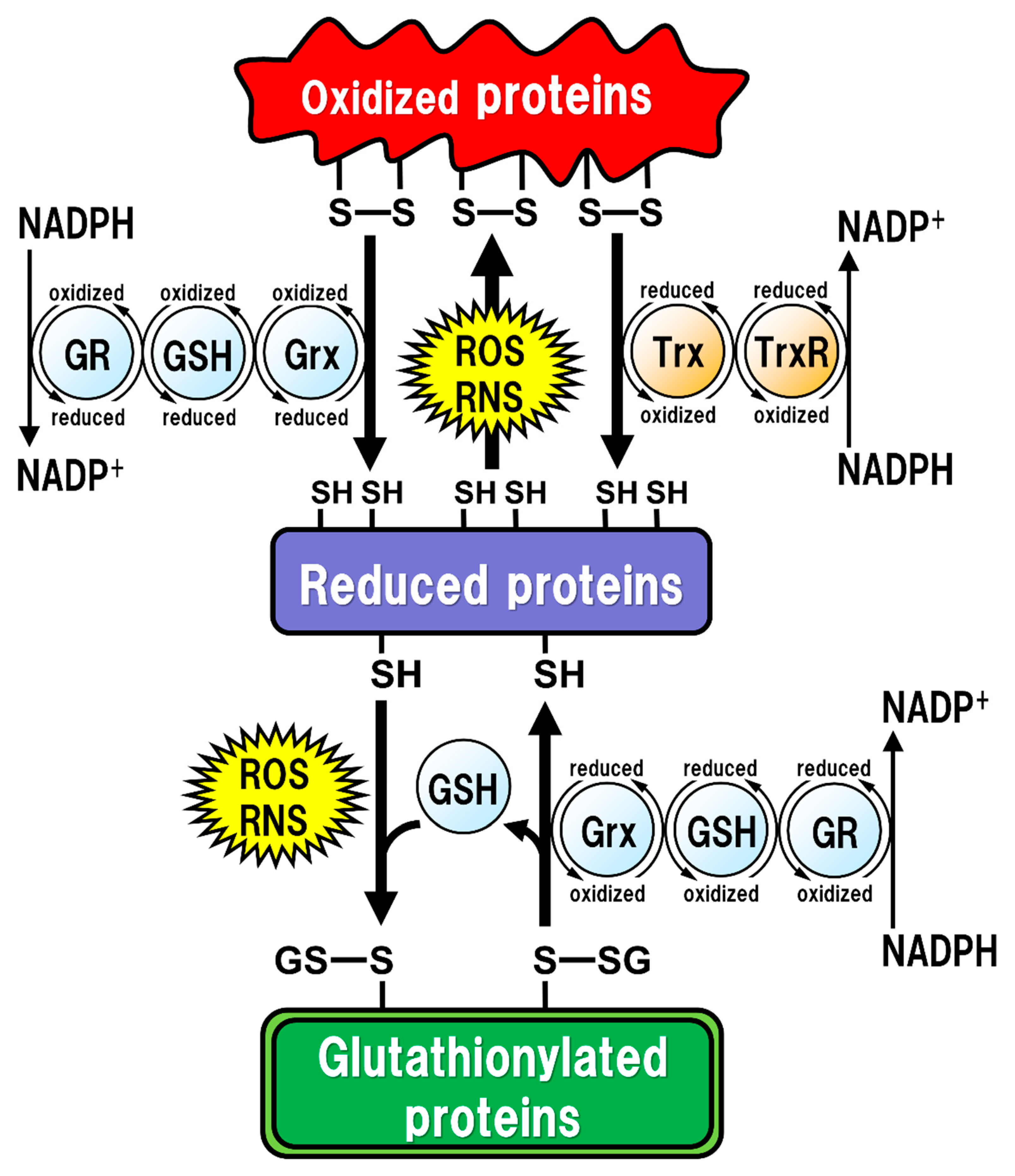
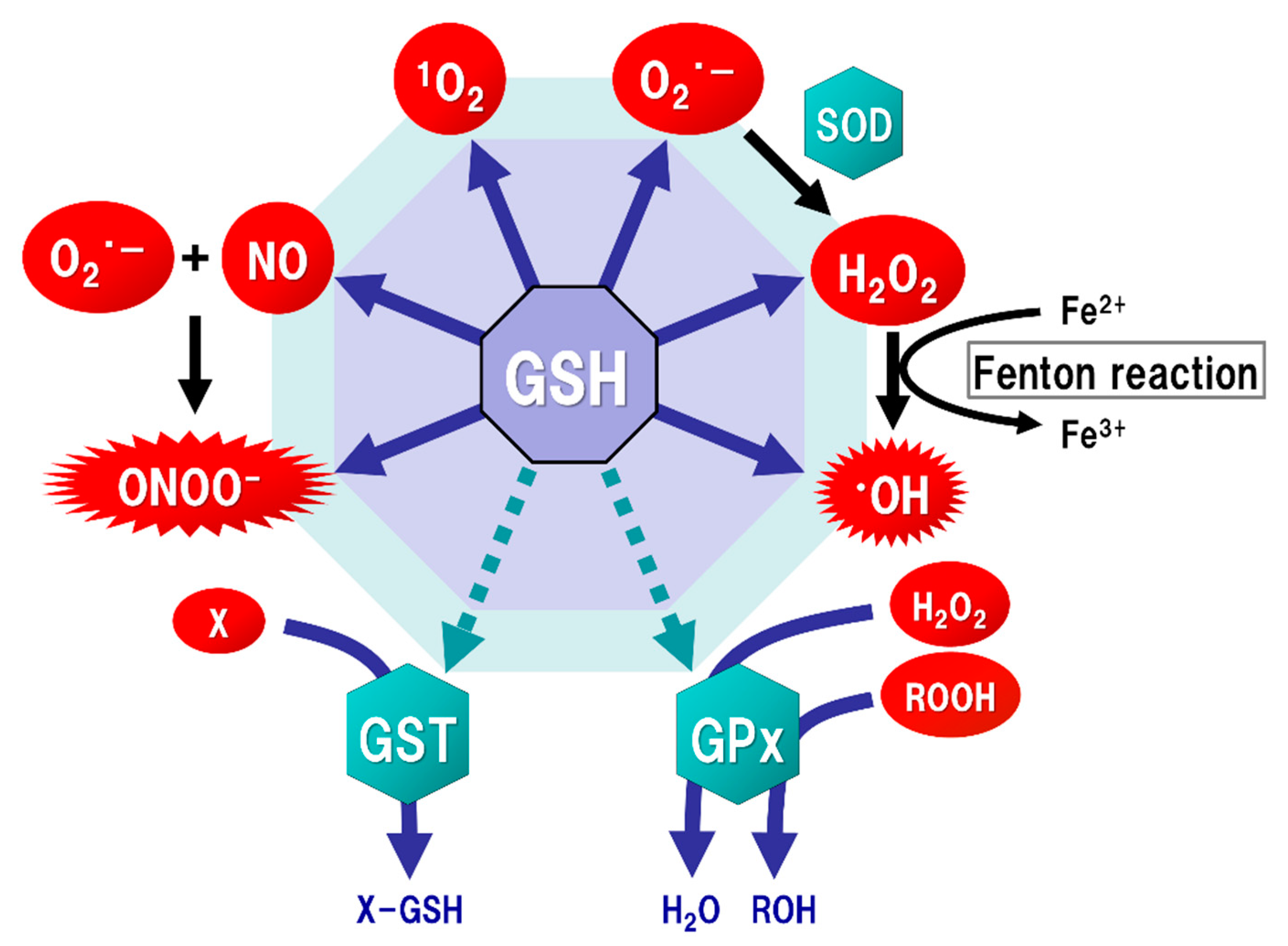
2. GSH Function
3. Oxidative Stress in the Brain
4. GSH Synthesis in Neurons
5. Regulatory Mechanism of EAAC1 Expression in Neurons
6. GSH Synthesis in Astrocytes
7. GSH Synthesis in Microglia
8. Brain GSH Levels in Neurodegenerative Diseases
9. GSH Treatment for Neurodegenerative Diseases
Funding
Conflicts of Interest
References
- De Rey-Pailhade, M.J. Sur un corps d’origine organique hydrogénant le soufre á froid. C. R. Hebd. Séances Acad. Sci. 1888, 106, 1683–1684. [Google Scholar]
- de Rey-Pailhade, M.J. Sur un nouveau principe immédiat organique. le philothion. Bull. Soc. Hist Nat. Toulouse 1888, 173–180. [Google Scholar]
- Meister, A. On the discovery of glutathione. Trends Biochem. Sci. 1988, 13, 185–188. [Google Scholar] [CrossRef]
- Heffter, A. Die Reduzierenden Bestandteile der Zellen. Mediz Nat. Arch. 1908, 1, 81–103. [Google Scholar]
- Hopkins, F.G. On an Autoxidisable Constituent of the Cell. Biochem. J. 1921, 15, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Hunter, G.; Eagles, B.A. Glutathione. A critical study. J. Biol. Chem. 1927, 72, 147–166. [Google Scholar] [CrossRef]
- Hopkins, F.G. On glutathione: A reinvestigation. J. Biol. Chem. 1929, 84, 269–320. [Google Scholar] [CrossRef]
- Kendall, E.C.; McKenzie, B.F.; Mason, H.L. A study of glutathione. I. Its preparation in crystalline form and its identification. J. Biol. Chem. 1929, 84, 657–674. [Google Scholar] [CrossRef]
- Vigneaud, V.D.; Mikller, G.L. A synthesis of glutathione. J. Biol. Chem. 1936, 116, 469–476. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. The sulfur-containing amino acids: An overview. J. Nutr. 2006, 136 (Suppl. 6), 1636s–1640s. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Commandeur, J.N.; Stijntjes, G.J.; Vermeulen, N.P. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharm. Rev. 1995, 47, 271–330. [Google Scholar]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzym. 1995, 251, 8–28. [Google Scholar]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758. [Google Scholar] [CrossRef]
- Ahsan, M.K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox regulation of cell survival by the thioredoxin superfamily: An implication of redox gene therapy in the heart. Antioxid Redox Signal. 2009, 11, 2741–2758. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrao, R.C. Thiol redox homeostasis in neurodegenerative disease. Redox Biol. 2015, 5, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med. 2004, 8, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Chock, P.B. Posttranslational modification of cysteine in redox signaling and oxidative stress: Focus on s-glutathionylation. Antioxid Redox Signal. 