
SUL-151 Decreases Airway Neutrophilia as a Prophylactic and Therapeutic Treatment in Mice after Cigarette Smoke Exposure

 , , , , , and
, , , , , and 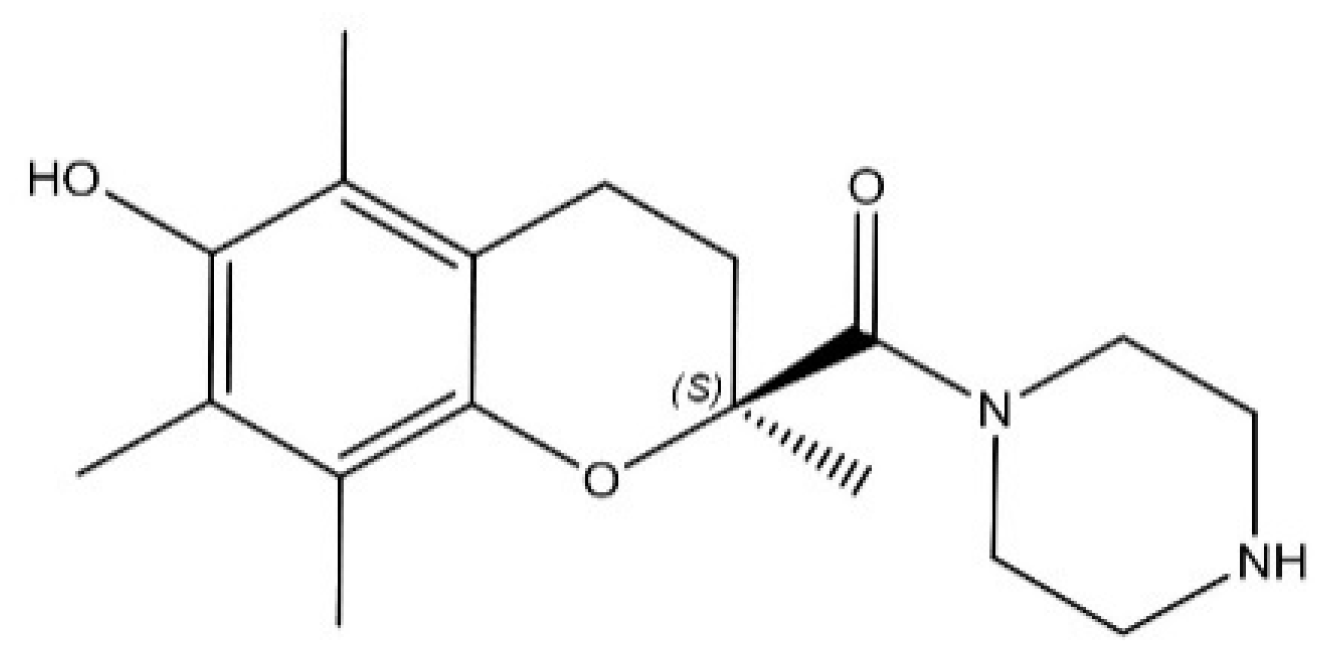{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
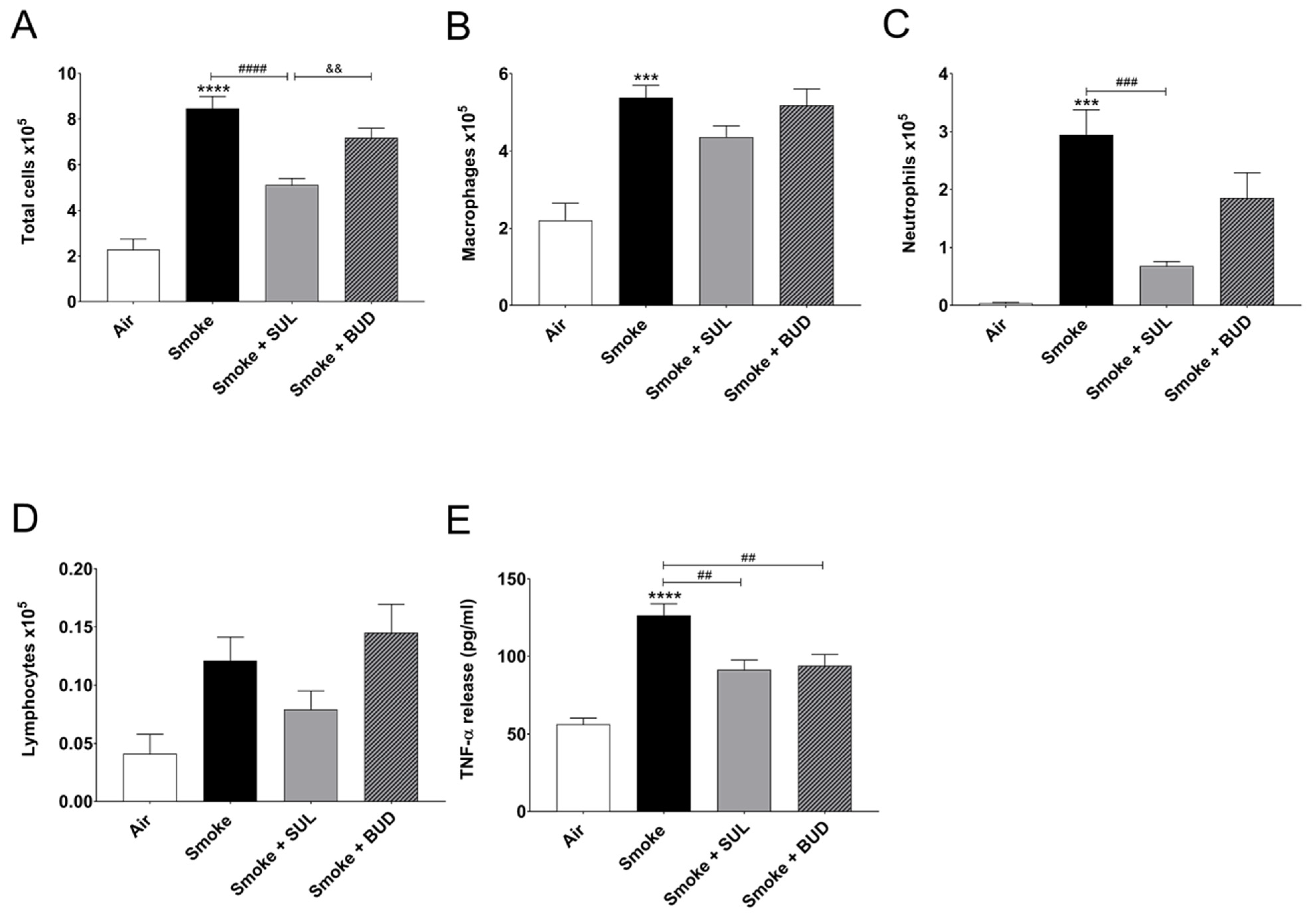
2.1. Prophylactic Treatment of SUL-151 Prevents