
Neurons and Glia Interplay in α-Synucleinopathies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Alpha-Synuclein in Neurons: A Multifaceted Protein
2.1. A Role at the Synapse
2.2. Association with Membranes and Lipid Trafficking
2.3. Aggregation and Post-Translational Modifications
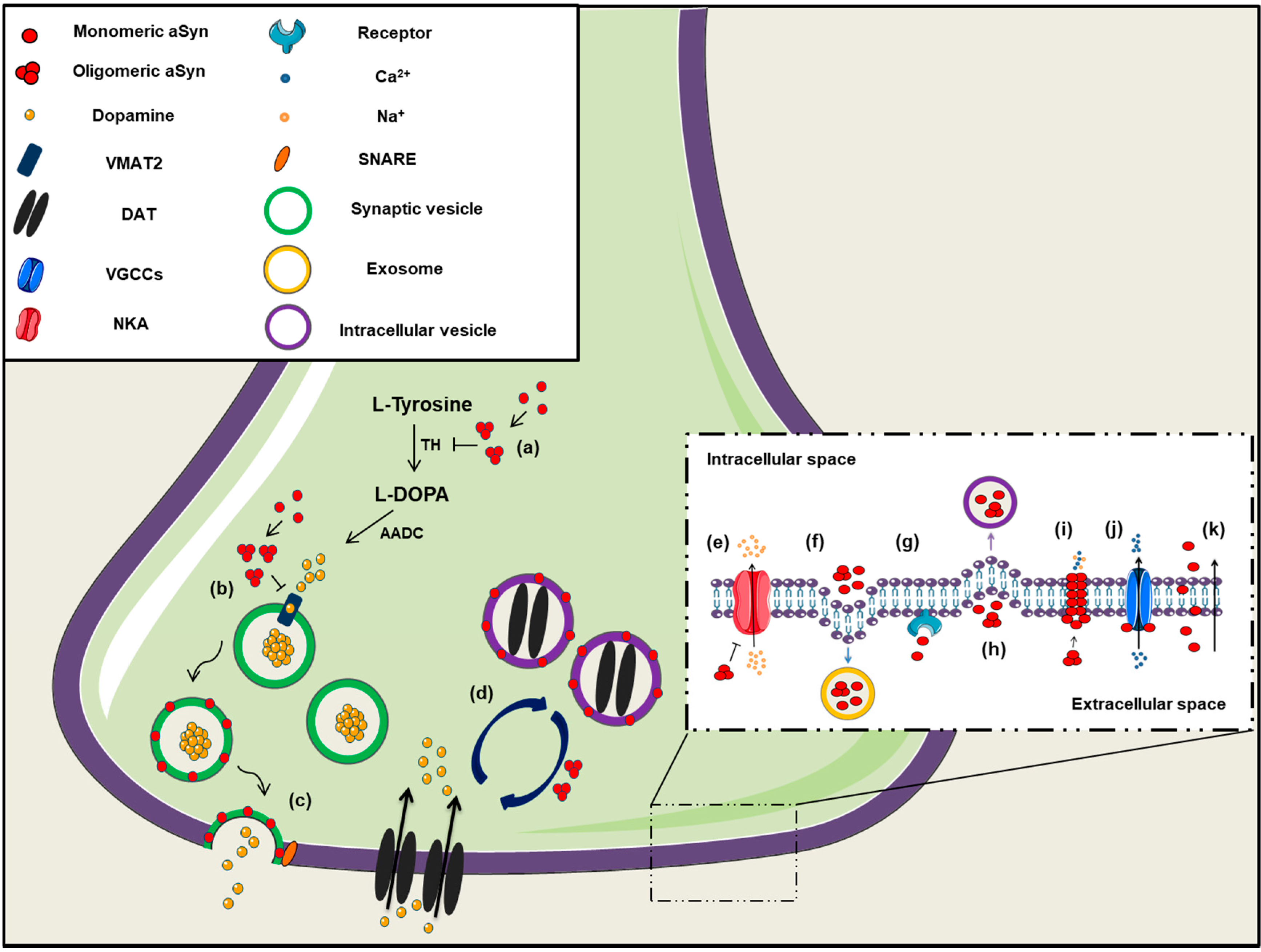
2.4. Channel Formation/Channel Interactions
2.5. Dopamine Metabolism
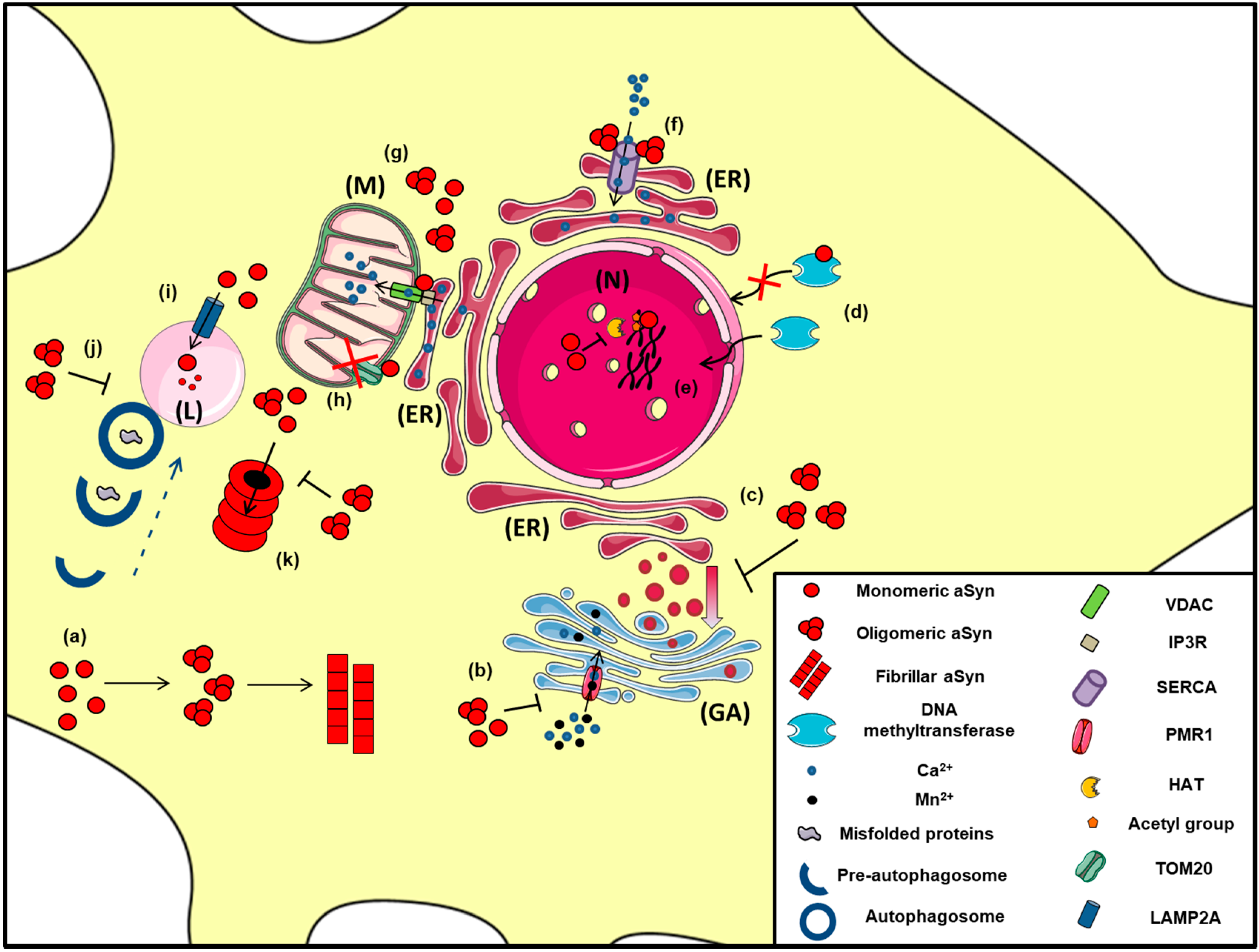
2.6. Interaction with Mitochondria and ER
2.7. Unfolded Protein Response, Regulation of ER/Golgi Trafficking and Ca2+ Homeostasis
2.8. a-Synuclein in the Nucleus
2.9. Alpha-Synuclein and Protein Degradation Pathways: An Intricate Interplay
2.10. Alpha-Synuclein in the Extracellular Space
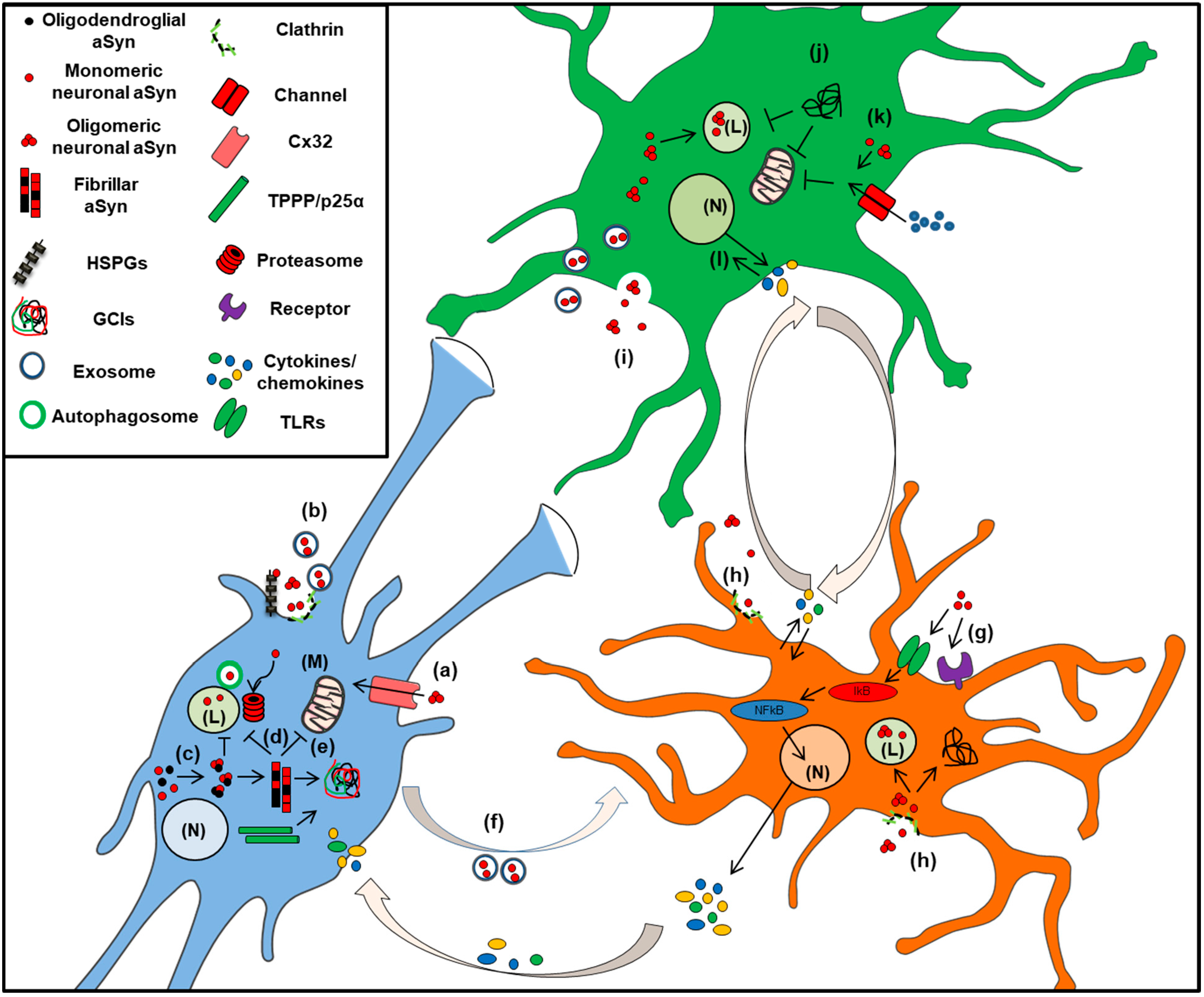
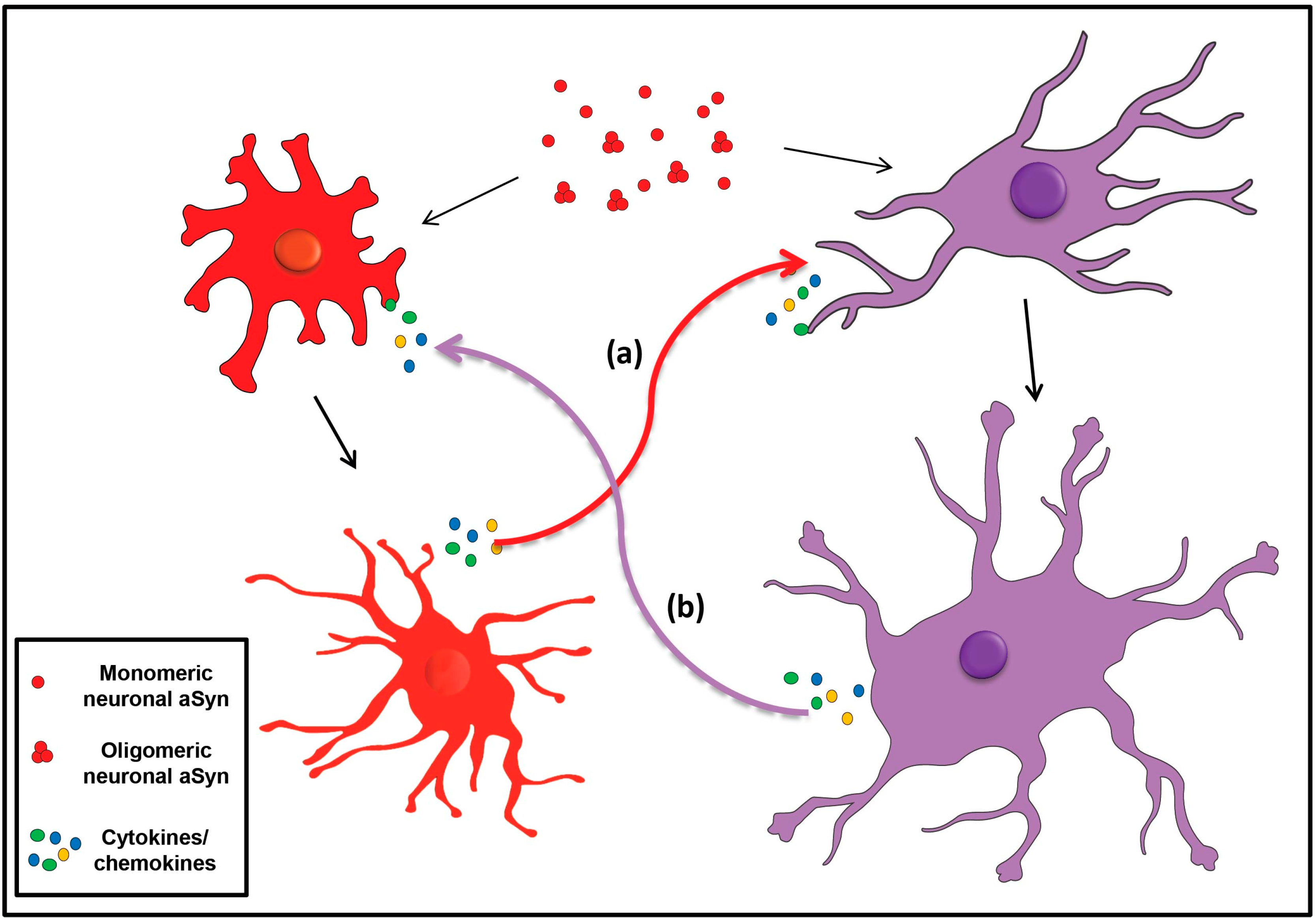
3. Glia in the CNS: Scavengers of Extracellular aSyn
3.1. Role in Microglia Function and Dysfunction
3.1.1. Physiological Role of aSyn in Microglia Function
3.1.2. Microgliosis in a-Synucleinopathies
3.1.3. Activation of Microglia and Clearance of Toxic aSyn Species
3.2. Astrocytes in a-Synucleinopathies
3.2.1. Astrocytes in PD: Friend or Foe?
3.2.2. Implication of astrocytes in MSA pathology
3.3. Alpha-Synuclein in Oligodendrocytes: The Pathologic Hallmark of MSA, A Unique Oligodendrogliopathy
Alpha-Synuclein Accumulation in Oligodendrocytes, Propagation and Spread of Pathology
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Pals, P.; Lincoln, S.; Manning, J.; Heckman, M.; Skipper, L.; Hulihan, M.; Van den Broeck, M.; De Pooter, T.; Cras, P.; Crook, J.; et al. alpha-Synuclein promoter confers susceptibility to Parkinson’s disease. Ann. Neurol. 2004, 56, 591–595. [Google Scholar] [CrossRef]
- Fuchs, J.; Tichopad, A.; Golub, Y.; Munz, M.; Schweitzer, K.J.; Wolf, B.; Berg, D.; Mueller, J.C.; Gasser, T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 2008, 22, 1327–1334. [Google Scholar] [CrossRef]
- Fanciulli, A.; Stankovic, I.; Krismer, F.; Seppi, K.; Levin, J.; Wenning, G.K. Multiple system atrophy. Int. Rev. Neurobiol. 2019, 149, 137–192. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Song, Y.J.; Lundvig, D.M.; Huang, Y.; Gai, W.P.; Blumbergs, P.C.; Hojrup, P.; Otzen, D.; Halliday, G.M.; Jensen, P.H. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am. J. Pathol. 2007, 171, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.J.; et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef]
- Wenning, G.K.; Jellinger, K.A. The role of alpha-synuclein in the pathogenesis of multiple system atrophy. Acta Neuropathol. 2005, 109, 129–140. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Bockmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Candelise, N.; Schmitz, M.; Llorens, F.; Villar-Pique, A.; Cramm, M.; Thom, T.; da Silva Correia, S.M.; da Cunha, J.E.G.; Mobius, W.; Outeiro, T.F.; et al. Seeding variability of different alpha synuclein strains in synucleinopathies. Ann. Neurol. 2019, 85, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Hashioka, S.; Klegeris, A.; Schwab, C.; McGeer, P.L. Interferon-gamma-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiol. Aging 2009, 30, 1924–1935. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.; Lucassen, P.J.; van Dam, A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Withers, G.S.; George, J.M.; Banker, G.A.; Clayton, D.F. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res. 1997, 99, 87–94. [Google Scholar] [CrossRef]
- Andringa, G.; Du, F.; Chase, T.N.; Bennett, M.C. Mapping of rat brain using the Synuclein-1 monoclonal antibody reveals somatodendritic expression of alpha-synuclein in populations of neurons homologous to those vulnerable to Lewy body formation in human synucleopathies. J. Neuropathol. Exp. Neurol. 2003, 62, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K.; Alvarez, R.B.; McFerrin, J.; Broccolini, A. Novel immunolocalization of alpha-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J. Neuropathol. Exp. Neurol. 2000, 59, 592–598. [Google Scholar] [CrossRef]
- Bottner, M.; Zorenkov, D.; Hellwig, I.; Barrenschee, M.; Harde, J.; Fricke, T.; Deuschl, G.; Egberts, J.H.; Becker, T.; Fritscher-Ravens, A.; et al. Expression pattern and localization of alpha-synuclein in the human enteric nervous system. Neurobiol. Dis. 2012, 48, 474–480. [Google Scholar] [CrossRef]
- Aldecoa, I.; Navarro-Otano, J.; Stefanova, N.; Sprenger, F.S.; Seppi, K.; Poewe, W.; Cuatrecasas, M.; Valldeoriola, F.; Gelpi, E.; Tolosa, E. Alpha-synuclein immunoreactivity patterns in the enteric nervous system. Neurosci. Lett. 2015, 602, 145–149. [Google Scholar] [CrossRef]
- Li, J.; Henning Jensen, P.; Dahlstrom, A. Differential localization of alpha-, beta- and gamma-synucleins in the rat CNS. Neuroscience 2002, 113, 463–478. [Google Scholar] [CrossRef]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Expression of alpha-synuclein is regulated in a neuronal cell type-dependent manner. Anat. Sci. Int. 2019, 94, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Brain region-dependent differential expression of alpha-synuclein. J. Comp. Neurol. 2016, 524, 1236–1258. [Google Scholar] [CrossRef] [PubMed]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M.; et al. alpha-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 2015, 128, 2231–2243. [Google Scholar] [CrossRef]
- Chen, R.H.C.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449. [Google Scholar] [CrossRef]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Sudhof, T.C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [PubMed]
- Lautenschlager, J.; Stephens, A.D.; Fusco, G.; Strohl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Sudhof, T.C. Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef] [PubMed]
- Betzer, C.; Movius, A.J.; Shi, M.; Gai, W.P.; Zhang, J.; Jensen, P.H. Identification of synaptosomal proteins binding to monomeric and oligomeric alpha-synuclein. PLoS ONE 2015, 10, e0116473. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Roy, S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef] [PubMed]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Zhu, Y.; Cai, Q.; Chan, P.; Ueda, K.; Yu, S.; Yang, H. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: An immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008, 1244, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova, N.; Petrucelli, L.; Farrer, M.; Mehta, N.; Choi, P.; Hardy, J.; Wolozin, B. alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J. Neurosci. 1999, 19, 5782–5791. [Google Scholar] [CrossRef]
- Kim, T.D.; Choi, E.; Rhim, H.; Paik, S.R.; Yang, C.H. Alpha-synuclein has structural and functional similarities to small heat shock proteins. Biochem. Biophys. Res. Commun. 2004, 324, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Kim, T.D.; Paik, S.R.; Yang, C.H. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef]
- Kim, T.D.; Paik, S.R.; Yang, C.H.; Kim, J. Structural changes in alpha-synuclein affect its chaperone-like activity in vitro. Protein Sci. 2000, 9, 2489–2496. [Google Scholar] [CrossRef]
- Park, S.M.; Jung, H.Y.; Kim, T.D.; Park, J.H.; Yang, C.H.; Kim, J. Distinct roles of the N-terminal-binding domain and the C-terminal-solubilizing domain of alpha-synuclein, a molecular chaperone. J. Biol. Chem. 2002, 277, 28512–28520. [Google Scholar] [CrossRef] [PubMed]
- Rekas, A.; Ahn, K.J.; Kim, J.; Carver, J.A. The chaperone activity of alpha-synuclein: Utilizing deletion mutants to map its interaction with target proteins. Proteins 2012, 80, 1316–1325. [Google Scholar] [CrossRef]
- Srivastava, T.; Raj, R.; Dubey, A.; Kumar, D.; Chaturvedi, R.K.; Sharma, S.K.; Priya, S. Fast kinetics of environmentally induced alpha-synuclein aggregation mediated by structural alteration in NAC region and result in structure dependent cytotoxicity. Sci. Rep. 2020, 10, 18412. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Sudhof, T.C. Properties of native brain alpha-synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.M.; Raussens, V. Toxic prefibrillar alpha-synuclein amyloid oligomers adopt a distinctive antiparallel beta-sheet structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Li, H.T.; Du, H.N.; Tang, L.; Hu, J.; Hu, H.Y. Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: Non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers 2002, 64, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, H.; Subramaniam, V.; Claessens, M. Direct visualization of model membrane remodeling by alpha-synuclein fibrillization. ChemPhysChem 2017, 18, 1620–1626. [Google Scholar] [CrossRef]
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef]
- Galvagnion, C. The Role of Lipids Interacting with alpha-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, V.; Scarlata, S. Membrane binding and self-association of alpha-synucleins. Biochemistry 2001, 40, 9927–9934. [Google Scholar] [CrossRef]
- Zhu, M.; Fink, A.L. Lipid binding inhibits alpha-synuclein fibril formation. J. Biol. Chem. 2003, 278, 16873–16877. [Google Scholar] [CrossRef]
- Lee, H.J.; Choi, C.; Lee, S.J. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Muller, M.B.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.; Cascella, R.; et al. A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovicic, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates alpha-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Sharikov, Y.; Kouznetsova, V.L.; Greenberg, J.P.; Wrasidlo, W.; Overk, C.; Gonzalez, T.; Trejo, M.; Spencer, B.; Kosberg, K.; et al. Molecular determinants of alpha-synuclein mutants’ oligomerization and membrane interactions. ACS Chem. Neurosci. 2015, 6, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Killinger, B.A.; Melki, R.; Brundin, P.; Kordower, J.H. Endogenous alpha-synuclein monomers, oligomers and resulting pathology: Let’s talk about the lipids in the room. NPJ Parkinsons Dis. 2019, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef]
- Jo, E.; McLaurin, J.; Yip, C.M.; St George-Hyslop, P.; Fraser, P.E. Alpha-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef]
- Trexler, A.J.; Rhoades, E. Alpha-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry 2009, 48, 2304–2306. [Google Scholar] [CrossRef] [PubMed]
- Jao, C.C.; Der-Sarkissian, A.; Chen, J.; Langen, R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA 2004, 101, 8331–8336. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Sudhof, T.C. A broken alpha -helix in folded alpha -Synuclein. J. Biol. Chem. 2003, 278, 15313–15318. [Google Scholar] [CrossRef]
- Drescher, M.; Veldhuis, G.; van Rooijen, B.D.; Milikisyants, S.; Subramaniam, V.; Huber, M. Antiparallel arrangement of the helices of vesicle-bound alpha-synuclein. J. Am. Chem. Soc. 2008, 130, 7796–7797. [Google Scholar] [CrossRef]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef]
- Lucke, C.; Gantz, D.L.; Klimtchuk, E.; Hamilton, J.A. Interactions between fatty acids and alpha-synuclein. J. Lipid Res. 2006, 47, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Doig, A.J.; Middleton, D.A. A study of the regional effects of alpha-synuclein on the organization and stability of phospholipid bilayers. Biochemistry 2006, 45, 5783–5792. [Google Scholar] [CrossRef]
- Adamczyk, A.; Kacprzak, M.; Kazmierczak, A. Alpha-synuclein decreases arachidonic acid incorporation into rat striatal synaptoneurosomes. Folia Neuropathol. 2007, 45, 230–235. [Google Scholar] [PubMed]
- Castagnet, P.I.; Golovko, M.Y.; Barcelo-Coblijn, G.C.; Nussbaum, R.L.; Murphy, E.J. Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J. Neurochem. 2005, 94, 839–849. [Google Scholar] [CrossRef]
- Barcelo-Coblijn, G.; Golovko, M.Y.; Weinhofer, I.; Berger, J.; Murphy, E.J. Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. J. Neurochem. 2007, 101, 132–141. [Google Scholar] [CrossRef]
- Alza, N.P.; Conde, M.A.; Scodelaro-Bilbao, P.G.; Salvador, G.A. Neutral lipids as early biomarkers of cellular fate: The case of alpha-synuclein overexpression. Cell Death Dis. 2021, 12, 52. [Google Scholar] [CrossRef]
- Ruf, V.C.; Nubling, G.S.; Willikens, S.; Shi, S.; Schmidt, F.; Levin, J.; Botzel, K.; Kamp, F.; Giese, A. Different effects of alpha-synuclein mutants on lipid binding and aggregation detected by single molecule fluorescence spectroscopy and ThT fluorescence-based measurements. ACS Chem. Neurosci. 2019, 10, 1649–1659. [Google Scholar] [CrossRef]
- Conde, M.A.; Alza, N.P.; Iglesias Gonzalez, P.A.; Scodelaro Bilbao, P.G.; Sanchez Campos, S.; Uranga, R.M.; Salvador, G.A. Phospholipase D1 downregulation by alpha-synuclein: Implications for neurodegeneration in Parkinson’s disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 639–650. [Google Scholar] [CrossRef]
- Ahn, B.H.; Rhim, H.; Kim, S.Y.; Sung, Y.M.; Lee, M.Y.; Choi, J.Y.; Wolozin, B.; Chang, J.S.; Lee, Y.H.; Kwon, T.K.; et al. alpha-Synuclein interacts with phospholipase D isozymes and inhibits pervanadate-induced phospholipase D activation in human embryonic kidney-293 cells. J. Biol. Chem. 2002, 277, 12334–12342. [Google Scholar] [CrossRef] [PubMed]
- Jenco, J.M.; Rawlingson, A.; Daniels, B.; Morris, A.J. Regulation of phospholipase D2: Selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 1998, 37, 4901–4909. [Google Scholar] [CrossRef] [PubMed]
- Payton, J.E.; Perrin, R.J.; Woods, W.S.; George, J.M. Structural determinants of PLD2 inhibition by alpha-synuclein. J. Mol. Biol. 2004, 337, 1001–1009. [Google Scholar] [CrossRef]