2012, 16, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Regulation of protein function by glutathionylation. Free Radic. Res. 2005, 39, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P., Jr. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem. Pharm. 2008, 75, 2234–2244. [Google Scholar] [CrossRef]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999, 72, 2523–2530. [Google Scholar] [CrossRef]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Impaired glutathione synthesis in neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021–21044. [Google Scholar] [CrossRef]
- Tateishi, N.; Higashi, T.; Shinya, S.; Naruse, A.; Sakamoto, Y. Studies on the regulation of glutathione level in rat liver. J. Biochem. 1974, 75, 93–103. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Seelig, G.F.; Simondsen, R.P.; Meister, A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J. Biol. Chem. 1984, 259, 9345–9347. [Google Scholar] [CrossRef]
- Meister, A. Mitochondrial changes associated with glutathione deficiency. Biochim. Biophys. Acta 1995, 1271, 35–42. [Google Scholar] [CrossRef][Green Version]
- Meredith, M.J.; Reed, D.J. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J. Biol. Chem. 1982, 257, 3747–3753. [Google Scholar] [CrossRef]
- Dalton, T.P.; Dieter, M.Z.; Yang, Y.; Shertzer, H.G.; Nebert, D.W. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: Embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem. Biophys. Res. Commun. 2000, 279, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Winkler, A.; Njalsson, R.; Carlsson, K.; Elgadi, A.; Rozell, B.; Abraham, L.; Ercal, N.; Shi, Z.Z.; Lieberman, M.W.; Larsson, A.; et al. Glutathione is essential for early embryogenesis—Analysis of a glutathione synthetase knockout mouse. Biochem. Biophys. Res. Commun. 2011, 412, 121–126. [Google Scholar] [CrossRef]
- Yang, Y.; Dieter, M.Z.; Chen, Y.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 2002, 277, 49446–49452. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Mari, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S. Peroxynitrite versus hydroxyl radical: The role of nitric oxide in superoxide-dependent cerebral injury. Ann. N. Y. Acad. Sci. 1994, 738, 69–75. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharm. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Kranich, O.; Hamprecht, B.; Dringen, R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett. 1996, 219, 211–214. [Google Scholar] [CrossRef]
- Dringen, R.; Hamprecht, B. N-acetylcysteine, but not methionine or 2-oxothiazolidine-4-carboxylate, serves as cysteine donor for the synthesis of glutathione in cultured neurons derived from embryonal rat brain. Neurosci. Lett. 1999, 259, 79–82. [Google Scholar] [CrossRef]