Pulmonary Inflammation in a CS Exposure Model
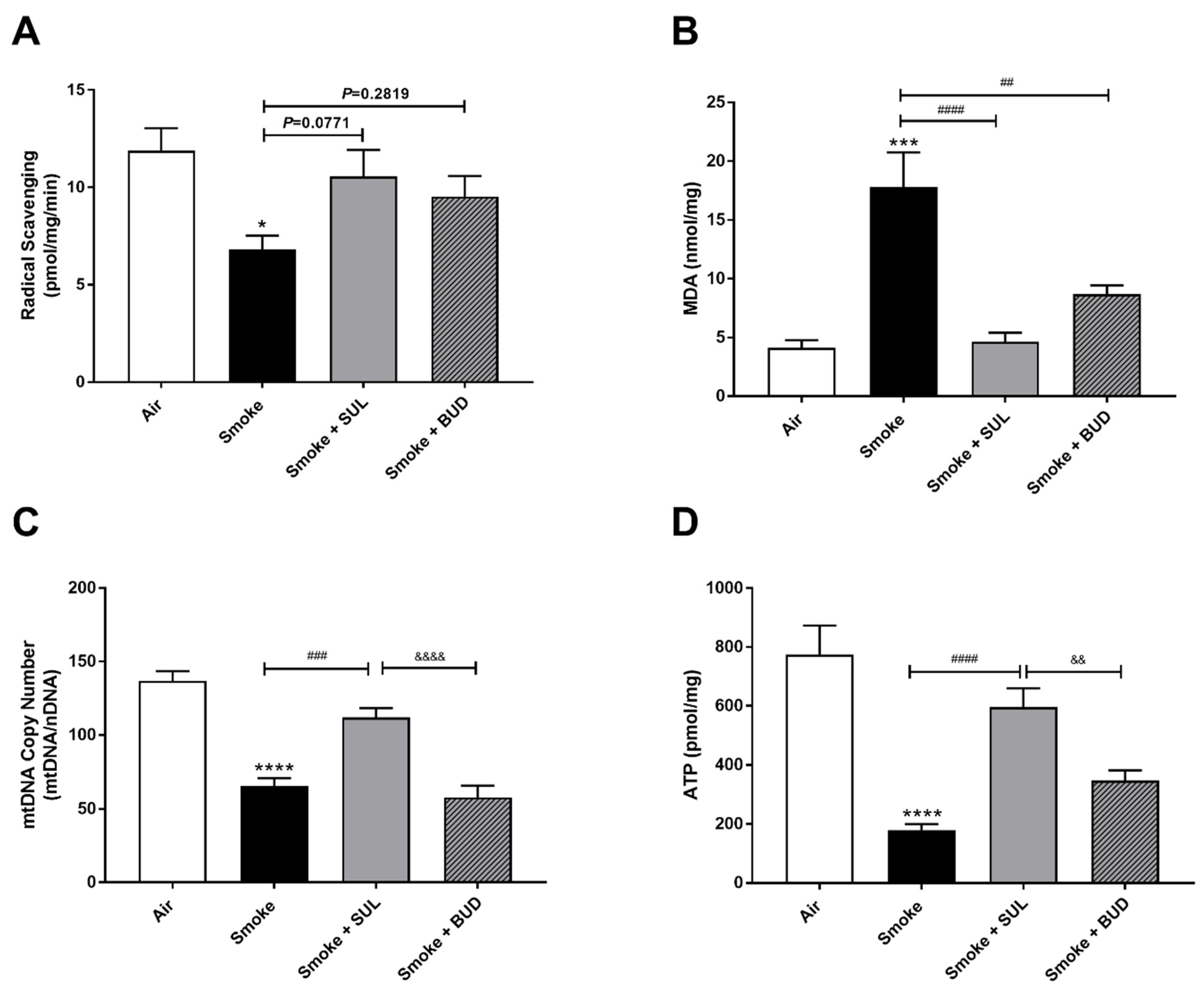
2.2. Prophylactic Treatment of SUL-151 Prevents Oxidative Stress in the Lungs of CS-Exposed Mice