- Rappley, I.; Gitler, A.D.; Selvy, P.E.; LaVoie, M.J.; Levy, B.D.; Brown, H.A.; Lindquist, S.; Selkoe, D.J. Evidence that alpha-synuclein does not inhibit phospholipase D. Biochemistry 2009, 48, 1077–1083. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Sode, K.; Ochiai, S.; Kobayashi, N.; Usuzaka, E. Effect of reparation of repeat sequences in the human alpha-synuclein on fibrillation ability. Int. J. Biol. Sci. 2006, 3, 1–7. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Longhena, F.; Faustini, G.; Missale, C.; Pizzi, M.; Spano, P.; Bellucci, A. The Contribution of alpha-Synuclein Spreading to Parkinson’s Disease Synaptopathy. Neural. Plast. 2017, 2017, 5012129. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. alpha-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Zacharska, D.; Koziorowski, D.; Ross, O.A.; Milewski, M.; Poznanski, J.A.; Jurek, M.; Wszolek, Z.K.; Soto-Ortolaza, A.; Awek, J.A.S.; Janik, P.; et al. Novel A18T and pA29S substitutions in alpha-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 1057–1060. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Bussell, R., Jr.; Eliezer, D. Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J. Biol. Chem. 2001, 276, 45996–46003. [Google Scholar] [CrossRef]
- Lazaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Krohnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef]
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T., Jr. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118. [Google Scholar] [CrossRef]
- Rospigliosi, C.C.; McClendon, S.; Schmid, A.W.; Ramlall, T.F.; Barre, P.; Lashuel, H.A.; Eliezer, D. E46K Parkinson’s-linked mutation enhances C-terminal-to-N-terminal contacts in alpha-synuclein. J. Mol. Biol. 2009, 388, 1022–1032. [Google Scholar] [CrossRef]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.K.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 2014, 289, 21856–21876. [Google Scholar] [CrossRef]
- Sanjeev, A.; Mattaparthi, V.S.K. Computational investigation on the effects of H50Q and G51D mutations on the alpha-Synuclein aggregation propensity. J. Biomol. Struct. Dyn. 2018, 36, 2224–2236. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J. Neurochem. 2014, 131, 859–867. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Giasson, B.I. The A53E alpha-synuclein pathological mutation demonstrates reduced aggregation propensity in vitro and in cell culture. Neurosci. Lett. 2015, 597, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Takano, H.; Onodera, O.; Kobayashi, H.; Ikeuchi, T.; Koide, R.; Okuizumi, K.; Shimohata, T.; Wakabayashi, K.; Takahashi, H.; et al. No mutation in the entire coding region of the alpha-synuclein gene in pathologically confirmed cases of multiple system atrophy. Neurosci. Lett. 1999, 270, 110–112. [Google Scholar] [CrossRef]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef]
- Pandey, N.; Schmidt, R.E.; Galvin, J.E. The alpha-synuclein mutation E46K promotes aggregation in cultured cells. Exp. Neurol. 2006, 197, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Margolis, R.L.; Li, X.; Troncoso, J.C.; Lee, M.K.; Dawson, V.L.; Dawson, T.M.; Iwatsubo, T.; Ross, C.A. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 2005, 25, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 2008, 283, 23179–23188. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.R.; Hu, Z.W.; Zhao, Y.F.; Chen, Y.X.; Li, Y.M. Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 2016, 6, 37130. [Google Scholar] [CrossRef]
- Sato, H.; Arawaka, S.; Hara, S.; Fukushima, S.; Koga, K.; Koyama, S.; Kato, T. Authentically phosphorylated alpha-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J. Neurosci. 2011, 31, 16884–16894. [Google Scholar] [CrossRef]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Investig. 2009, 119, 3257–3265. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Azeredo da Silveira, S.; Schneider, B.L.; Cifuentes-Diaz, C.; Sage, D.; Abbas-Terki, T.; Iwatsubo, T.; Unser, M.; Aebischer, P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 872–887. [Google Scholar] [CrossRef]
- Mbefo, M.K.; Paleologou, K.E.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822. [Google Scholar] [CrossRef]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fredenburg, R.A.; Lansbury, P.T., Jr.; Fernandez, C.O.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, Q.; Duan, C.; Li, Y.; Yu, S.; Chan, P.; Ueda, K.; Yang, H. Phosphorylation of alpha-synuclein upregulates tyrosine hydroxylase activity in MN9D cells. Acta Histochem. 2011, 113, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Tonegawa, R.; Ito, G.; Mitani, S.; Iwatsubo, T. Phosphorylation of alpha-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 7098–7109. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kato, T.; Arawaka, S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: A review of in vivo models. Rev. Neurosci. 2013, 24, 115–123. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Sabbagh, M.N.; Akiyama, H.; Serrano, G.E.; Sue, L.I.; Beach, T.G. Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 2013, 240, 190–204. [Google Scholar] [CrossRef]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef]
- Xu, Y.; Deng, Y.; Qing, H. The phosphorylation of alpha-synuclein: Development and implication for the mechanism and therapy of the Parkinson’s disease. J. Neurochem. 2015, 135, 4–18. [Google Scholar] [CrossRef]
- Lu, Y.; Prudent, M.; Fauvet, B.; Lashuel, H.A.; Girault, H.H. Phosphorylation of alpha-Synuclein at Y125 and S129 alters its metal binding properties: Implications for understanding the role of alpha-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675. [Google Scholar] [CrossRef]
- Fayyad, M.; Erskine, D.; Majbour, N.K.; Vaikath, N.N.; Ghanem, S.S.; Sudhakaran, I.P.; Abdesselem, H.; Lamprokostopoulou, A.; Vekrellis, K.; Morris, C.M.; et al. Investigating the presence of doubly phosphorylated alpha-synuclein at tyrosine 125 and serine 129 in idiopathic Lewy body diseases. Brain Pathol. 2020, 30, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Danielson, S.R.; Held, J.M.; Schilling, B.; Oo, M.; Gibson, B.W.; Andersen, J.K. Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 2009, 81, 7823–7828. [Google Scholar] [CrossRef]
- McCormack, A.L.; Mak, S.K.; Di Monte, D.A. Increased alpha-synuclein phosphorylation and nitration in the aging primate substantia nigra. Cell Death Dis. 2012, 3, e315. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xu, X.; Xiang, Z.; Zhou, J.; Zhang, Z.; Hu, C.; He, C. Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS ONE 2010, 5, e9956. [Google Scholar] [CrossRef]
- Souza, J.M.; Peluffo, G.; Radi, R. Protein tyrosine nitration—Functional alteration or just a biomarker? Free Radic. Biol. Med. 2008, 45, 357–366. [Google Scholar] [CrossRef]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamashita, H.; Nakamura, T.; Nagano, Y.; Nakamura, S. Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 2002, 938, 73–80. [Google Scholar] [CrossRef]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef]
- Fernandez, E.; Garcia-Moreno, J.M.; Martin de Pablos, A.; Chacon, J. May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosine proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum alpha-synuclein serve for diagnosis of sporadic Parkinson’s disease? Antioxid. Redox. Signal. 2013, 19, 912–918. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef]
- Liani, E.; Eyal, A.; Avraham, E.; Shemer, R.; Szargel, R.; Berg, D.; Bornemann, A.; Riess, O.; Ross, C.A.; Rott, R.; et al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 5500–5505. [Google Scholar] [CrossRef]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2008, 17, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K.; Chau, H.; Lozano, A.M.; Hyman, B.T.; McLean, P.J. Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS ONE 2011, 6, e14695. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, site-specific ubiquitin modification of alpha-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471. [Google Scholar] [CrossRef] [PubMed]
- Weetman, J.; Wong, M.B.; Sharry, S.; Rcom-H’cheo-Gauthier, A.; Gai, W.P.; Meedeniya, A.; Pountney, D.L. Increased SUMO-1 expression in the unilateral rotenone-lesioned mouse model of Parkinson’s disease. Neurosci. Lett. 2013, 544, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.B.; Goodwin, J.; Norazit, A.; Meedeniya, A.C.; Richter-Landsberg, C.; Gai, W.P.; Pountney, D.L. SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox. Res. 2013, 23, 1–21. [Google Scholar] [CrossRef]
- Pountney, D.L.; Chegini, F.; Shen, X.; Blumbergs, P.C.; Gai, W.P. SUMO-1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neurosci. Lett. 2005, 381, 74–79. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and ubiquitination reciprocally regulate alpha-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef]
- Abeywardana, T.; Pratt, M.R. Extent of inhibition of alpha-synuclein aggregation in vitro by SUMOylation is conjugation site- and SUMO isoform-selective. Biochemistry 2015, 54, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kugler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef]
- Vinueza-Gavilanes, R.; Inigo-Marco, I.; Larrea, L.; Lasa, M.; Carte, B.; Santamaria, E.; Fernandez-Irigoyen, J.; Bugallo, R.; Aragon, T.; Aldabe, R.; et al. N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity. Neurobiol. Dis. 2020, 137, 104781. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Roeters, S.J.; Schilderink, N.; Hommersom, B.; Heeren, R.M.; Woutersen, S.; Claessens, M.M.; Subramaniam, V. The impact of N-terminal acetylation of alpha-synuclein on phospholipid membrane binding and fibril structure. J. Biol. Chem. 2016, 291, 21110–21122. [Google Scholar] [CrossRef]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef]
- Runfola, M.; De Simone, A.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. The N-terminal acetylation of alpha-synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci. Rep. 2020, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Bu, B.; Tong, X.; Li, D.; Hu, Y.; He, W.; Zhao, C.; Hu, R.; Li, X.; Shao, Y.; Liu, C.; et al. N-terminal acetylation preserves alpha-synuclein from oligomerization by blocking intermolecular hydrogen bonds. ACS Chem. Neurosci. 2017, 8, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of alpha-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727. [Google Scholar] [CrossRef]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; Gritsenko, M.A.; Rodland, K.D.; Smith, R.D.; Qian, W.J.; Gong, C.X.; et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol. 2017, 243, 78–88. [Google Scholar] [CrossRef]
- Alfaro, J.F.; Gong, C.X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.; Purvine, S.O.; Wang, Z.; Camp, D.G., 2nd; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-specific GlcNAcylation of human erythrocyte proteins: Potential biomarker(s) for diabetes. Diabetes 2009, 58, 309–317. [Google Scholar] [CrossRef]
- Wang, Z.; Udeshi, N.D.; O’Malley, M.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell Proteom. 2010, 9, 153–160. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, H.; Chen, Y.; Ma, Y.T.; Jiang, F.; Tan, J.; Zhang, Y.; Li, J.D. Enzymatic O-GlcNAcylation of alpha-synuclein reduces aggregation and increases SDS-resistant soluble oligomers. Neurosci. Lett. 2017, 655, 90–94. [Google Scholar] [CrossRef]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. alpha-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef]
- Lewis, Y.E.; Galesic, A.; Levine, P.M.; De Leon, C.A.; Lamiri, N.; Brennan, C.K.; Pratt, M.R. O-GlcNAcylation of alpha-synuclein at serine 87 reduces aggregation without affecting membrane binding. ACS Chem. Biol. 2017, 12, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc modification blocks the aggregation and toxicity of the protein alpha-synuclein associated with Parkinson’s disease. Nat. Chem. 2015, 7, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jakala, P.; Hartmann, T.; Price, D.L.; et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167. [Google Scholar] [CrossRef]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Ulusoy, A.; Febbraro, F.; Jensen, P.H.; Kirik, D.; Romero-Ramos, M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur. J. Neurosci. 2010, 32, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.; Yang, S.; Sauchanka, O.; Spillantini, M.G.; Anichtchik, O. Behavioural deficits in transgenic mice expressing human truncated (1-120 amino acid) alpha-synuclein. Exp. Neurol. 2015, 264, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nguyen, L.T.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O.S.; Miller, D.W.; Datta, D.; et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein alpha-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 9587–9592. [Google Scholar] [CrossRef]
- Iyer, A.; Roeters, S.J.; Kogan, V.; Woutersen, S.; Claessens, M.; Subramaniam, V. C-terminal truncated alpha-synuclein fibrils contain strongly twisted beta-sheets. J. Am. Chem. Soc. 2017, 139, 15392–15400. [Google Scholar] [CrossRef]
- Ma, L.; Yang, C.; Zhang, X.; Li, Y.; Wang, S.; Zheng, L.; Huang, K. C-terminal truncation exacerbates the aggregation and cytotoxicity of alpha-Synuclein: A vicious cycle in Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3714–3725. [Google Scholar] [CrossRef] [PubMed]