- Gegg, M.E.; Clark, J.B.; Heales, S.J. Co-culture of neurones with glutathione deficient astrocytes leads to increased neuronal susceptibility to nitric oxide and increased glutamate-cysteine ligase activity. Brain Res. 2005, 1036, 1–6. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef]
- Bendahan, A.; Armon, A.; Madani, N.; Kavanaugh, M.P.; Kanner, B.I. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem. 2000, 275, 37436–37442. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001, 902, 156–163. [Google Scholar] [CrossRef]
- Aoyama, K.; Nakaki, T. Neuroprotective properties of the excitatory amino acid carrier 1 (EAAC1). Amino Acids 2013, 45, 133–142. [Google Scholar] [CrossRef]
- Nieoullon, A.; Canolle, B.; Masmejean, F.; Guillet, B.; Pisano, P.; Lortet, S. The neuronal excitatory amino acid transporter EAAC1/EAAT3: Does it represent a major actor at the brain excitatory synapse? J. Neurochem. 2006, 98, 1007–1018. [Google Scholar] [CrossRef]
- Lin, C.I.; Orlov, I.; Ruggiero, A.M.; Dykes-Hoberg, M.; Lee, A.; Jackson, M.; Rothstein, J.D. Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3–18. Nature 2001, 410, 84–88. [Google Scholar] [CrossRef]
- Aoyama, K.; Nakaki, T. Inhibition of GTRAP3–18 May Increase Neuroprotective Glutathione (GSH) Synthesis. Int. J. Mol. Sci. 2012, 13, 12017–12035. [Google Scholar] [CrossRef]
- Watabe, M.; Aoyama, K.; Nakaki, T. A dominant role of GTRAP3–18 in neuronal glutathione synthesis. J. Neurosci. 2008, 28, 9404–9413. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Wang, F.; Matsumura, N.; Kiyonari, H.; Shioi, G.; Tanaka, K.; Kinoshita, C.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Increased neuronal glutathione and neuroprotection in GTRAP3–18-deficient mice. Neurobiol. Dis. 2012, 45, 973–982. [Google Scholar] [CrossRef]
- Maier, S.; Reiterer, V.; Ruggiero, A.M.; Rothstein, J.D.; Thomas, S.; Dahm, R.; Sitte, H.H.; Farhan, H. GTRAP3–18 serves as a negative regulator of Rab1 in protein transport and neuronal differentiation. J. Cell. Mol. Med. 2009, 13, 114–124. [Google Scholar] [CrossRef]
- Sha, S.; Xu, J.; Lu, Z.H.; Hong, J.; Qu, W.J.; Zhou, J.W.; Chen, L. Lack of JWA Enhances Neurogenesis and Long-Term Potentiation in Hippocampal Dentate Gyrus Leading to Spatial Cognitive Potentiation. Mol. Neurobiol. 2016, 53, 355–368. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Requejo-Aguilar, R.; Gonzalez-Fernandez, S.; Resch, M.; Carabias-Carrasco, M.; Ros, J.; Almeida, A.; Bolanos, J.P. Hippocampal neurons require a large pool of glutathione to sustain dendrite integrity and cognitive function. Redox Biol. 2018, 19, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Joon Won, S.; Malgorn, C.; Auregan, G.; Berman, A.E.; Chen, P.C.; Deglon, N.; Johnson, J.A.; Won Suh, S.; Swanson, R.A. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino Acid transporter 3 expression. J. Neurosci. 2011, 31, 7392–7401. [Google Scholar] [CrossRef]