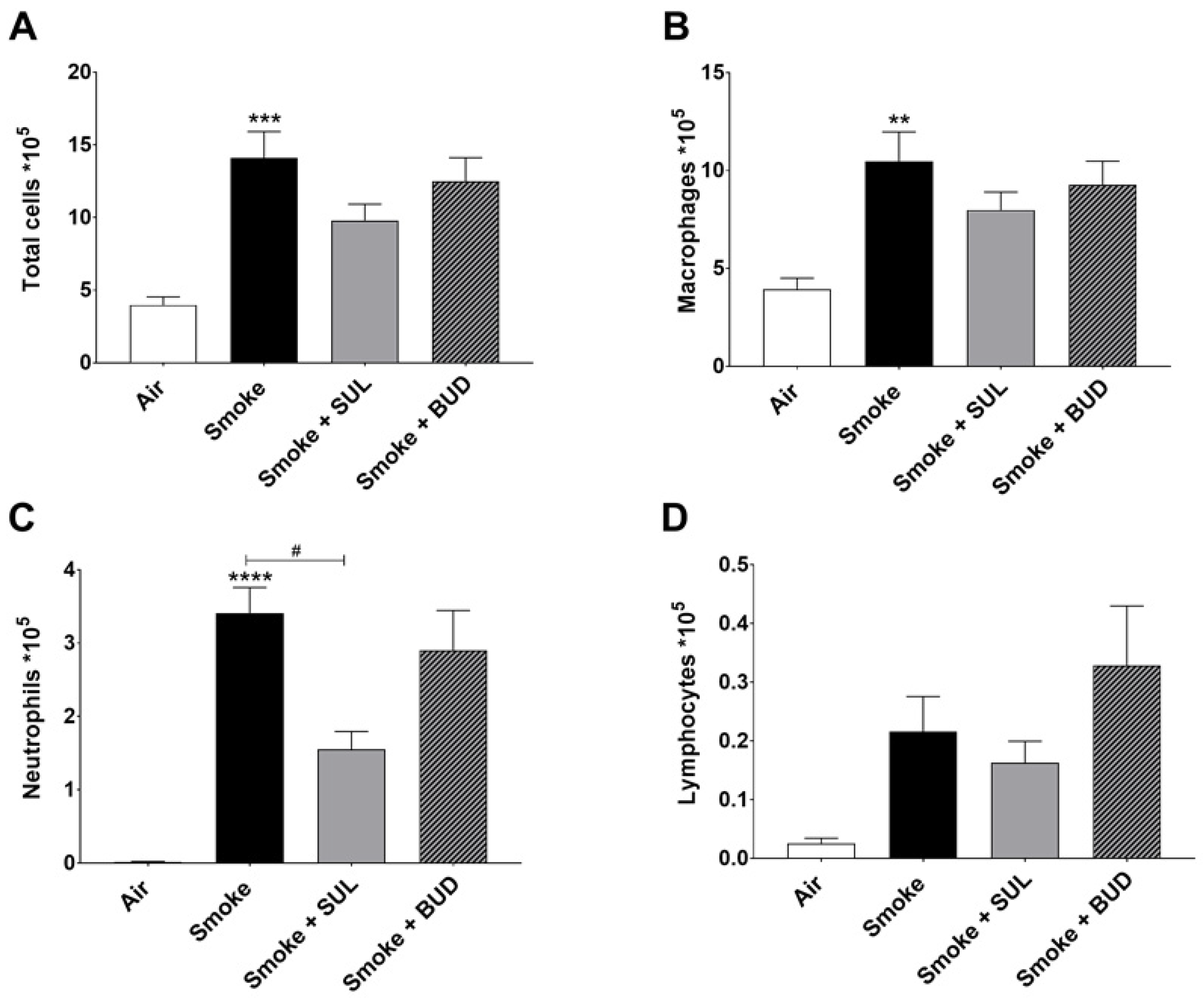
2.3. SUL-151 Reduces the Influx of Neutrophils in the BALF after the Development of CS-Induced Pulmonary Inflammation