- Terada, M.; Suzuki, G.; Nonaka, T.; Kametani, F.; Tamaoka, A.; Hasegawa, M. The effect of truncation on prion-like properties of alpha-synuclein. J. Biol. Chem. 2018, 293, 13910–13920. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Garcia Reitbock, P.; Humby, T.; Lambourne, S.L.; O’Connell, M.; Ghetti, B.; Gossage, H.; Emson, P.C.; Wilkinson, L.S.; Goedert, M.; et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): Implications for Lewy body disorders. J. Neurosci. 2006, 26, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 2004, 43, 16233–16242. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.V.; Giasson, B.I.; Quinn, S.M.; Koppaka, V.; Axelsen, P.H.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 2003, 42, 8530–8540. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. The driving force of alpha-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: Key role of ganglioside- and cholesterol-binding domains. Adv. Exp. Med. Biol. 2013, 991, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, S.D.; Hulleman, J.D.; Dutseva, E.A.; Antonenko, Y.N.; Rochet, J.C.; Cramer, W.A. Helical alpha-synuclein forms highly conductive ion channels. Biochemistry 2007, 46, 14369–14379. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef]
- Volles, M.J.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- Stockl, M.T.; Zijlstra, N.; Subramaniam, V. alpha-Synuclein oligomers: An amyloid pore? Insights into mechanisms of alpha-synuclein oligomer-lipid interactions. Mol. Neurobiol. 2013, 47, 613–621. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Fantini, J.; Chahinian, H.; Maresca, M.; Taieb, N.; Yahi, N. Altered ion channel formation by the Parkinson’s-disease-linked E46K mutant of alpha-synuclein is corrected by GM3 but not by GM1 gangliosides. J. Mol. Biol. 2010, 397, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Matsuzaki-Kobayashi, M.; Hasegawa, T.; Kikuchi, A.; Sugeno, N.; Itoyama, Y.; Wang, Y.; Yao, P.J.; Bushlin, I.; Takeda, A. Plasma membrane ion permeability induced by mutant alpha-synuclein contributes to the degeneration of neural cells. J. Neurochem. 2006, 97, 1071–1077. [Google Scholar] [CrossRef]
- Feng, L.R.; Federoff, H.J.; Vicini, S.; Maguire-Zeiss, K.A. Alpha-synuclein mediates alterations in membrane conductance: A potential role for alpha-synuclein oligomers in cell vulnerability. Eur. J. Neurosci. 2010, 32, 10–17. [Google Scholar] [CrossRef]
- Schmidt, F.; Levin, J.; Kamp, F.; Kretzschmar, H.; Giese, A.; Botzel, K. Single-channel electrophysiology reveals a distinct and uniform pore complex formed by alpha-synuclein oligomers in lipid membranes. PLoS ONE 2012, 7, e42545. [Google Scholar] [CrossRef]
- Mironov, S.L. alpha-Synuclein forms non-selective cation channels and stimulates ATP-sensitive potassium channels in hippocampal neurons. J. Physiol. 2015, 593, 145–159. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Lena, C.; et al. Alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423. [Google Scholar] [CrossRef]
- Ronzitti, G.; Bucci, G.; Emanuele, M.; Leo, D.; Sotnikova, T.D.; Mus, L.V.; Soubrane, C.H.; Dallas, M.L.; Thalhammer, A.; Cingolani, L.A.; et al. Exogenous alpha-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 2014, 34, 10603–10615. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 2010, 67, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, B.; Svanstrom, H.; Nielsen, N.M.; Fugger, L.; Melbye, M.; Hviid, A. Use of calcium channel blockers and Parkinson’s disease. Am. J. Epidemiol. 2012, 175, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, T.M.; Patzlaff, N.E.; Grady, S.R.; Heinemann, S.F.; Booker, T.K. alpha4beta2 nicotinic acetylcholine receptors on dopaminergic neurons mediate nicotine reward and anxiety relief. J. Neurosci. 2011, 31, 10891–10902. [Google Scholar] [CrossRef]
- Swant, J.; Goodwin, J.S.; North, A.; Ali, A.A.; Gamble-George, J.; Chirwa, S.; Khoshbouei, H. alpha-Synuclein stimulates a dopamine transporter-dependent chloride current and modulates the activity of the transporter. J. Biol. Chem. 2011, 286, 43933–43943. [Google Scholar] [CrossRef]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine transporter activity is modulated by alpha-synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Wersinger, C.; Vernier, P. alpha-Synuclein regulation of the dopaminergic transporter: A possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 2004, 565, 1–5. [Google Scholar] [CrossRef]
- Dean, E.D.; Li, Y.; Torres, G.E.; Miller, G.W. Identification of a novel interaction between α-synuclein and VMAT2. FASEB J. 2008, 22, 715.6. [Google Scholar]
- Guo, J.T.; Chen, A.Q.; Kong, Q.; Zhu, H.; Ma, C.M.; Qin, C. Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell Mol. Neurobiol. 2008, 28, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef]
- Baptista, M.J.; O’Farrell, C.; Daya, S.; Ahmad, R.; Miller, D.W.; Hardy, J.; Farrer, M.J.; Cookson, M.R. Co-ordinate transcriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastoma cell lines. J. Neurochem. 2003, 85, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zuo, X.; Li, Y.; Zhang, C.; Zhou, M.; Zhang, Y.A.; Ueda, K.; Chan, P. Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci. Lett. 2004, 367, 34–39. [Google Scholar] [CrossRef]
- Li, Y.H.; Gao, N.; Ye, Y.W.; Li, X.; Yu, S.; Yang, H.; Ueda, K.; Chan, P. Alpha-synuclein functions as a negative regulator for expression of tyrosine hydroxylase. Acta Neurol. Belg. 2011, 111, 130–135. [Google Scholar] [PubMed]
- Liu, D.; Jin, L.; Wang, H.; Zhao, H.; Zhao, C.; Duan, C.; Lu, L.; Wu, B.; Yu, S.; Chan, P.; et al. Silencing alpha-synuclein gene expression enhances tyrosine hydroxylase activity in MN9D cells. Neurochem. Res. 2008, 33, 1401–1409. [Google Scholar] [CrossRef]
- Peng, X.; Tehranian, R.; Dietrich, P.; Stefanis, L.; Perez, R.G. Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 2005, 118, 3523–3530. [Google Scholar] [CrossRef]
- Alerte, T.N.; Akinfolarin, A.A.; Friedrich, E.E.; Mader, S.A.; Hong, C.S.; Perez, R.G. Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: Lessons from viral transduction of knockout mice. Neurosci. Lett. 2008, 435, 24–29. [Google Scholar] [CrossRef]
- Lou, H.; Montoya, S.E.; Alerte, T.N.; Wang, J.; Wu, J.; Peng, X.; Hong, C.S.; Friedrich, E.E.; Mader, S.A.; Pedersen, C.J.; et al. Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 2010, 285, 17648–17661. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kordower, J.H. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Chadchankar, H.; Ihalainen, J.; Tanila, H.; Yavich, L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res. 2011, 1382, 37–44. [Google Scholar] [CrossRef]
- Al-Wandi, A.; Ninkina, N.; Millership, S.; Williamson, S.J.; Jones, P.A.; Buchman, V.L. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 2010, 31, 796–804. [Google Scholar] [CrossRef]
- Robertson, D.C.; Schmidt, O.; Ninkina, N.; Jones, P.A.; Sharkey, J.; Buchman, V.L. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J. Neurochem. 2004, 89, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Du, T.; Wang, X.; Duan, C.; Gao, G.; Zhang, J.; Lu, L.; Yang, H. alpha-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res. 2014, 1591, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Robotta, M.; Gerding, H.R.; Vogel, A.; Hauser, K.; Schildknecht, S.; Karreman, C.; Leist, M.; Subramaniam, V.; Drescher, M. Alpha-synuclein binds to the inner membrane of mitochondria in an alpha-helical conformation. ChemBioChem 2014, 15, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Marchesan, E.; Zonta, L.; Bono, F.; Bottani, E.; Longhena, F.; Ziviani, E.; Valerio, A.; Bellucci, A. Alpha-synuclein preserves mitochondrial fusion and function in neuronal cells. Oxid. Med. Cell Longev. 2019, 2019, 4246350. [Google Scholar] [CrossRef]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barcelo-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef]
- Hu, D.; Sun, X.; Liao, X.; Zhang, X.; Zarabi, S.; Schimmer, A.; Hong, Y.; Ford, C.; Luo, Y.; Qi, X. Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol. 2019, 137, 939–960. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. Alpha-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Bir, A.; Sen, O.; Anand, S.; Khemka, V.K.; Banerjee, P.; Cappai, R.; Sahoo, A.; Chakrabarti, S. alpha-Synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: Implications in the pathogenesis of Parkinson’s disease. J. Neurochem. 2014, 131, 868–877. [Google Scholar] [CrossRef]
- Perfeito, R.; Lazaro, D.F.; Outeiro, T.F.; Rego, A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell Neurosci. 2014, 62, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.F. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef]
- Fornai, F.; Schluter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002, 277, 1641–1644. [Google Scholar] [CrossRef]
- Radad, K.; Al-Shraim, M.; Al-Emam, A.; Wang, F.; Kranner, B.; Rausch, W.D.; Moldzio, R. Rotenone: From modelling to implication in Parkinson’s disease. Folia Neuropathol. 2019, 57, 317–326. [Google Scholar] [CrossRef]
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef]
- Loeb, V.; Yakunin, E.; Saada, A.; Sharon, R. The transgenic overexpression of alpha-synuclein and not its related pathology associates with complex I inhibition. J. Biol. Chem. 2010, 285, 7334–7343. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xie, Z.; Turkson, S.; Zhuang, X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci. 2015, 35, 890–905. [Google Scholar] [CrossRef]
- Poon, H.F.; Frasier, M.; Shreve, N.; Calabrese, V.; Wolozin, B.; Butterfield, D.A. Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice—A model of familial Parkinson’s disease. Neurobiol. Dis. 2005, 18, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Nistico, R.; Mehdawy, B.; Piccirilli, S.; Mercuri, N. Paraquat- and rotenone-induced models of Parkinson’s disease. Int. J. Immunopathol. Pharmacol. 2011, 24, 313–322. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Lee, H.J.; Shin, S.Y.; Choi, C.; Lee, Y.H.; Lee, S.J. Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J. Biol. Chem. 2002, 277, 5411–5417. [Google Scholar] [CrossRef] [PubMed]
- Kowall, N.W.; Hantraye, P.; Brouillet, E.; Beal, M.F.; McKee, A.C.; Ferrante, R.J. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 2000, 11, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Chen, Q.; Vila, M.; Giasson, B.I.; Djaldatti, R.; Vukosavic, S.; Souza, J.M.; Jackson-Lewis, V.; Lee, V.M.; Ischiropoulos, H. Oxidative post-translational modifications of alpha-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J. Neurochem. 2001, 76, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Hu, M.; Liu, J.; Zhang, B.; Zhang, Z.; Zhou, F.H.; Wang, L.; Dong, J. Phosphorylation of tau and alpha-synuclein induced neurodegeneration in MPTP mouse model of Parkinson’s disease. Neuropsychiatry Dis. Treat. 2020, 16, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Oh, S.T.; Jeong, H.J.; Pak, S.C.; Park, H.J.; Kim, J.; Cho, H.S.; Jeon, S. MPTP-induced vulnerability of dopamine neurons in A53T alpha-synuclein overexpressed mice with the potential involvement of DJ-1 downregulation. Korean J. Physiol. Pharmacol. 2017, 21, 625–632. [Google Scholar] [CrossRef]
- Bazzu, G.; Calia, G.; Puggioni, G.; Spissu, Y.; Rocchitta, G.; Debetto, P.; Grigoletto, J.; Zusso, M.; Migheli, R.; Serra, P.A.; et al. alpha-Synuclein- and MPTP-generated rodent models of Parkinson’s disease and the study of extracellular striatal dopamine dynamics: A microdialysis approach. CNS Neurol. Disord. Drug Targets 2010, 9, 482–490. [Google Scholar] [CrossRef]
- Grunewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Munchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1843.e1. [Google Scholar] [CrossRef]
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [PubMed]
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. A new role for alpha-synuclein in Parkinson’s disease: Alteration of ER-mitochondrial communication. Mov. Disord. 2015, 30, 1026–1033. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.; Hoogerheide, D.P.; Rovini, A.; Jiang, Z.; Lee, J.C.; Rostovtseva, T.K.; Bezrukov, S.M. Probing membrane association of alpha-synuclein domains with VDAC nanopore reveals unexpected binding pattern. Sci. Rep. 2019, 9, 4580. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. Alpha-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef]
- Hoogerheide, D.P.; Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Mechanism of alpha-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale 2017, 9, 183–192. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, C.; Cai, Q.; Lu, Q.; Duan, C.; Zhu, Y.; Yang, H. Voltage-dependent anion channel involved in the alpha-synuclein-induced dopaminergic neuron toxicity in rats. Acta Biochim. Biophys. Sin. 2013, 45, 170–178. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Zalk, R.; Gincel, D.; Vardi, N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim. Biophys. Acta 2004, 1657, 105–114. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Israelson, A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: Characterization, modulation and possible function. J. Membr. Biol. 2005, 204, 57–66. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef]