- Ma, K.; Zheng, S.; Zuo, Z. The transcription factor regulatory factor X1 increases the expression of neuronal glutamate transporter type 3. J. Biol. Chem. 2006, 281, 21250–21255. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.G.; Gazzola, G.C.; Cagnin, S.; Kagechika, H.; Bussolati, O. The ATRA-dependent overexpression of the glutamate transporter EAAC1 requires RARbeta induction. Biochim. Biophys. Acta 2009, 1788, 1861–1868. [Google Scholar] [CrossRef][Green Version]
- Kinoshita, C.; Aoyama, K.; Matsumura, N.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Rhythmic oscillations of the microRNA miR-96–5p play a neuroprotective role by indirectly regulating glutathione levels. Nat. Commun 2014, 5, 3823. [Google Scholar] [CrossRef]
- Kinoshita, C.; Aoyama, K.; Nakaki, T. Neuroprotection afforded by circadian regulation of intracellular glutathione levels: A key role for miRNAs. Free Radic. Biol. Med. 2018, 119, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Almilaji, A.; Pakladok, T.; Guo, A.; Munoz, C.; Foller, M.; Lang, F. Regulation of the glutamate transporter EAAT3 by mammalian target of rapamycin mTOR. Biochem. Biophys. Res. Commun. 2012, 421, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, Z.; Bhavsar, S.K.; Sopjani, M.; Alesutan, I.; Saxena, A.; Dermaku-Sopjani, M.; Lang, F. Regulation of the glutamate transporters by JAK2. Cell. Physiol. Biochem. 2011, 28, 693–702. [Google Scholar] [CrossRef]
- Sopjani, M.; Alesutan, I.; Dermaku-Sopjani, M.; Fraser, S.; Kemp, B.E.; Foller, M.; Lang, F. Down-regulation of Na+-coupled glutamate transporter EAAT3 and EAAT4 by AMP-activated protein kinase. J. Neurochem. 2010, 113, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K.; Norenberg, M.D. Astrocytes. Sci. Am. 1989, 260, 66–72. [Google Scholar] [CrossRef] [PubMed]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Drukarch, B.; Schepens, E.; Jongenelen, C.A.; Stoof, J.C.; Langeveld, C.H. Astrocyte-mediated enhancement of neuronal survival is abolished by glutathione deficiency. Brain Res. 1997, 770, 123–130. [Google Scholar] [CrossRef]
- Chen, Y.; Vartiainen, N.E.; Ying, W.; Chan, P.H.; Koistinaho, J.; Swanson, R.A. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J. Neurochem. 2001, 77, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef]
- McGann, J.C.; Mandel, G. Neuronal activity induces glutathione metabolism gene expression in astrocytes. Glia 2018, 66, 2024–2039. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- McBean, G.J. The transsulfuration pathway: A source of cysteine for glutathione in astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef]