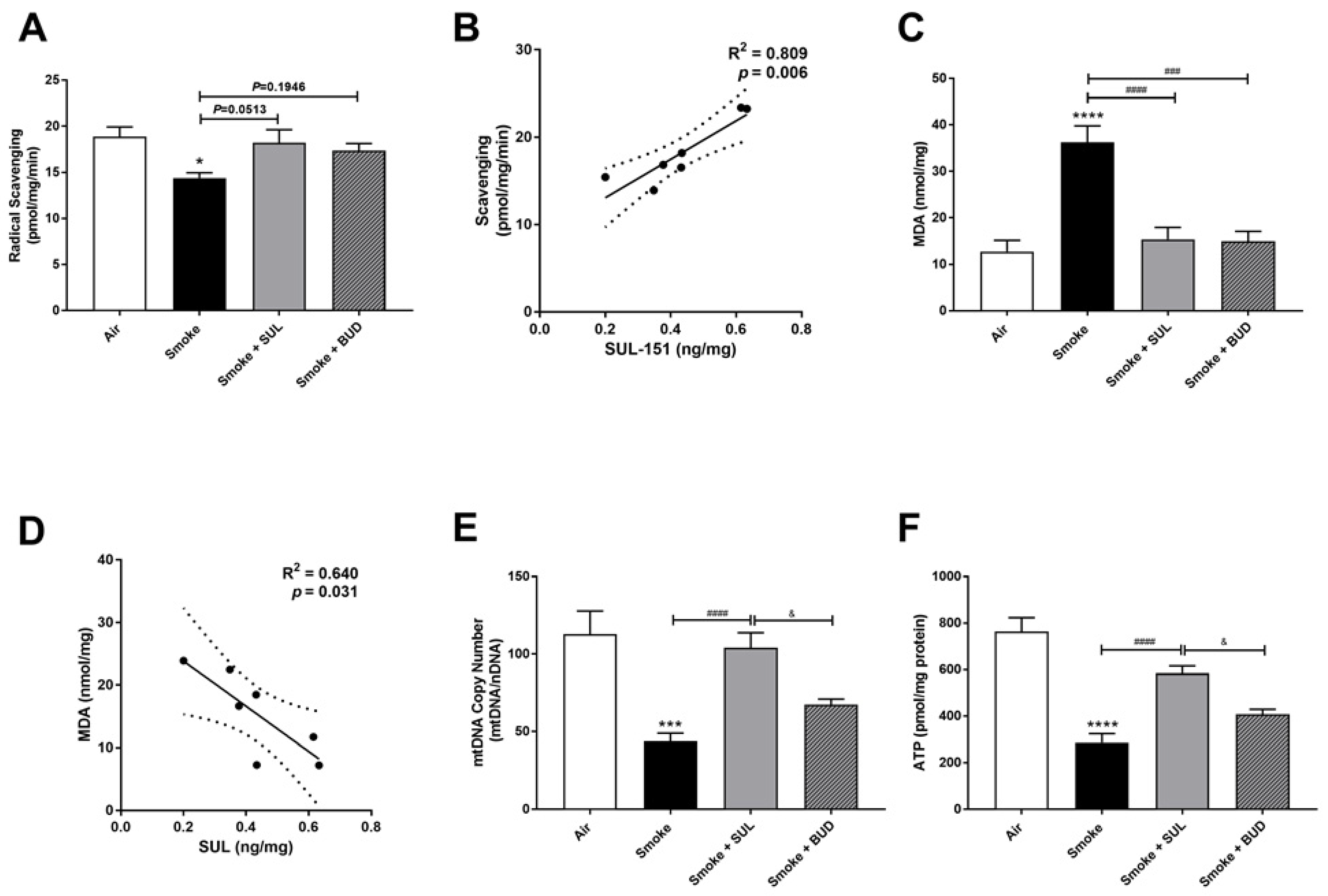
2.4. SUL-151 Reduces the Oxidative Stress in the Lungs after the Development of CS-Induced Pulmonary Inflammation