- Chu, Y.; Goldman, J.G.; Kelly, L.; He, Y.; Waliczek, T.; Kordower, J.H. Abnormal alpha-synuclein reduces nigral voltage-dependent anion channel 1 in sporadic and experimental Parkinson’s disease. Neurobiol. Dis. 2014, 69, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. Alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Semenkow, S.; Hanaford, A.; Wong, M. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant alpha-synuclein transgenic mice. Neurobiol. Aging 2014, 35, 1132–1152. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox. Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef]
- Baek, J.H.; Whitfield, D.; Howlett, D.; Francis, P.; Bereczki, E.; Ballard, C.; Hortobagyi, T.; Attems, J.; Aarsland, D. Unfolded protein response is activated in Lewy body dementias. Neuropathol. Appl. Neurobiol. 2016, 42, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Yagi-Utsumi, M.; Satoh, T.; Kato, K. Structural basis of redox-dependent substrate binding of protein disulfide isomerase. Sci. Rep. 2015, 5, 13909. [Google Scholar] [CrossRef]
- Honjo, Y.; Ito, H.; Horibe, T.; Takahashi, R.; Kawakami, K. Protein disulfide isomerase immunopositive glial cytoplasmic inclusions in patients with multiple system atrophy. Int. J. Neurosci. 2011, 121, 543–550. [Google Scholar] [CrossRef]
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172. [Google Scholar] [CrossRef]
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol. Cell Proteom. 2007, 6, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Smith, M.; Macer, D.; Webster, P.; Mortara, R. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J. Cell Sci. 1986, 86, 217–232. [Google Scholar] [CrossRef]
- Labrador-Garrido, A.; Cejudo-Guillen, M.; Daturpalli, S.; Leal, M.M.; Klippstein, R.; De Genst, E.J.; Villadiego, J.; Toledo-Aral, J.J.; Dobson, C.M.; Jackson, S.E.; et al. Chaperome screening leads to identification of Grp94/Gp96 and FKBP4/52 as modulators of the alpha-synuclein-elicited immune response. FASEB J. 2016, 30, 564–577. [Google Scholar] [CrossRef]
- Lee, A.S. Mammalian stress response: Induction of the glucose-regulated protein family. Curr. Opin. Cell Biol. 1992, 4, 267–273. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef]
- Dalfo, E.; Gomez-Isla, T.; Rosa, J.L.; Nieto Bodelon, M.; Cuadrado Tejedor, M.; Barrachina, M.; Ambrosio, S.; Ferrer, I. Abnormal alpha-synuclein interactions with Rab proteins in alpha-synuclein A30P transgenic mice. J. Neuropathol. Exp. Neurol. 2004, 63, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Lopes da Fonseca, T.; Eisbach, S.E.; Anduaga, A.M.; Breda, C.; Orcellet, M.L.; Szego, E.M.; Guerreiro, P.; Lazaro, D.F.; Braus, G.H.; et al. alpha-Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation-dependent manner. Neurobiol. Dis. 2014, 70, 149–161. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Breda, C.; Nugent, M.L.; Estranero, J.G.; Kyriacou, C.P.; Outeiro, T.F.; Steinert, J.R.; Giorgini, F. Rab11 modulates alpha-synuclein-mediated defects in synaptic transmission and behaviour. Hum. Mol. Genet. 2015, 24, 1077–1091. [Google Scholar] [CrossRef]
- Fujita, Y.; Ohama, E.; Takatama, M.; Al-Sarraj, S.; Okamoto, K. Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006, 112, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur. J. Neurosci. 2006, 24, 3153–3162. [Google Scholar] [CrossRef]
- Gosavi, N.; Lee, H.J.; Lee, J.S.; Patel, S.; Lee, S.J. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 2002, 277, 48984–48992. [Google Scholar] [CrossRef]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Cali, T.; Gai, W.P.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. PMR1/SPCA Ca2+ pumps and the role of the Golgi apparatus as a Ca2+ store. Pflugers Arch. 2003, 446, 148–153. [Google Scholar] [CrossRef]
- Buttner, S.; Faes, L.; Reichelt, W.N.; Broeskamp, F.; Habernig, L.; Benke, S.; Kourtis, N.; Ruli, D.; Carmona-Gutierrez, D.; Eisenberg, T.; et al. The Ca2+/Mn2+ ion-pump PMR1 links elevation of cytosolic Ca(2+) levels to alpha-synuclein toxicity in Parkinson’s disease models. Cell Death Differ. 2013, 20, 465–477. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Tavernarakis, N. The PMR1 pump in alpha-synuclein toxicity and neurodegeneration. Neurosci. Lett. 2018, 663, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res. 2002, 941, 118–126. [Google Scholar] [CrossRef]
- Yu, S.; Li, X.; Liu, G.; Han, J.; Zhang, C.; Li, Y.; Xu, S.; Liu, C.; Gao, Y.; Yang, H.; et al. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience 2007, 145, 539–555. [Google Scholar] [CrossRef]
- Huang, Z.; Xu, Z.; Wu, Y.; Zhou, Y. Determining nuclear localization of alpha-synuclein in synuclein mouse brains. Neuroscience 2011, 199, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef]
- Ma, K.L.; Song, L.K.; Yuan, Y.H.; Zhang, Y.; Han, N.; Gao, K.; Chen, N.H. The nuclear accumulation of alpha-synuclein is mediated by importin alpha and promotes neurotoxicity by accelerating the cell cycle. Neuropharmacology 2014, 82, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Davidi, D.; Schechter, M.; Elhadi, S.A.; Matatov, A.; Nathanson, L.; Sharon, R. Alpha-synuclein translocates to the nucleus to activate retinoic-acid-dependent gene transcription. iScience 2020, 23, 100910. [Google Scholar] [CrossRef]
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 2009, 160, 796–804. [Google Scholar] [CrossRef]
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Pique, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.B.I.; Liew, H. Sumoylated α-synuclein translocates into the nucleus by karyopherin α6. Mol. Cell. Toxicol. 2019, 15, 103–109. [Google Scholar] [CrossRef]
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The mechanistic role of alpha-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 2020, 29, 3107–3121. [Google Scholar] [CrossRef] [PubMed]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef]
- Liu, X.; Lee, Y.J.; Liou, L.C.; Ren, Q.; Zhang, Z.; Wang, S.; Witt, S.N. Alpha-synuclein functions in the nucleus to protect against hydroxyurea-induced replication stress in yeast. Hum. Mol. Genet. 2011, 20, 3401–3414. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.C.; Bishop, J.F.; Leng, Y.; Chock, P.B.; Chase, T.N.; Mouradian, M.M. Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 1999, 274, 33855–33858. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef]
- Haj-Yahya, M.; Fauvet, B.; Herman-Bachinsky, Y.; Hejjaoui, M.; Bavikar, S.N.; Karthikeyan, S.V.; Ciechanover, A.; Lashuel, H.A.; Brik, A. Synthetic polyubiquitinated alpha-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. USA 2013, 110, 17726–17731. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Layfield, R.; Spillantini, M.G. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001, 509, 22–26. [Google Scholar] [CrossRef]
- Liu, C.W.; Corboy, M.J.; DeMartino, G.N.; Thomas, P.J. Endoproteolytic activity of the proteasome. Science 2003, 299, 408–411. [Google Scholar] [CrossRef]
- Rideout, H.J.; Larsen, K.E.; Sulzer, D.; Stefanis, L. Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J. Neurochem. 2001, 78, 899–908. [Google Scholar] [CrossRef]
- Rideout, H.J.; Stefanis, L. Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol. Cell. Neurosci. 2002, 21, 223–238. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. alpha-Synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Patel, S.; Lee, S.J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 2004, 24, 1888–1896. [Google Scholar] [CrossRef]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [PubMed]
- Issa, A.R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Cherif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Valina, L.D.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef] [PubMed]
- Martin-Clemente, B.; Alvarez-Castelao, B.; Mayo, I.; Sierra, A.B.; Diaz, V.; Milan, M.; Farinas, I.; Gomez-Isla, T.; Ferrer, I.; Castano, J.G. alpha-Synuclein expression levels do not significantly affect proteasome function and expression in mice and stably transfected PC12 cell lines. J. Biol. Chem. 2004, 279, 52984–52990. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by alpha-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef]
- Bentea, E.; Verbruggen, L.; Massie, A. The proteasome inhibition model of Parkinson’s disease. J. Parkinsons Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. Alpha-synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant alpha-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant alpha-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis 2019, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease—Links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef]
- Liu, G.; Chen, M.; Mi, N.; Yang, W.; Li, X.; Wang, P.; Yin, N.; Li, Y.; Yue, F.; Chan, P.; et al. Increased oligomerization and phosphorylation of alpha-synuclein are associated with decreased activity of glucocerebrosidase and protein phosphatase 2A in aging monkey brains. Neurobiol. Aging 2015, 36, 2649–2659. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained systemic glucocerebrosidase inhibition induces brain alpha-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid. Redox. Signal. 2015, 23, 550–564. [Google Scholar] [CrossRef]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Widner, H.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Brundin, P. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov. Disord. 2010, 25, 1091–1096. [Google Scholar] [CrossRef]
- Kordower, J.H.; Dodiya, H.B.; Kordower, A.M.; Terpstra, B.; Paumier, K.; Madhavan, L.; Sortwell, C.; Steece-Collier, K.; Collier, T.J. Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol. Dis. 2011, 43, 552–557. [Google Scholar] [CrossRef]
- Kurowska, Z.; Englund, E.; Widner, H.; Lindvall, O.; Li, J.Y.; Brundin, P. Signs of degeneration in 12–22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [Google Scholar] [CrossRef]
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Abdelmotilib, H.; Maltbie, T.; Delic, V.; Liu, Z.; Hu, X.; Fraser, K.B.; Moehle, M.S.; Stoyka, L.; Anabtawi, N.; Krendelchtchikova, V.; et al. alpha-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol. Dis. 2017, 105, 84–98. [Google Scholar] [CrossRef]
- Gustafsson, G.; Loov, C.; Persson, E.; Lazaro, D.F.; Takeda, S.; Bergstrom, J.; Erlandsson, A.; Sehlin, D.; Balaj, L.; Gyorgy, B.; et al. Secretion and uptake of alpha-synuclein via extracellular vesicles in cultured cells. Cell. Mol. Neurobiol. 2018, 38, 1539–1550. [Google Scholar] [CrossRef]
- Fussi, N.; Hollerhage, M.; Chakroun, T.; Nykanen, N.P.; Rosler, T.W.; Koeglsperger, T.; Wurst, W.; Behrends, C.; Hoglinger, G.U. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018, 9, 757. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef]
- Delenclos, M.; Trendafilova, T.; Mahesh, D.; Baine, A.M.; Moussaud, S.; Yan, I.K.; Patel, T.; McLean, P.J. Investigation of endocytic pathways for the internalization of exosome-associated oligomeric alpha-synuclein. Front. Neurosci. 2017, 11, 172. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Kim, J.; Paik, S.R.; Park, J.H.; Ahn, Y.S.; Chung, K.C. Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J. Biol. Chem. 2001, 276, 27441–27448. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.J.; Paik, S.R.; Chung, K.C.; Kim, J. Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J. Neurochem. 2006, 97, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.C.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Ihse, E.; Yamakado, H.; van Wijk, X.M.; Lawrence, R.; Esko, J.D.; Masliah, E. Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 2017, 7, 9008. [Google Scholar] [CrossRef]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef] [PubMed]
- Karampetsou, M.; Ardah, M.T.; Semitekolou, M.; Polissidis, A.; Samiotaki, M.; Kalomoiri, M.; Majbour, N.; Xanthou, G.; El-Agnaf, O.M.A.; Vekrellis, K. Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 2017, 7, 16533. [Google Scholar] [CrossRef]
- Karpowicz, R.J., Jr.; Haney, C.M.; Mihaila, T.S.; Sandler, R.M.; Petersson, E.J.; Lee, V.M. Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J. Biol. Chem. 2017, 292, 13482–13497. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Decker, S.C.; Riddle, D.M.; Caputo, A.; Zhang, B.; Cole, T.; Caswell, C.; Xie, S.X.; Lee, V.M.Y.; Luk, K.C. Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 2018, 135, 855–875. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef]
- Sacino, A.N.; Brooks, M.; McKinney, A.B.; Thomas, M.A.; Shaw, G.; Golde, T.E.; Giasson, B.I. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J. Neurosci. 2014, 34, 12368–12378. [Google Scholar] [CrossRef]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. Alpha-synuclein (alphaSyn) preformed fibrils induce endogenous alphaSyn aggregation, compromise synaptic activity and enhance synapse loss in cultured excitatory hippocampal neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef]
- Chu, Y.; Kordower, J.H. The prion hypothesis of Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2015, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef]
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.C.; Citron, M.; Biere, A.L. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 1999, 274, 19509–19512. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Kubo, M.; Shimozawa, A.; Akiyama, H.; Hasegawa, M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol. Commun. 2014, 2, 88. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef]
- Mezias, C.; Rey, N.; Brundin, P.; Raj, A. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 2020, 134, 104623. [Google Scholar] [CrossRef]