- Sagara, J.I.; Miura, K.; Bannai, S. Maintenance of neuronal glutathione by glial cells. J. Neurochem. 1993, 61, 1672–1676. [Google Scholar] [CrossRef]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Gros, C.; Hirrlinger, J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res. 2001, 66, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Lindenau, J.; Noack, H.; Asayama, K.; Wolf, G. Enhanced cellular glutathione peroxidase immunoreactivity in activated astrocytes and in microglia during excitotoxin induced neurodegeneration. Glia 1998, 24, 252–256. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Gutterer, J.M.; Kussmaul, L.; Hamprecht, B.; Dringen, R. Microglial cells in culture express a prominent glutathione system for the defense against reactive oxygen species. Dev. Neurosci. 2000, 22, 384–392. [Google Scholar] [CrossRef]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anions by a CNS macrophage, the microglia. Febs Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef]
- Minghetti, L.; Levi, G. Microglia as effector cells in brain damage and repair: Focus on prostanoids and nitric oxide. Prog. Neurobiol. 1998, 54, 99–125. [Google Scholar] [CrossRef]
- Persson, M.; Sandberg, M.; Hansson, E.; Ronnback, L. Microglial glutamate uptake is coupled to glutathione synthesis and glutamate release. Eur. J. Neurosci. 2006, 24, 1063–1070. [Google Scholar] [CrossRef]
- Chretien, F.; Vallat-Decouvelaere, A.V.; Bossuet, C.; Rimaniol, A.C.; Le Grand, R.; Le Pavec, G.; Creminon, C.; Dormont, D.; Gray, F.; Gras, G. Expression of excitatory amino acid transporter-2 (EAAT-2) and glutamine synthetase (GS) in brain macrophages and microglia of SIVmac251-infected macaques. Neuropathol. Appl. Neurobiol. 2002, 28, 410–417. [Google Scholar] [CrossRef]
- Persson, M.; Ronnback, L. Microglial self-defence mediated through GLT-1 and glutathione. Amino Acids 2012, 42, 207–219. [Google Scholar] [CrossRef]
- Slivka, A.; Spina, M.B.; Cohen, G. Reduced and oxidized glutathione in human and monkey brain. Neurosci. Lett. 1987, 74, 112–118. [Google Scholar] [CrossRef]
- Venkateshappa, C.; Harish, G.; Mahadevan, A.; Srinivas Bharath, M.M.; Shankar, S.K. Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: Implications for neurodegeneration in Alzheimer’s disease. Neurochem. Res. 2012, 37, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Mandal, P.K.; Ersland, L.; Gruner, E.R.; Tripathi, M.; Raghunathan, P.; Sharma, A.; Chaithya, G.R.; Punjabi, K.; Splaine, C. A Multi-Center Study on Human Brain Glutathione Conformation using Magnetic Resonance Spectroscopy. J. Alzheimers Dis. 2018, 66, 517–532. [Google Scholar] [CrossRef]