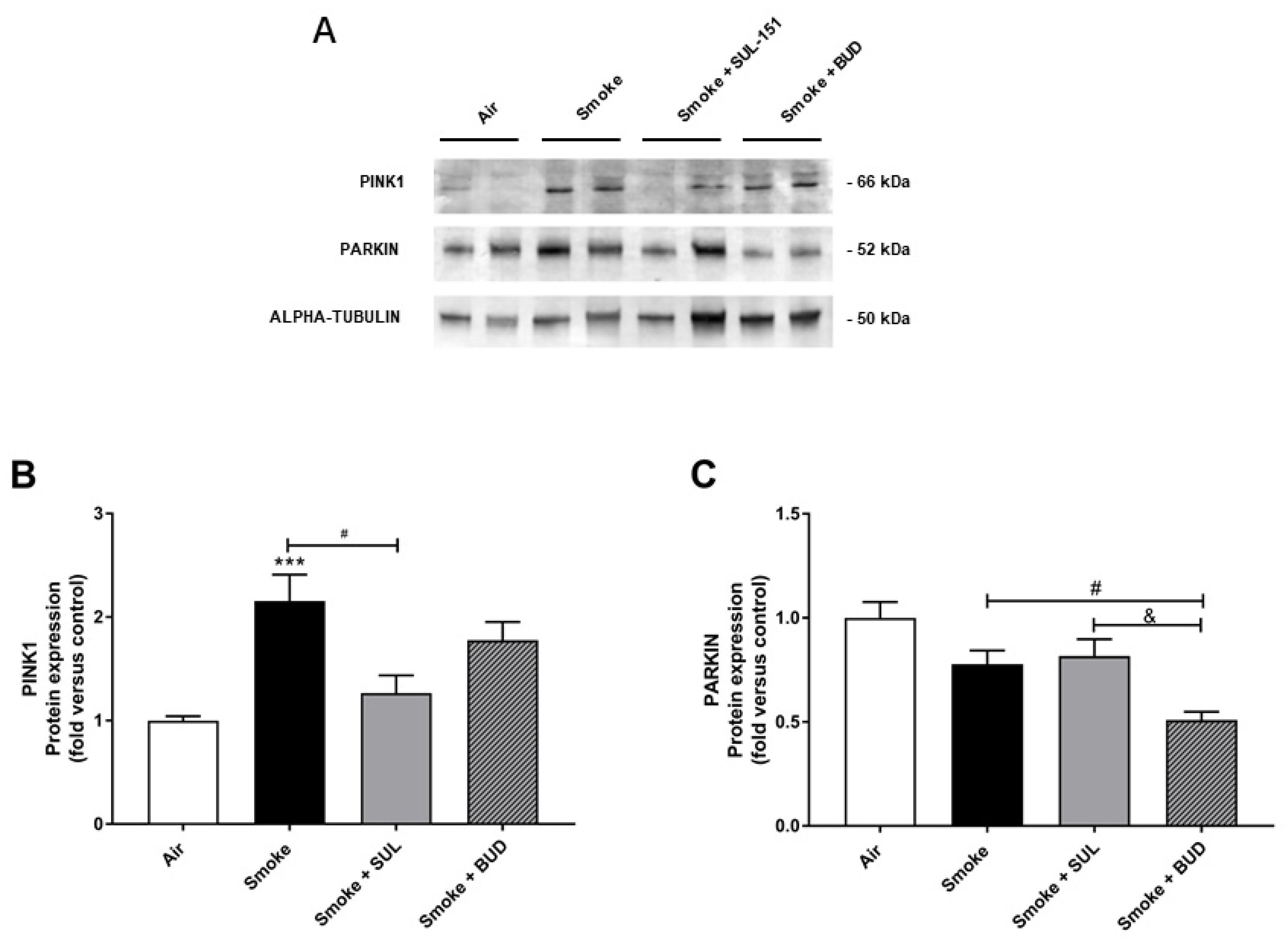
2.5. SUL-151 Inhibits the Increase in PINK1-Expression in the Lungs after the Development of CS-Induced Pulmonary Inflammation