- Tang, Y.; Das, U.; Scott, D.A.; Roy, S. The slow axonal transport of alpha-synuclein--mechanistic commonalities amongst diverse cytosolic cargoes. Cytoskeleton 2012, 69, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.R.; Hill, J.; Utton, M.A.; Asuni, A.A.; Ackerley, S.; Grierson, A.J.; Miller, C.C.; Davies, A.M.; Buchman, V.L.; Anderton, B.H.; et al. Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J. Cell Sci. 2004, 117, 1017–1024. [Google Scholar] [CrossRef]
- Li, W.; Hoffman, P.N.; Stirling, W.; Price, D.L.; Lee, M.K. Axonal transport of human alpha-synuclein slows with aging but is not affected by familial Parkinson’s disease-linked mutations. J. Neurochem. 2004, 88, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Bohm, K.J.; Winner, B. alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 2013, 288, 21742–21754. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073. [Google Scholar] [CrossRef]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Holec, S.A.M.; Woerman, A.L. Evidence of distinct alpha-synuclein strains underlying disease heterogeneity. Acta Neuropathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; So, R.W.L.; Lau, H.H.C.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.C.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. alpha-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 2020, 23, 21–31. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. Alpha-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Rey, N.L.; Bousset, L.; George, S.; Madaj, Z.; Meyerdirk, L.; Schulz, E.; Steiner, J.A.; Melki, R.; Brundin, P. alpha-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol. Commun. 2019, 7, 221. [Google Scholar] [CrossRef]
- Wake, H.; Fields, R.D. Physiological function of microglia. Neuron Glia Biol. 2011, 7, 1–3. [Google Scholar] [CrossRef][Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Colton, C.; Wilcock, D.M. Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 2010, 9, 174–191. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Streit, W.J.; Conde, J.R.; Fendrick, S.E.; Flanary, B.E.; Mariani, C.L. Role of microglia in the central nervous system’s immune response. Neurol. Res. 2005, 27, 685–691. [Google Scholar] [CrossRef]
- Van Rossum, D.; Hanisch, U.K. Microglia. Metab. Brain Dis. 2004, 19, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Paolo, T.D.; Tremblay, M.E. Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions? Front. Cell Neurosci. 2018, 12, 282. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Vieira, B.D.; Radford, R.A.; Chung, R.S.; Guillemin, G.J.; Pountney, D.L. Neuroinflammation in multiple system atrophy: Response to and cause of alpha-synuclein aggregation. Front. Cell Neurosci. 2015, 9, 437. [Google Scholar] [CrossRef]
- Surendranathan, A.; Su, L.; Mak, E.; Passamonti, L.; Hong, Y.T.; Arnold, R.; Vazquez Rodriguez, P.; Bevan-Jones, W.R.; Brain, S.A.E.; Fryer, T.D.; et al. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 2018, 141, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R. Activated microglia in dementia with Lewy bodies. Neurology 2000, 55, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Mosley, R.L.; Benner, E.J.; Kadiu, I.; Thomas, M.; Boska, M.D.; Hasan, K.; Laurie, C.; Gendelman, H.E. Neuroinflammation, oxidative stress and the pathogenesis of Parkinson’s disease. Clin. Neurosci. Res. 2006, 6, 261–281. [Google Scholar] [CrossRef]
- Appel, S.H.; Beers, D.R.; Henkel, J.S. T cell-microglial dialogue in Parkinson’s disease and amyotrophic lateral sclerosis: Are we listening? Trends Immunol. 2010, 31, 7–17. [Google Scholar] [CrossRef]
- Peterson, L.J.; Flood, P.M. Oxidative stress and microglial cells in Parkinson’s disease. Mediat. Inflamm. 2012, 2012, 401264. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Liu, C.Y.; Wang, X.; Liu, C.; Zhang, H.L. Pharmacological targeting of microglial activation: New therapeutic approach. Front. Cell Neurosci. 2019, 13, 514. [Google Scholar] [CrossRef]
- Austin, S.A.; Floden, A.M.; Murphy, E.J.; Combs, C.K. Alpha-synuclein expression modulates microglial activation phenotype. J. Neurosci. 2006, 26, 10558–10563. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Rojanathammanee, L.; Golovko, M.Y.; Murphy, E.J.; Combs, C.K. Lack of alpha-synuclein modulates microglial phenotype in vitro. Neurochem. Res. 2011, 36, 994–1004. [Google Scholar] [CrossRef]
- Rojanathammanee, L.; Murphy, E.J.; Combs, C.K. Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J. Neuroinflamm. 2011, 8, 44. [Google Scholar] [CrossRef]
- Gardai, S.J.; Mao, W.; Schule, B.; Babcock, M.; Schoebel, S.; Lorenzana, C.; Alexander, J.; Kim, S.; Glick, H.; Hilton, K.; et al. Elevated alpha-synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson’s disease. PLoS ONE 2013, 8, e71634. [Google Scholar] [CrossRef]
- Haenseler, W.; Zambon, F.; Lee, H.; Vowles, J.; Rinaldi, F.; Duggal, G.; Houlden, H.; Gwinn, K.; Wray, S.; Luk, K.C.; et al. Excess alpha-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci. Rep. 2017, 7, 9003. [Google Scholar] [CrossRef]
- Kim, S.; Cho, S.H.; Kim, K.Y.; Shin, K.Y.; Kim, H.S.; Park, C.H.; Chang, K.A.; Lee, S.H.; Cho, D.; Suh, Y.H. Alpha-synuclein induces migration of BV-2 microglial cells by up-regulation of CD44 and MT1-MMP. J. Neurochem. 2009, 109, 1483–1496. [Google Scholar] [CrossRef]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis. Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Sullivan, A.M.; Nolan, Y.M. The influence of microglia on the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2009, 89, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef]
- De Araujo, F.M.; Cuenca-Bermejo, L.; Fernandez-Villalba, E.; Costa, S.L.; Silva, V.D.A.; Herrero, M.T. Role of microgliosis and NLRP3 inflammasome in Parkinson’s disease pathogenesis and therapy. Cell Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Jayaram, S.; Krishnamurthy, P.T. Role of microgliosis, oxidative stress and associated neuroinflammation in the pathogenesis of Parkinson’s disease: The therapeutic role of Nrf2 activators. Neurochem. Int. 2021, 145, 105014. [Google Scholar] [CrossRef]
- Marinova-Mutafchieva, L.; Sadeghian, M.; Broom, L.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: A time course study in a 6-hydroxydopamine model of Parkinson’s disease. J. Neurochem. 2009, 110, 966–975. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.; Doorduin, J.; de Vries, E.F.; Dierckx, R.A.; Leenders, K.L. [11C]-PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat. Disord. 2010, 16, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Barkholt, P.; Sanchez-Guajardo, V.; Kirik, D.; Romero-Ramos, M. Long-term polarization of microglia upon alpha-synuclein overexpression in nonhuman primates. Neuroscience 2012, 208, 85–96. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5, e8784. [Google Scholar] [CrossRef] [PubMed]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef]
- Gomez-Isla, T.; Irizarry, M.C.; Mariash, A.; Cheung, B.; Soto, O.; Schrump, S.; Sondel, J.; Kotilinek, L.; Day, J.; Schwarzschild, M.A.; et al. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol. Aging 2003, 24, 245–258. [Google Scholar] [CrossRef]
- Emmer, K.L.; Waxman, E.A.; Covy, J.P.; Giasson, B.I. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J. Biol. Chem. 2011, 286, 35104–35118. [Google Scholar] [CrossRef]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.F. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Esparcia, P.; Llorens, F.; Carmona, M.; Ferrer, I. Complex deregulation and expression of cytokines and mediators of the immune response in Parkinson’s disease brain is region dependent. Brain Pathol. 2014, 24, 584–598. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhou, Y.; Hong, J.S.; Zhang, J. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, K.; Komori, T.; Sasaki, S.; Arai, N.; Mizutani, T.; Hirose, T. Microglial activation parallels system degeneration in multiple system atrophy. J. Neuropathol. Exp. Neurol. 2004, 63, 43–52. [Google Scholar] [CrossRef]
- Kubler, D.; Wachter, T.; Cabanel, N.; Su, Z.; Turkheimer, F.E.; Dodel, R.; Brooks, D.J.; Oertel, W.H.; Gerhard, A. Widespread microglial activation in multiple system atrophy. Mov. Disord. 2019, 34, 564–568. [Google Scholar] [CrossRef]
- Harms, A.S.; Kordower, J.H.; Sette, A.; Lindestam Arlehamn, C.S.; Sulzer, D.; Mach, R.H. Inflammation in experimental models of alpha-synucleinopathies. Mov. Disord. 2021, 36, 37–49. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Faucheux, B. Glial cells and inflammation in Parkinson’s disease: A role in neurodegeneration? Ann. Neurol. 1998, 44, S115–S120. [Google Scholar] [CrossRef]
- Shavali, S.; Combs, C.K.; Ebadi, M. Reactive macrophages increase oxidative stress and alpha-synuclein nitration during death of dopaminergic neuronal cells in co-culture: Relevance to Parkinson’s disease. Neurochem. Res. 2006, 31, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dallas, S.; Zhang, D.; Guo, J.P.; Pang, H.; Wilson, B.; Miller, D.S.; Chen, B.; McGeer, P.L.; Hong, J.S.; et al. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 2007, 55, 1178–1188. [Google Scholar] [CrossRef]
- Bick, R.J.; Poindexter, B.J.; Kott, M.M.; Liang, Y.A.; Dinh, K.; Kaur, B.; Bick, D.L.; Doursout, M.F.; Schiess, M.C. Cytokines disrupt intracellular patterns of Parkinson’s disease-associated proteins alpha-synuclein, tau and ubiquitin in cultured glial cells. Brain Res. 2008, 1217, 203–212. [Google Scholar] [CrossRef]
- Schiess, M.C.; Barnes, J.L.; Ellmore, T.M.; Poindexter, B.J.; Dinh, K.; Bick, R.J. CSF from Parkinson disease patients differentially affects cultured microglia and astrocytes. BMC Neurosci. 2010, 11, 151. [Google Scholar] [CrossRef]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.; Casaceli, C.; Colla, E.; Lee, M.K.; Palaniappan, C.; Federoff, H.J. Wild-type and mutant alpha-synuclein induce a multi-component gene expression profile consistent with shared pathophysiology in different transgenic mouse models of PD. Exp. Neurol. 2007, 204, 421–432. [Google Scholar] [CrossRef]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.; Anthony, D.C. The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J. Neuroinflamm. 2011, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Wilms, H.; Rosenstiel, P.; Romero-Ramos, M.; Arlt, A.; Schafer, H.; Seegert, D.; Kahle, P.J.; Odoy, S.; Claasen, J.H.; Holzknecht, C.; et al. Suppression of MAP kinases inhibits microglial activation and attenuates neuronal cell death induced by alpha-synuclein protofibrils. Int. J. Immunopathol. Pharmacol. 2009, 22, 897–909. [Google Scholar] [CrossRef]
- Jin, J.; Shie, F.S.; Liu, J.; Wang, Y.; Davis, J.; Schantz, A.M.; Montine, K.S.; Montine, T.J.; Zhang, J. Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J. Neuroinflamm. 2007, 4, 2. [Google Scholar] [CrossRef]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef]
- St Martin, J.L.; Klucken, J.; Outeiro, T.F.; Nguyen, P.; Keller-McGandy, C.; Cantuti-Castelvetri, I.; Grammatopoulos, T.N.; Standaert, D.G.; Hyman, B.T.; McLean, P.J. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J. Neurochem. 2007, 100, 1449–1457. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Couch, Y.; Richardson, J.; Cooper, J.M.; Wood, M.J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 2011, 69, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Paik, S.R.; Jou, I.; Park, S.M. Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: Implications for Parkinson’s disease. Glia 2008, 56, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Park, S.M.; Ahn, K.J.; Chung, K.C.; Paik, S.R.; Kim, J. Identification of the amino acid sequence motif of alpha-synuclein responsible for macrophage activation. Biochem. Biophys. Res. Commun. 2009, 381, 39–43. [Google Scholar] [CrossRef]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef]
- Lee, E.J.; Woo, M.S.; Moon, P.G.; Baek, M.C.; Choi, I.Y.; Kim, W.K.; Junn, E.; Kim, H.S. Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Roodveldt, C.; Labrador-Garrido, A.; Gonzalez-Rey, E.; Fernandez-Montesinos, R.; Caro, M.; Lachaud, C.C.; Waudby, C.A.; Delgado, M.; Dobson, C.M.; Pozo, D. Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: Differences between wild-type and Parkinson’s disease-linked mutants. PLoS ONE 2010, 5, e13481. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Stewart, T.; Sheng, L.; Shi, M.; Cilento, E.M.; Wu, Y.; Hong, J.S.; Zhang, J. Immunoregulation of microglial polarization: An unrecognized physiological function of alpha-synuclein. J. Neuroinflamm. 2020, 17, 272. [Google Scholar] [CrossRef]
- Reynolds, A.D.; Glanzer, J.G.; Kadiu, I.; Ricardo-Dukelow, M.; Chaudhuri, A.; Ciborowski, P.; Cerny, R.; Gelman, B.; Thomas, M.P.; Mosley, R.L.; et al. Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J. Neurochem. 2008, 104, 1504–1525. [Google Scholar] [CrossRef]
- Beraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Ton, V.; Neitzke, K.; Chasovskikh, S.; Mhyre, T.R.; Maguire-Zeiss, K.A. Alpha-synuclein alters toll-like receptor expression. Front. Neurosci. 2011, 5, 80. [Google Scholar] [CrossRef]