- Rae, C.D.; Williams, S.R. Glutathione in the human brain: Review of its roles and measurement by magnetic resonance spectroscopy. Anal. Biochem. 2017, 529, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Chiang, G.C.; Mao, X.; Kang, G.; Chang, E.; Pandya, S.; Vallabhajosula, S.; Isaacson, R.; Ravdin, L.D.; Shungu, D.C. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with 1H-MRS and Pittsburgh Compound-B PET. AJNR Am. J. Neuroradiol. 2017, 38, 1130–1137. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef]
- Duerson, K.; Woltjer, R.L.; Mookherjee, P.; Leverenz, J.B.; Montine, T.J.; Bird, T.D.; Pow, D.V.; Rauen, T.; Cook, D.G. Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol. 2009, 19, 267–278. [Google Scholar] [CrossRef]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative damage in neurodegenerative disease. Lancet 1994, 344, 796–798. [Google Scholar] [CrossRef]
- Langston, J.W. The etiology of Parkinson’s disease with emphasis on the MPTP story. Neurology 1996, 47, S153–S160. [Google Scholar] [CrossRef]
- Kamel, F. Epidemiology. Paths from pesticides to Parkinson’s. Science 2013, 341, 722–723. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Aoyama, K.; Matsumura, N.; Watabe, M.; Nakaki, T. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur. J. Neurosci. 2008, 27, 20–30. [Google Scholar] [CrossRef]
- Mischley, L.K.; Conley, K.E.; Shankland, E.G.; Kavanagh, T.J.; Rosenfeld, M.E.; Duda, J.E.; White, C.C.; Wilbur, T.K.; De La Torre, P.U.; Padowski, J.M. Central nervous system uptake of intranasal glutathione in Parkinson’s disease. NPJ Parkinsons Dis. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Graff, C.L.; Pollack, G.M. Nasal drug administration: Potential for targeted central nervous system delivery. J. Pharm. Sci. 2005, 94, 1187–1195. [Google Scholar] [CrossRef]
- Aoyama, K.; Matsubara, K.; Fujikawa, Y.; Nagahiro, Y.; Shimizu, K.; Umegae, N.; Hayase, N.; Shiono, H.; Kobayashi, S. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann. Neurol. 2000, 47, 524–527. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Nicholson, K.; Jafari-Khouzani, K.; Bogner, W.; Wang, J.; Chan, J.; Macklin, E.A.; Levine-Weinberg, M.; Breen, C.; Schwarzschild, M.A.; et al. Imaging Neurochemistry and Brain Structure Tracks Clinical Decline and Mechanisms of ALS in Patients. Front. Neurol. 2020, 11, 590573. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Killoy, K.M.; Harlan, B.A.; Pehar, M.; Helke, K.L.; Johnson, J.A.; Vargas, M.R. Decreased glutathione levels cause overt motor neuron degeneration in hSOD1(WT) over-expressing mice. Exp. Neurol. 2018, 302, 129–135. [Google Scholar] [CrossRef]