- Thomas, M.P.; Chartrand, K.; Reynolds, A.; Vitvitsky, V.; Banerjee, R.; Gendelman, H.E. Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: Relevance for the pathogenesis of Parkinson’s disease. J. Neurochem. 2007, 100, 503–519. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ettle, B.; Bruno, A.; Kulinich, A.; Hoffmann, A.C.; von Wittgenstein, J.; Winkler, J.; Xiang, W.; Schlachetzki, J.C.M. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem. Biophys. Res. Commun. 2016, 479, 881–886. [Google Scholar] [CrossRef]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-synuclein proteins promote pro-inflammatory cascades in microglia: Stronger effects of the A53T mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Niu, M.; Zhao, A.; Kang, W.; Chen, Z.; Luo, N.; Zhou, L.; Zhu, X.; Lu, L.; Liu, J. CXCL12 is involved in alpha-synuclein-triggered neuroinflammation of Parkinson’s disease. J. Neuroinflamm. 2019, 16, 263. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Wang, Y.; Fong, H.; Murray, T.M.; Zhang, J. Identification of proteins involved in microglial endocytosis of alpha-synuclein. J. Proteome Res. 2007, 6, 3614–3627. [Google Scholar] [CrossRef]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox. Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Hou, L.; Qu, X.; Qiu, X.; Huang, R.; Zhao, X.; Wang, Q. Integrin CD11b mediates locus coeruleus noradrenergic neurodegeneration in a mouse Parkinson’s disease model. J. Neuroinflamm. 2020, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chu, C.H.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.S.; Zhang, J. alpha-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Federoff, H.J.; Maguire-Zeiss, K.A. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res. 2009, 16, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in alpha-synuclein—Mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef]
- Cao, S.; Standaert, D.G.; Harms, A.S. The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflamm. 2012, 9, 259. [Google Scholar] [CrossRef]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef]
- Venezia, S.; Refolo, V.; Polissidis, A.; Stefanis, L.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 stimulation with monophosphoryl lipid A ameliorates motor deficits and nigral neurodegeneration triggered by extraneuronal alpha-synucleinopathy. Mol. Neurodegener. 2017, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017, 133, 303–319. [Google Scholar] [CrossRef]
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.J.; et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha-synuclein transmission and neuroinflammation. Mol. Neurodegener. 2018, 13, 43. [Google Scholar] [CrossRef]
- Kwon, S.; Iba, M.; Masliah, E.; Kim, C. Targeting microglial and neuronal toll-like receptor 2 in synucleinopathies. Exp. Neurobiol. 2019, 28, 547–553. [Google Scholar] [CrossRef]
- Caplan, I.F.; Maguire-Zeiss, K.A. Toll-like receptor 2 signaling and current approaches for therapeutic modulation in synucleinopathies. Front. Pharmacol. 2018, 9, 417. [Google Scholar] [CrossRef]
- Roodveldt, C.; Labrador-Garrido, A.; Gonzalez-Rey, E.; Lachaud, C.C.; Guilliams, T.; Fernandez-Montesinos, R.; Benitez-Rondan, A.; Robledo, G.; Hmadcha, A.; Delgado, M.; et al. Preconditioning of microglia by alpha-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS ONE 2013, 8, e79160. [Google Scholar] [CrossRef]
- Reynolds, A.D.; Kadiu, I.; Garg, S.K.; Glanzer, J.G.; Nordgren, T.; Ciborowski, P.; Banerjee, R.; Gendelman, H.E. Nitrated alpha-synuclein and microglial neuroregulatory activities. J. Neuroimmune Pharmacol. 2008, 3, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Beraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, K.; Chapman, G.D.; Gunasekar, P.G. alpha-Synuclein overexpression enhances manganese-induced neurotoxicity through the NF-kappaB-mediated pathway. Toxicol. Mech. Methods 2011, 21, 435–443. [Google Scholar] [CrossRef]
- Maekawa, T.; Sasaoka, T.; Azuma, S.; Ichikawa, T.; Melrose, H.L.; Farrer, M.J.; Obata, F. Leucine-rich repeat kinase 2 (LRRK2) regulates alpha-synuclein clearance in microglia. BMC Neurosci. 2016, 17, 77. [Google Scholar] [CrossRef]
- Daher, J.P.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef]
- Beraud, D.; Hathaway, H.A.; Trecki, J.; Chasovskikh, S.; Johnson, D.A.; Johnson, J.A.; Federoff, H.J.; Shimoji, M.; Mhyre, T.R.; Maguire-Zeiss, K.A. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein alpha-synuclein. J. Neuroimmune Pharmacol. 2013, 8, 94–117. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rabano, A.; Kirik, D.; Cuadrado, A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Hwang, V.; Ando, D.M.; Daub, A.; Lee, A.K.; Ravisankar, A.; Modan, S.; Finucane, M.M.; Shaby, B.A.; Finkbeiner, S. Nrf2 mitigates LRRK2- and alpha-synuclein-induced neurodegeneration by modulating proteostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 1165–1170. [Google Scholar] [CrossRef]
- Subbarayan, M.S.; Hudson, C.; Moss, L.D.; Nash, K.R.; Bickford, P.C. T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an alpha-synuclein rat model of Parkinson’s disease. J. Neuroinflamm. 2020, 17, 242. [Google Scholar] [CrossRef]
- Harms, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standaert, D.G. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef]
- Williams, G.P.; Marmion, D.J.; Schonhoff, A.M.; Jurkuvenaite, A.; Won, W.J.; Standaert, D.G.; Kordower, J.H.; Harms, A.S. T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol. 2020, 139, 855–874. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. Alpha-synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef]
- Jimenez-Ferrer, I.; Backstrom, F.; Duenas-Rey, A.; Jewett, M.; Boza-Serrano, A.; Luk, K.C.; Deierborg, T.; Swanberg, M. The MHC class II transactivator modulates seeded alpha-synuclein pathology and dopaminergic neurodegeneration in an in vivo rat model of Parkinson’s disease. Brain Behav. Immun. 2021, 91, 369–382. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef]
- Wu, K.C.; Liou, H.H.; Kao, Y.H.; Lee, C.Y.; Lin, C.J. The critical role of Nramp1 in degrading alpha-synuclein oligomers in microglia under iron overload condition. Neurobiol. Dis. 2017, 104, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Bussi, C.; Peralta Ramos, J.M.; Arroyo, D.S.; Gallea, J.I.; Ronchi, P.; Kolovou, A.; Wang, J.M.; Florey, O.; Celej, M.S.; Schwab, Y.; et al. Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef]
- Scheffold, A.; Holtman, I.R.; Dieni, S.; Brouwer, N.; Katz, S.F.; Jebaraj, B.M.; Kahle, P.J.; Hengerer, B.; Lechel, A.; Stilgenbauer, S.; et al. Telomere shortening leads to an acceleration of synucleinopathy and impaired microglia response in a genetic mouse model. Acta Neuropathol. Commun. 2016, 4, 87. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On the mechanism of internalization of alpha-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Deitmer, J.W.; Theparambil, S.M.; Ruminot, I.; Noor, S.I.; Becker, H.M. Energy dynamics in the brain: Contributions of astrocytes to metabolism and pH homeostasis. Front. Neurosci. 2019, 13, 1301. [Google Scholar] [CrossRef]
- Dienel, G.A.; Carlson, G.M. Major advances in brain glycogen research: Understanding of the roles of glycogen Have evolved from emergency fuel reserve to dynamic, regulated participant in diverse brain functions. Adv. Neurobiol. 2019, 23, 1–16. [Google Scholar] [CrossRef]
- Halassa, M.M.; Haydon, P.G. Integrated brain circuits: Astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 2010, 72, 335–355. [Google Scholar] [CrossRef]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K.; Nedergaard, M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 2010, 7, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Seth, P.; Koul, N. Astrocyte, the star avatar: Redefined. J. Biosci. 2008, 33, 405–421. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Mulder, M. Sterols in the central nervous system. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 152–158. [Google Scholar] [CrossRef]
- Kiray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283, 541–549. [Google Scholar] [CrossRef]
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and microglia as major players of myelin production in normal and pathological conditions. Front. Cell. Neurosci. 2020, 14, 79. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter release from astrocytes: Functional, developmental, and pathological implications in the brain. Front. Neurosci. 2015, 9, 499. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Sun, M.Y.; Murphy, T.; Myers, T.; Lauderdale, K.; Fiacco, T.A. Calcium signaling and gliotransmission in normal vs. reactive astrocytes. Front. Pharmacol. 2012, 3, 139. [Google Scholar] [CrossRef]
- Tabata, H. Diverse subtypes of astrocytes and their development during corticogenesis. Front. Neurosci. 2015, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, A.; Baltazar, G.; Santos, P.; Carvalho, C.M.; Duarte, E.P. Selective injury to dopaminergic neurons up-regulates GDNF in substantia nigra postnatal cell cultures: Role of neuron-glia crosstalk. Neurobiol. Dis. 2006, 23, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Gardaneh, M.; Iwasiow, R.; Lanthier, P.; Gangaraju, S.; Ribecco-Lutkiewicz, M.; Tremblay, R.; Kiuchi, K.; Sikorska, M. Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol. Dis. 2009, 33, 405–414. [Google Scholar] [CrossRef]
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp. Neurol. 2002, 176, 98–104. [Google Scholar] [CrossRef]
- Tanji, K.; Imaizumi, T.; Yoshida, H.; Mori, F.; Yoshimoto, M.; Satoh, K.; Wakabayashi, K. Expression of alpha-synuclein in a human glioma cell line and its up-regulation by interleukin-1beta. Neuroreport 2001, 12, 1909–1912. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Trombetta, L.D. The induction of amyloid precursor protein and alpha-synuclein in rat hippocampal astrocytes by diethyldithiocarbamate and copper with or without glutathione. Toxicol. Lett. 2004, 146, 139–149. [Google Scholar] [CrossRef]
- Stevenson, T.J.; Murray, H.C.; Turner, C.; Faull, R.L.M.; Dieriks, B.V.; Curtis, M.A. alpha-synuclein inclusions are abundant in non-neuronal cells in the anterior olfactory nucleus of the Parkinson’s disease olfactory bulb. Sci. Rep. 2020, 10, 6682. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. alpha-Synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. 2019, 138, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.S.; Mori, F.; Hayashi, S.; Tanji, K.; Yoshimoto, M.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Alpha-synuclein pathology affecting Bergmann glia of the cerebellum in patients with alpha-synucleinopathies. Acta Neuropathol. 2003, 105, 403–409. [Google Scholar] [CrossRef]
- Hishikawa, N.; Hashizume, Y.; Yoshida, M.; Sobue, G. Widespread occurrence of argyrophilic glial inclusions in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 362–372. [Google Scholar] [CrossRef]
- Terada, S.; Ishizu, H.; Yokota, O.; Tsuchiya, K.; Nakashima, H.; Ishihara, T.; Fujita, D.; Ueda, K.; Ikeda, K.; Kuroda, S. Glial involvement in diffuse Lewy body disease. Acta Neuropathol. 2003, 105, 163–169. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Goodwin, M.S.; Riffe, C.J.; Dhillon, J.S.; Xia, Y.; Gorion, K.M.; Vijayaraghavan, N.; McFarland, K.N.; Golbe, L.I.; Yachnis, A.T.; et al. Unique alpha-synuclein pathology within the amygdala in Lewy body dementia: Implications for disease initiation and progression. Acta Neuropathol. Commun. 2019, 7, 142. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Ding, Z.B.; Song, L.J.; Wang, Q.; Kumar, G.; Yan, Y.Q.; Ma, C.G. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural. Regen. Res. 2021, 16, 1702–1710. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-synuclein: From early synaptic dysfunction to neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.M.; Chakrabarty, P.; Saha, K.; Khoshbouei, H.; Golde, T.E.; Giasson, B.I. Proteolysis of alpha-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J. Neurochem. 2017, 140, 662–678. [Google Scholar] [CrossRef]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.T.; Ureshino, R.P.; et al. Overexpression of alpha-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Knott, C.; Stern, G.; Kingsbury, A.; Welcher, A.A.; Wilkin, G.P. Elevated glial brain-derived neurotrophic factor in Parkinson’s diseased nigra. Parkinsonism Relat. Disord. 2002, 8, 329–341. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef]
- Mythri, R.B.; Venkateshappa, C.; Harish, G.; Mahadevan, A.; Muthane, U.B.; Yasha, T.C.; Srinivas Bharath, M.M.; Shankar, S.K. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem. Res. 2011, 36, 1452–1463. [Google Scholar] [CrossRef]
- Hua, J.; Yin, N.; Xu, S.; Chen, Q.; Tao, T.; Zhang, J.; Ding, J.; Fan, Y.; Hu, G. Enhancing the astrocytic clearance of extracellular alpha-synuclein aggregates by ginkgolides attenuates neural cell injury. Cell Mol. Neurobiol. 2019, 39, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Munoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Thomas, M.A.; Ceballos-Diaz, C.; Cruz, P.E.; Rosario, A.M.; Lewis, J.; Giasson, B.I.; Golde, T.E. Conformational templating of alpha-synuclein aggregates in neuronal-glial cultures. Mol. Neurodegener. 2013, 8, 17. [Google Scholar] [CrossRef]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F.; et al. In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Xia, Y.; Funk, C.; Riffe, C.J.; Rutherford, N.J.; Ceballos Diaz, C.; Sacino, A.N.; Price, N.D.; Golde, T.E.; Giasson, B.I.; et al. Motor neuron loss and neuroinflammation in a model of alpha-synuclein-induced neurodegeneration. Neurobiol. Dis. 2018, 120, 98–106. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Brooks, M.M.T.; Hudson, V., 3rd; Rutherford, N.J.; Golde, T.E.; Giasson, B.I.; Chakrabarty, P. Intrastriatal injection of alpha-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol. Neurodegener. 2017, 12, 40. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef]
- Rostami, J.; Holmqvist, S.; Lindstrom, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergstrom, J.; Roybon, L.; Erlandsson, A. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Karampetsou, M.; Sykioti, V.S.; Leandrou, E.; Melachroinou, K.; Lambiris, A.; Giannelos, A.; Emmanouilidou, E.; Vekrellis, K. Intrastriatal administration of exosome-associated pathological alpha-synuclein is not sufficient by itself to cause pathology transmission. Front. Neurosci 2020, 14, 246. [Google Scholar] [CrossRef]
- Fan, R.Z.; Guo, M.; Luo, S.; Cui, M.; Tieu, K. Exosome release and neuropathology induced by alpha-synuclein: New insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol. Commun. 2019, 7, 184. [Google Scholar] [CrossRef]
- Niu, M.; Li, Y.; Li, G.; Zhou, L.; Luo, N.; Yao, M.; Kang, W.; Liu, J. A longitudinal study on alpha-synuclein in plasma neuronal exosomes as a biomarker for Parkinson’s disease development and progression. Eur. J. Neurol. 2020, 27, 967–974. [Google Scholar] [CrossRef]
- Pascua-Maestro, R.; Gonzalez, E.; Lillo, C.; Ganfornina, M.D.; Falcon-Perez, J.M.; Sanchez, D. Extracellular vesicles secreted by astroglial cells transport apolipoprotein D to neurons and mediate neuronal survival upon oxidative stress. Front. Cell Neurosci. 2018, 12, 526. [Google Scholar] [CrossRef]
- Shakespear, N.; Ogura, M.; Yamaki, J.; Homma, Y. Astrocyte-Derived Exosomal microRNA miR-200a-3p Prevents MPP(+)-induced apoptotic cell death through down-regulation of MKK4. Neurochem. Res. 2020, 45, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Ding, J.; Li, C.; Fan, H.; He, Y.; Qiu, P. Transfer of pathological alpha-synuclein from neurons to astrocytes via exosomes causes inflammatory responses after METH exposure. Toxicol. Lett. 2020, 331, 188–199. [Google Scholar] [CrossRef]
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R.A. Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006, 20, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Stefanova, N.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Reindl, M. Glial cell death induced by overexpression of alpha-synuclein. J. Neurosci. Res. 2001, 65, 432–438. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Koob, A.O.; Paulino, A.D.; Masliah, E. GFAP reactivity, apolipoprotein E redistribution and cholesterol reduction in human astrocytes treated with alpha-synuclein. Neurosci Lett 2010, 469, 11–14. [Google Scholar] [CrossRef]
- Chavarria, C.; Rodriguez-Bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169. [Google Scholar] [CrossRef]
- Choi, D.K.; Pennathur, S.; Perier, C.; Tieu, K.; Teismann, P.; Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Vonsattel, J.P.; Heinecke, J.W.; et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J. Neurosci. 2005, 25, 6594–6600. [Google Scholar] [CrossRef] [PubMed]
- Hashioka, S.; Klegeris, A.; Schwab, C.; Yu, S.; McGeer, P.L. Differential expression of interferon-gamma receptor on human glial cells in vivo and in vitro. J. Neuroimmunol. 2010, 225, 91–99. [Google Scholar] [CrossRef]
- Barcia, C.; Ros, C.M.; Annese, V.; Gomez, A.; Ros-Bernal, F.; Aguado-Yera, D.; Martinez-Pagan, M.E.; de Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. IFN-gamma signaling, with the synergistic contribution of TNF-alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2011, 2, e142. [Google Scholar] [CrossRef]
- Diaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzun, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the alpha-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar] [CrossRef]
- Sheng, L.; Stewart, T.; Yang, D.; Thorland, E.; Soltys, D.; Aro, P.; Khrisat, T.; Xie, Z.; Li, N.; Liu, Z.; et al. Erythrocytic alpha-synuclein contained in microvesicles regulates astrocytic glutamate homeostasis: A new perspective on Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Qin, L.; Wang, L.; Tan, J.; Zhang, H.; Tang, J.; Shen, X.; Tan, L.; Wang, C. alphasynuclein induces apoptosis of astrocytes by causing dysfunction of the endoplasmic reticulumGolgi compartment. Mol. Med. Rep. 2018, 18, 322–332. [Google Scholar] [CrossRef]
- Lindstrom, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergstrom, J.; Erlandsson, A. Extensive uptake of alpha-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef]
- Jo, M.; Kim, J.H.; Song, G.J.; Seo, M.; Hwang, E.M.; Suk, K. Astrocytic Orosomucoid-2 Modulates Microglial Activation and Neuroinflammation. J. Neurosci. 2017, 37, 2878–2894. [Google Scholar] [CrossRef]
- Rohl, C.; Sievers, J. Microglia is activated by astrocytes in trimethyltin intoxication. Toxicol. Appl. Pharmacol. 2005, 204, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, C.; Lee, S.J. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxid. Med. Cell Longev. 2010, 3, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Di Marco Vieira, B.; Radford, R.A.W.; Hayashi, J.; Eaton, E.D.; Greenaway, B.; Jambas, M.; Petcu, E.B.; Chung, R.S.; Pountney, D.L. Extracellular alpha-synuclein promotes a neuroinhibitory secretory phenotype in astrocytes. Life 2020, 10, 183. [Google Scholar] [CrossRef]
- Miklossy, J.; Doudet, D.D.; Schwab, C.; Yu, S.; McGeer, E.G.; McGeer, P.L. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 2006, 197, 275–283. [Google Scholar] [CrossRef]
- Barry, M.; Bradford, L.I.M.; David, A.; Hume, C.P.; Neil, A. Mabbott. Complete microglia deficiency accelerates prion disease without enhancing CNS prion accumulation. Cell Rep. 2021. [Google Scholar] [CrossRef]
- Watanabe, H.; Saito, Y.; Terao, S.; Ando, T.; Kachi, T.; Mukai, E.; Aiba, I.; Abe, Y.; Tamakoshi, A.; Doyu, M.; et al. Progression and prognosis in multiple system atrophy: An analysis of 230 Japanese patients. Brain 2002, 125, 1070–1083. [Google Scholar] [CrossRef]
- Ozawa, T.; Paviour, D.; Quinn, N.P.; Josephs, K.A.; Sangha, H.; Kilford, L.; Healy, D.G.; Wood, N.W.; Lees, A.J.; Holton, J.L.; et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: Clinicopathological correlations. Brain 2004, 127, 2657–2671. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Seppi, K.; Wenning, G.K. Grading of neuropathology in multiple system atrophy: Proposal for a novel scale. Mov. Disord. 2005, 20 (Suppl. S12), S29–S36. [Google Scholar] [CrossRef]
- Radford, R.; Rcom-H’cheo-Gauthier, A.; Wong, M.B.; Eaton, E.D.; Quilty, M.; Blizzard, C.; Norazit, A.; Meedeniya, A.; Vickers, J.C.; Gai, W.P.; et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to alpha-synuclein inclusions. Mol. Cell Neurosci. 2015, 65, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Mori, F.; Kon, T.; Tanji, K.; Miki, Y.; Tomiyama, M.; Kurotaki, H.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; et al. Accumulation of phosphorylated alpha-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology 2016, 36, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Rockenstein, E.; Crews, L.; Adame, A.; Mante, M.; Larrea, G.; Hashimoto, M.; Song, D.; Iwatsubo, T.; Tsuboi, K.; et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: Implications for multiple system atrophy. J. Neurosci. 2005, 25, 10689–10699. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Ubhi, K.; Mante, M.; Rockenstein, E.; Masliah, E. Antidepressants reduce neuroinflammatory responses and astroglial alpha-synuclein accumulation in a transgenic mouse model of multiple system atrophy. Glia 2014, 62, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, I.; Giasson, B.I.; Sasaki, R.; Zhang, B.; Joyce, S.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 2005, 45, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Reindl, M.; Neumann, M.; Haass, C.; Poewe, W.; Kahle, P.J.; Wenning, G.K. Oxidative stress in transgenic mice with oligodendroglial alpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am. J. Pathol. 2005, 166, 869–876. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef]
- Bean, B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8, 451–465. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in development, myelin generation and beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and axonal support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Rodriguez-Rodriguez, R.; Gaebler, A.; Casals, N.; Scheller, A.; Kuerschner, L. Astrocytes and oligodendrocytes in grey and white matter regions of the brain metabolize fatty acids. Sci. Rep. 2017, 7, 10779. [Google Scholar] [CrossRef]
- Dulamea, A.O. Role of oligodendrocyte dysfunction in demyelination, remyelination and neurodegeneration in multiple sclerosis. Adv. Exp. Med. Biol. 2017, 958, 91–127. [Google Scholar] [CrossRef]
- Wong, J.H.; Halliday, G.M.; Kim, W.S. Exploring myelin dysfunction in multiple system atrophy. Exp. Neurobiol. 2014, 23, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Jellinger, K.A.; Wenning, G.K.; Stefanova, N. Glial dysfunction in the pathogenesis of alpha-synucleinopathies: Emerging concepts. Acta Neuropathol. 2011, 121, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; McLean, C.A.; Culvenor, J.G.; Gai, W.P.; Blumbergs, P.C.; Jakala, P.; Beyreuther, K.; Masters, C.L.; Li, Q.X. The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J. Neurochem. 2001, 76, 87–96. [Google Scholar] [CrossRef]
- Yamada, T.; McGeer, P.L.; McGeer, E.G. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992, 84, 100–104. [Google Scholar] [CrossRef]
- Yamada, T.; McGeer, P.L.; McGeer, E.G. Some immunohistochemical features of argyrophilic grain dementia with normal cortical choline acetyltransferase levels but extensive subcortical pathology and markedly reduced dopamine. J. Geriatr. Psychiatry Neurol. 1992, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol. Aging 2004, 25, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 2009, 201, 1–119. [Google Scholar]
- Takagi, S.; Hayakawa, N.; Kimoto, H.; Kato, H.; Araki, T. Damage to oligodendrocytes in the striatum after MPTP neurotoxicity in mice. J. Neural. Transm. 2007, 114, 1553–1557. [Google Scholar] [CrossRef]
- Wenning, G.K.; Stefanova, N.; Jellinger, K.A.; Poewe, W.; Schlossmacher, M.G. Multiple system atrophy: A primary oligodendrogliopathy. Ann. Neurol. 2008, 64, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Okuizumi, K.; Ikeuchi, T.; Wakabayashi, K.; Takahashi, H.; Tsuji, S. Analysis of the expression level of alpha-synuclein mRNA using postmortem brain samples from pathologically confirmed cases of multiple system atrophy. Acta Neuropathol. 2001, 102, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.I.; Lantos, P.L. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain 1994, 117, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Lantos, P.L. Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: An update. Acta Neuropathol. 2010, 119, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.I.; Lantos, P.L. Accumulation of tubular structures in oligodendroglial and neuronal cells as the basic alteration in multiple system atrophy. J. Neurol. Sci. 1992, 107, 172–182. [Google Scholar] [CrossRef]
- Yoshida, M. Multiple system atrophy: Alpha-synuclein and neuronal degeneration. Neuropathology 2007, 27, 484–493. [Google Scholar] [CrossRef]
- Takeda, A.; Arai, N.; Komori, T.; Kato, S.; Oda, M. Neuronal inclusions in the dentate fascia in patients with multiple system atrophy. Neurosci. Lett. 1997, 227, 157–160. [Google Scholar] [CrossRef]
- Papp, M.I.; Kahn, J.E.; Lantos, P.L. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J. Neurol. Sci. 1989, 94, 79–100. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Gai, W.P.; Pountney, D.L.; Power, J.H.; Li, Q.X.; Culvenor, J.G.; McLean, C.A.; Jensen, P.H.; Blumbergs, P.C. alpha-Synuclein fibrils constitute the central core of oligodendroglial inclusion filaments in multiple system atrophy. Exp. Neurol. 2003, 181, 68–78. [Google Scholar] [CrossRef]
- McCormack, A.; Chegeni, N.; Chegini, F.; Colella, A.; Power, J.; Keating, D.; Chataway, T. Purification of alpha-synuclein containing inclusions from human post mortem brain tissue. J. Neurosci. Methods 2016, 266, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Riedel, M.; Goldbaum, O.; Richter-Landsberg, C. alpha-Synuclein promotes the recruitment of tau to protein inclusions in oligodendroglial cells: Effects of oxidative and proteolytic stress. J. Mol. Neurosci. 2009, 39, 226–234. [Google Scholar] [CrossRef]
- Skjoerringe, T.; Lundvig, D.M.; Jensen, P.H.; Moos, T. P25alpha/Tubulin polymerization promoting protein expression by myelinating oligodendrocytes of the developing rat brain. J. Neurochem. 2006, 99, 333–342. [Google Scholar] [CrossRef]
- Takahashi, M.; Tomizawa, K.; Fujita, S.C.; Sato, K.; Uchida, T.; Imahori, K. A brain-specific protein p25 is localized and associated with oligodendrocytes, neuropil, and fiber-like structures of the CA3 hippocampal region in the rat brain. J. Neurochem. 1993, 60, 228–235. [Google Scholar] [CrossRef]
- Otzen, D.E.; Lundvig, D.M.; Wimmer, R.; Nielsen, L.H.; Pedersen, J.R.; Jensen, P.H. p25alpha is flexible but natively folded and binds tubulin with oligomeric stoichiometry. Protein Sci. 2005, 14, 1396–1409. [Google Scholar] [CrossRef]
- Lindersson, E.; Lundvig, D.; Petersen, C.; Madsen, P.; Nyengaard, J.R.; Hojrup, P.; Moos, T.; Otzen, D.; Gai, W.P.; Blumbergs, P.C.; et al. p25alpha Stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synucleinopathies. J. Biol. Chem. 2005, 280, 5703–5715. [Google Scholar] [CrossRef] [PubMed]