- Marx, J. Neurodegenerative diseases. Picking apart the causes of mysterious dementias. Science 2006, 314, 42–43. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Baloh, R.H. TDP-43-based animal models of neurodegeneration: New insights into ALS pathology and pathophysiology. Neurodegener. Dis. 2011, 8, 262–274. [Google Scholar] [CrossRef]
- Chen, T.; Turner, B.J.; Beart, P.M.; Sheehan-Hennessy, L.; Elekwachi, C.; Muyderman, H. Glutathione monoethyl ester prevents TDP-43 pathology in motor neuronal NSC-34 cells. Neurochem. Int. 2018, 112, 278–287. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Fanciulli, A.; Wenning, G.K. Multiple-system atrophy. N. Engl. J. Med. 2015, 372, 249–263. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Kinoshita, C.; Aoyama, K.; Nakaki, T. MicroRNA as a new agent for regulating neuronal glutathione synthesis and metabolism. Aims Mol. Sci 2015. [Google Scholar] [CrossRef]
- Ubhi, K.; Rockenstein, E.; Kragh, C.; Inglis, C.; Spencer, B.; Michael, S.; Mante, M.; Adame, A.; Galasko, D.; Masliah, E. Widespread microRNA dysregulation in multiple system atrophy—Disease-related alteration in miR-96. Eur. J. Neurosci. 2014, 39, 1026–1041. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, C.; Kikuchi-Utsumi, K.; Aoyama, K.; Suzuki, R.; Okamoto, Y.; Matsumura, N.; Omata, D.; Maruyama, K.; Nakaki, T. Inhibition of miR-96–5p in the mouse brain increases glutathione levels by altering NOVA1 expression. Commun. Biol. 2021, 4, 182. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Braun, L.D.; Crane, P.D.; Oldendorf, W.H. Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology 1978, 103, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Aebi, S.; Assereto, R.; Lauterburg, B.H. High-dose intravenous glutathione in man. Pharmacokinetics and effects on cyst(e)ine in plasma and urine. Eur. J. Clin. Investig. 1991, 21, 103–110. [Google Scholar] [CrossRef]
- Grunewald, R.A. Ascorbic acid in the brain. Brain Res. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Arteel, G.E.; Briviba, K.; Sies, H. Protection against peroxynitrite. FEBS Lett. 1999, 445, 226–230. [Google Scholar] [CrossRef]
- Metcalfe, T.; Bowen, D.M.; Muller, D.P. Vitamin E concentrations in human brain of patients with Alzheimer’s disease, fetuses with Down’s syndrome, centenarians, and controls. Neurochem. Res. 1989, 14, 1209–1212. [Google Scholar] [CrossRef]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer Disease: A Randomized Clinical Trial With Cerebrospinal Fluid Biomarker Measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef]
- Logroscino, G.; Marder, K.; Cote, L.; Tang, M.X.; Shea, S.; Mayeux, R. Dietary lipids and antioxidants in Parkinson’s disease: A population-based, case-control study. Ann. Neurol. 1996, 39, 89–94. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Dexter, D.T.; Ward, R.J.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Peters, T.J.; Jenner, P.; Marsden, C.D. Alpha-tocopherol levels in brain are not altered in Parkinson’s disease. Ann. Neurol. 1992, 32, 591–593. [Google Scholar] [CrossRef]