- Ejlerskov, P.; Rasmussen, I.; Nielsen, T.T.; Bergstrom, A.L.; Tohyama, Y.; Jensen, P.H.; Vilhardt, F. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J. Biol. Chem. 2013, 288, 17313–17335. [Google Scholar] [CrossRef] [PubMed]
- Kragh, C.L.; Fillon, G.; Gysbers, A.; Hansen, H.D.; Neumann, M.; Richter-Landsberg, C.; Haass, C.; Zalc, B.; Lubetzki, C.; Gai, W.P.; et al. FAS-dependent cell death in alpha-synuclein transgenic oligodendrocyte models of multiple system atrophy. PLoS ONE 2013, 8, e55243. [Google Scholar] [CrossRef]
- Kragh, C.L.; Lund, L.B.; Febbraro, F.; Hansen, H.D.; Gai, W.P.; El-Agnaf, O.; Richter-Landsberg, C.; Jensen, P.H. Alpha-synuclein aggregation and Ser-129 phosphorylation-dependent cell death in oligodendroglial cells. J. Biol. Chem. 2009, 284, 10211–10222. [Google Scholar] [CrossRef]
- Mavroeidi, P.; Arvanitaki, F.; Karakitsou, A.K.; Vetsi, M.; Kloukina, I.; Zweckstetter, M.; Giller, K.; Becker, S.; Sorrentino, Z.A.; Giasson, B.I.; et al. Endogenous oligodendroglial alpha-synuclein and TPPP/p25alpha orchestrate alpha-synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol. 2019, 138, 415–441. [Google Scholar] [CrossRef] [PubMed]
- Ubhi, K.; Rockenstein, E.; Mante, M.; Inglis, C.; Adame, A.; Patrick, C.; Whitney, K.; Masliah, E. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J. Neurosci. 2010, 30, 6236–6246. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Takahashi, H. Cellular pathology in multiple system atrophy. Neuropathology 2006, 26, 338–345. [Google Scholar] [CrossRef]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.S.; Goldwurm, S.; Frisen, J.; Deierborg, T.; Roybon, L. Alpha-synuclein expression in the oligodendrocyte lineage: An in vitro and in vivo study using rodent and human models. Stem Cell Rep. 2015, 5, 174–184. [Google Scholar] [CrossRef]
- Asi, Y.T.; Simpson, J.E.; Heath, P.R.; Wharton, S.B.; Lees, A.J.; Revesz, T.; Houlden, H.; Holton, J.L. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia 2014, 62, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014, 62, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.; Goldbaum, O.; Bergmann, M.; Probst-Cousin, S.; Richter-Landsberg, C. Involvement of macroautophagy in multiple system atrophy and protein aggregate formation in oligodendrocytes. J. Mol. Neurosci. 2012, 47, 256–266. [Google Scholar] [CrossRef]
- Rockenstein, E.; Ubhi, K.; Inglis, C.; Mante, M.; Patrick, C.; Adame, A.; Masliah, E. Neuronal to oligodendroglial alpha-synuclein redistribution in a double transgenic model of multiple system atrophy. Neuroreport 2012, 23, 259–264. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Ahn, W.J.; Ricarte, D.; Ortiz, D.; Shin, C.Y.; Lee, S.J.; Lee, H.J. Alpha-Synuclein Inclusion Formation in Human Oligodendrocytes. Biomol. Ther. 2021, 29, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergstrom, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. Alpha-synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef]
- Yamada, K.; Iwatsubo, T. Extracellular alpha-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 2018, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Kisos, H.; Pukass, K.; Ben-Hur, T.; Richter-Landsberg, C.; Sharon, R. Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS ONE 2012, 7, e46817. [Google Scholar] [CrossRef]
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.C.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38. [Google Scholar] [CrossRef]
- Pukass, K.; Richter-Landsberg, C. Oxidative stress promotes uptake, accumulation, and oligomerization of extracellular alpha-synuclein in oligodendrocytes. J. Mol. Neurosci. 2014, 52, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeis, C.; Frohlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Mobius, W.; Goebbels, S.; Nave, K.A.; et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef]
- Ghidoni, R.; Benussi, L.; Binetti, G. Exosomes: The Trojan horses of neurodegeneration. Med. Hypotheses 2008, 70, 1226–1227. [Google Scholar] [CrossRef]
- Nakamura, S.; Kawamoto, Y.; Nakano, S.; Akiguchi, I. Expression of the endocytosis regulatory proteins Rab5 and Rabaptin-5 in glial cytoplasmic inclusions from brains with multiple system atrophy. Clin. Neuropathol. 2000, 19, 51–56. [Google Scholar]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef]
- Woerman, A.L.; Oehler, A.; Kazmi, S.A.; Lee, J.; Halliday, G.M.; Middleton, L.T.; Gentleman, S.M.; Mordes, D.A.; Spina, S.; Grinberg, L.T.; et al. Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol. 2019, 137, 437–454. [Google Scholar] [CrossRef]
- Bernis, M.E.; Babila, J.T.; Breid, S.; Wusten, K.A.; Wullner, U.; Tamguney, G. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol. Commun. 2015, 3, 75. [Google Scholar] [CrossRef]
- Woerman, A.L.; Stohr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef] [PubMed]
- Kaji, S.; Maki, T.; Ishimoto, T.; Yamakado, H.; Takahashi, R. Insights into the pathogenesis of multiple system atrophy: Focus on glial cytoplasmic inclusions. Transl. Neurodegener. 2020, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.F.; Sackmann, C.; Hoffmann, A.; Svenningsson, P.; Winkler, J.; Ingelsson, M.; Hallbeck, M. Binding of alpha-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol. 2019, 138, 23–47. [Google Scholar] [CrossRef] [PubMed]
- Kaji, S.; Maki, T.; Kinoshita, H.; Uemura, N.; Ayaki, T.; Kawamoto, Y.; Furuta, T.; Urushitani, M.; Hasegawa, M.; Kinoshita, Y.; et al. Pathological endogenous alpha-synuclein accumulation in oligodendrocyte precursor cells potentially induces inclusions in multiple system atrophy. Stem Cell Rep. 2018, 10, 356–365. [Google Scholar] [CrossRef]
- Uemura, N.; Uemura, M.T.; Lo, A.; Bassil, F.; Zhang, B.; Luk, K.C.; Lee, V.M.; Takahashi, R.; Trojanowski, J.Q. Slow progressive accumulation of oligodendroglial alpha-synuclein (alpha-Syn) pathology in synthetic alpha-Syn Fibril-Induced Mouse Models of Synucleinopathy. J. Neuropathol Exp. Neurol 2019, 78, 877–890. [Google Scholar] [CrossRef]
- Ettle, B.; Reiprich, S.; Deusser, J.; Schlachetzki, J.C.; Xiang, W.; Prots, I.; Masliah, E.; Winner, B.; Wegner, M.; Winkler, J. Intracellular alpha-synuclein affects early maturation of primary oligodendrocyte progenitor cells. Mol. Cell Neurosci. 2014, 62, 68–78. [Google Scholar] [CrossRef]
- May, V.E.; Ettle, B.; Poehler, A.M.; Nuber, S.; Ubhi, K.; Rockenstein, E.; Winner, B.; Wegner, M.; Masliah, E.; Winkler, J. alpha-Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol. Aging 2014, 35, 2357–2368. [Google Scholar] [CrossRef]
- Ettle, B.; Kerman, B.E.; Valera, E.; Gillmann, C.; Schlachetzki, J.C.; Reiprich, S.; Buttner, C.; Ekici, A.B.; Reis, A.; Wegner, M.; et al. alpha-Synuclein-induced myelination deficit defines a novel interventional target for multiple system atrophy. Acta Neuropathol. 2016, 132, 59–75. [Google Scholar] [CrossRef]
- Kaji, S.; Maki, T.; Ueda, J.; Ishimoto, T.; Inoue, Y.; Yasuda, K.; Sawamura, M.; Hikawa, R.; Ayaki, T.; Yamakado, H.; et al. BCAS1-positive immature oligodendrocytes are affected by the alpha-synuclein-induced pathology of multiple system atrophy. Acta Neuropathol. Commun. 2020, 8, 120. [Google Scholar] [CrossRef]
- Terni, B.; Rey, M.J.; Boluda, S.; Torrejon-Escribano, B.; Sabate, M.P.; Calopa, M.; van Leeuwen, F.W.; Ferrer, I. Mutant ubiquitin and p62 immunoreactivity in cases of combined multiple system atrophy and Alzheimer’s disease. Acta Neuropathol. 2007, 113, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Odagiri, S.; Maruyama, A.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins in the brain of multiple system atrophy. Neurobiol. Dis. 2013, 49, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Odagiri, S.; Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in alpha-synucleinopathy. Acta Neuropathol. 2012, 124, 173–186. [Google Scholar] [CrossRef]
- Mori, F.; Nishie, M.; Piao, Y.S.; Kito, K.; Kamitani, T.; Takahashi, H.; Wakabayashi, K. Accumulation of NEDD8 in neuronal and glial inclusions of neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2005, 31, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology 2011, 31, 561–568. [Google Scholar] [CrossRef]
- Miki, Y.; Tanji, K.; Mori, F.; Tatara, Y.; Utsumi, J.; Sasaki, H.; Kakita, A.; Takahashi, H.; Fimia, G.M.; Wakabayashi, K. AMBRA1, a novel alpha-synuclein-binding protein, is implicated in the pathogenesis of multiple system atrophy. Brain Pathol. 2018, 28, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Pukass, K.; Richter-Landsberg, C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents alpha-synuclein aggregate formation by activating the autophagic pathway: Implications for multiple system atrophy. Front. Cell Neurosci. 2015, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Pukass, K.; Goldbaum, O.; Richter-Landsberg, C. Mitochondrial impairment and oxidative stress compromise autophagosomal degradation of alpha-synuclein in oligodendroglial cells. J. Neurochem. 2015, 135, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Valera, E.; Rockenstein, E.; Trejo-Morales, M.; Adame, A.; Masliah, E. A brain-targeted, modified neurosin (kallikrein-6) reduces alpha-synuclein accumulation in a mouse model of multiple system atrophy. Mol. Neurodegener. 2015, 10, 48. [Google Scholar] [CrossRef]
- Kiely, A.P.; Miners, J.S.; Courtney, R.; Strand, C.; Love, S.; Holton, J.L. Exploring the putative role of kallikrein-6, calpain-1 and cathepsin-D in the proteolytic degradation of alpha-synuclein in multiple system atrophy. Neuropathol. Appl. Neurobiol. 2019, 45, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Maruyama, M.; Akagi, T.; Hashikawa, T.; Kanazawa, I.; Tsuji, S.; Nukina, N. Alpha-synuclein degradation by serine protease neurosin: Implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 2003, 12, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Kaufmann, W.A.; Humpel, C.; Poewe, W.; Wenning, G.K. Systemic proteasome inhibition triggers neurodegeneration in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: Implications for multiple system atrophy. Acta Neuropathol. 2012, 124, 51–65. [Google Scholar] [CrossRef][Green Version]
- Cohlberg, J.A.; Li, J.; Uversky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Maiza, A.; Chantepie, S.; Vera, C.; Fifre, A.; Huynh, M.B.; Stettler, O.; Ouidja, M.O.; Papy-Garcia, D. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 2018, 592, 3806–3818. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, G.F.; Mendes, A.; Castro, R.A.; Bau, E.C.; Nader, H.B.; Dietrich, C.P. Distribution of sulfated glycosaminoglycans in the animal kingdom: Widespread occurrence of heparin-like compounds in invertebrates. Biochim. Biophys. Acta 2000, 1475, 287–294. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Skaanning, L.K.; Santoro, A.; Skamris, T.; Martinsen, J.H.; D’Ursi, A.M.; Bucciarelli, S.; Vestergaard, B.; Bugge, K.; Langkilde, A.E.; Kragelund, B.B. The non-fibrillating N-terminal of alpha-synuclein binds and co-fibrillates with heparin. Biomolecules 2020, 10, 1192. [Google Scholar] [CrossRef]
- Hudak, A.; Kusz, E.; Domonkos, I.; Josvay, K.; Kodamullil, A.T.; Szilak, L.; Hofmann-Apitius, M.; Letoha, T. Contribution of syndecans to cellular uptake and fibrillation of alpha-synuclein and tau. Sci. Rep. 2019, 9, 16543. [Google Scholar] [CrossRef] [PubMed]
- Lehri-Boufala, S.; Ouidja, M.O.; Barbier-Chassefiere, V.; Henault, E.; Raisman-Vozari, R.; Garrigue-Antar, L.; Papy-Garcia, D.; Morin, C. New roles of glycosaminoglycans in alpha-synuclein aggregation in a cellular model of Parkinson disease. PLoS ONE 2015, 10, e0116641. [Google Scholar] [CrossRef]
- Stefanova, N.; Schanda, K.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Reindl, M. Tumor necrosis factor-alpha-induced cell death in U373 cells overexpressing alpha-synuclein. J. Neurosci. Res. 2003, 73, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Grzesiak, J.J.; Bouvet, M.; Hashimoto, M.; Masliah, E.; Shults, C.W. Alpha-synuclein overexpression in oligodendrocytic cells results in impaired adhesion to fibronectin and cell death. Mol. Cell Neurosci. 2005, 29, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kuzdas, D.; Stemberger, S.; Gaburro, S.; Stefanova, N.; Singewald, N.; Wenning, G.K. Oligodendroglial alpha-synucleinopathy and MSA-like cardiovascular autonomic failure: Experimental evidence. Exp. Neurol. 2013, 247, 531–536. [Google Scholar] [CrossRef][Green Version]
- Stefanova, N.; Reindl, M.; Neumann, M.; Kahle, P.J.; Poewe, W.; Wenning, G.K. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: Implications for multiple system atrophy. Mov. Disord. 2007, 22, 2196–2203. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavroeidi, P.; Xilouri, M. Neurons and Glia Interplay in α-Synucleinopathies. Int. J. Mol. Sci. 2021, 22, 4994. https://doi.org/10.3390/ijms22094994
Mavroeidi P, Xilouri M. Neurons and Glia Interplay in α-Synucleinopathies. International Journal of Molecular Sciences. 2021; 22(9):4994. https://doi.org/10.3390/ijms22094994
Chicago/Turabian StyleMavroeidi, Panagiota, and Maria Xilouri. 2021. "Neurons and Glia Interplay in α-Synucleinopathies" International Journal of Molecular Sciences 22, no. 9: 4994. https://doi.org/10.3390/ijms22094994
APA StyleMavroeidi, P., & Xilouri, M. (2021). Neurons and Glia Interplay in α-Synucleinopathies. International Journal of Molecular Sciences, 22(9), 4994. https://doi.org/10.3390/ijms22094994