- Parkinson Study Group, Q.E.I.; Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 Gene Deletion Alters Zinc Homeostasis and Exacerbates Neuronal Injury after Transient Cerebral Ischemia. J. Neurosci. 2010, 30, 15409–15418. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Slikker, W., Jr.; Ali, S.F. Role of metallothionein and other antioxidants in scavenging superoxide radicals and their possible role in neuroprotection. Neurochem. Int. 1996, 29, 145–152. [Google Scholar] [CrossRef]
- De Flora, S.; Bennicelli, C.; Zanacchi, P.; Camoirano, A.; Morelli, A.; De Flora, A. In vitro effects of N-acetylcysteine on the mutagenicity of direct-acting compounds and procarcinogens. Carcinogenesis 1984, 5, 505–510. [Google Scholar] [CrossRef]
- Skrzydlewska, E.; Farbiszewski, R. Protective effect of N-acetylcysteine on reduced glutathione, reduced glutathione-related enzymes and lipid peroxidation in methanol intoxication. Drug Alcohol Depend. 1999, 57, 61–67. [Google Scholar] [CrossRef]
- Girgis, R.R.; Baker, S.; Mao, X.; Gil, R.; Javitt, D.C.; Kantrowitz, J.T.; Gu, M.; Spielman, D.M.; Ojeil, N.; Xu, X.; et al. Effects of acute N-acetylcysteine challenge on cortical glutathione and glutamate in schizophrenia: A pilot in vivo proton magnetic resonance spectroscopy study. Psychiatry Res. 2019, 275, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Oz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106. [Google Scholar] [CrossRef]
- Suzuki, R.; Oda, Y.; Utoguchi, N.; Namai, E.; Taira, Y.; Okada, N.; Kadowaki, N.; Kodama, T.; Tachibana, K.; Maruyama, K. A novel strategy utilizing ultrasound for antigen delivery in dendritic cell-based cancer immunotherapy. J. Control. Release 2009, 133, 198–205. [Google Scholar] [CrossRef]
- Dimcevski, G.; Kotopoulis, S.; Bjanes, T.; Hoem, D.; Schjott, J.; Gjertsen, B.T.; Biermann, M.; Molven, A.; Sorbye, H.; McCormack, E.; et al. A human clinical trial using ultrasound and microbubbles to enhance gemcitabine treatment of inoperable pancreatic cancer. J. Control. Release 2016, 243, 172–181. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. https://doi.org/10.3390/ijms22095010
Aoyama K. Glutathione in the Brain. International Journal of Molecular Sciences. 2021; 22(9):5010. https://doi.org/10.3390/ijms22095010
Chicago/Turabian StyleAoyama, Koji. 2021. "Glutathione in the Brain" International Journal of Molecular Sciences 22, no. 9: 5010. https://doi.org/10.3390/ijms22095010
APA StyleAoyama, K. (2021). Glutathione in the Brain. International Journal of Molecular Sciences, 22(9), 5010. https://doi.org/10.3390/ijms22095010