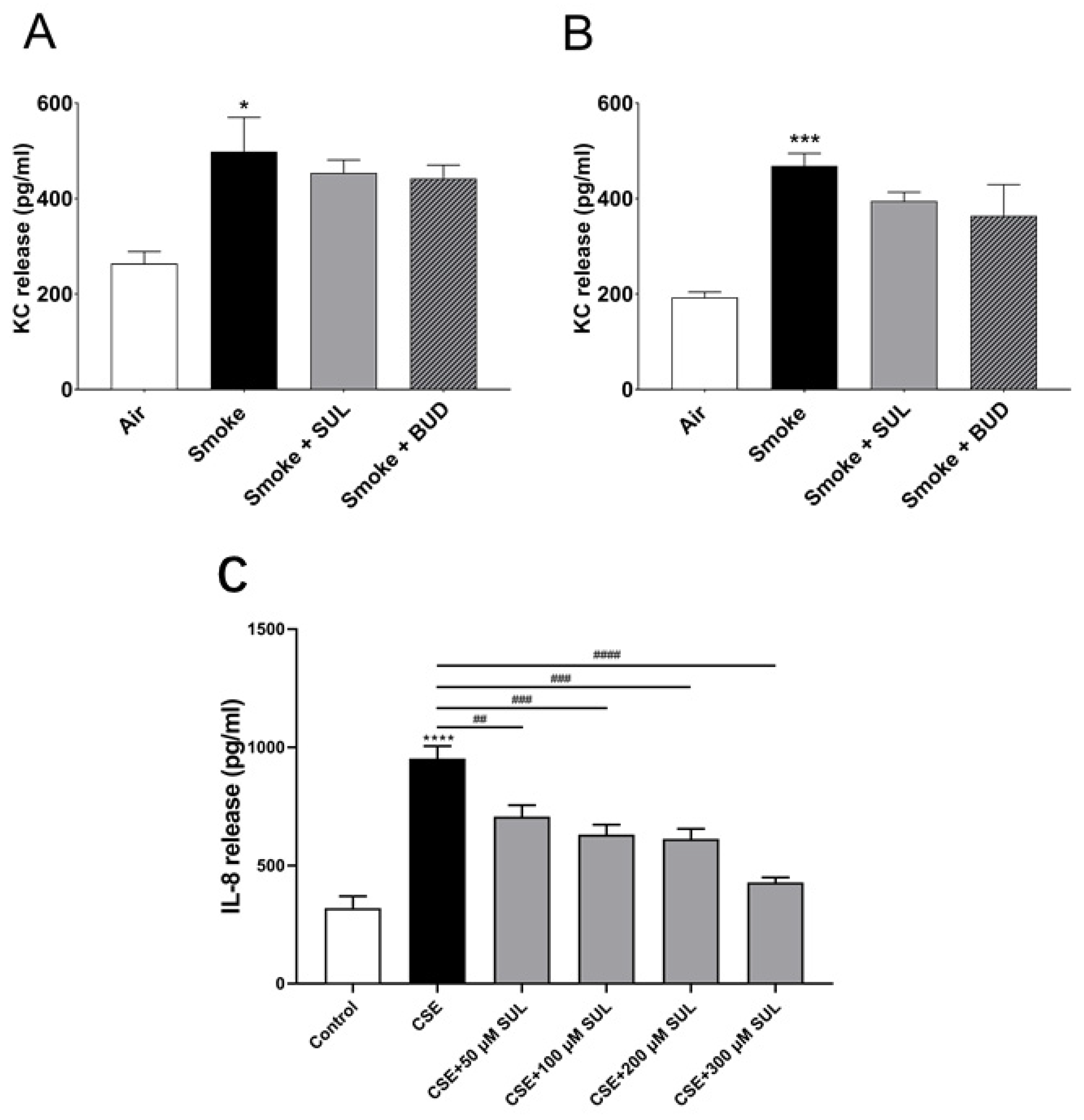
2.6. SUL-151 Hardly Affects KC Levels in Lung Homogenates but Concentration-Dependently Inhibits IL8-Production in Human Bronchial Epithelial Cells
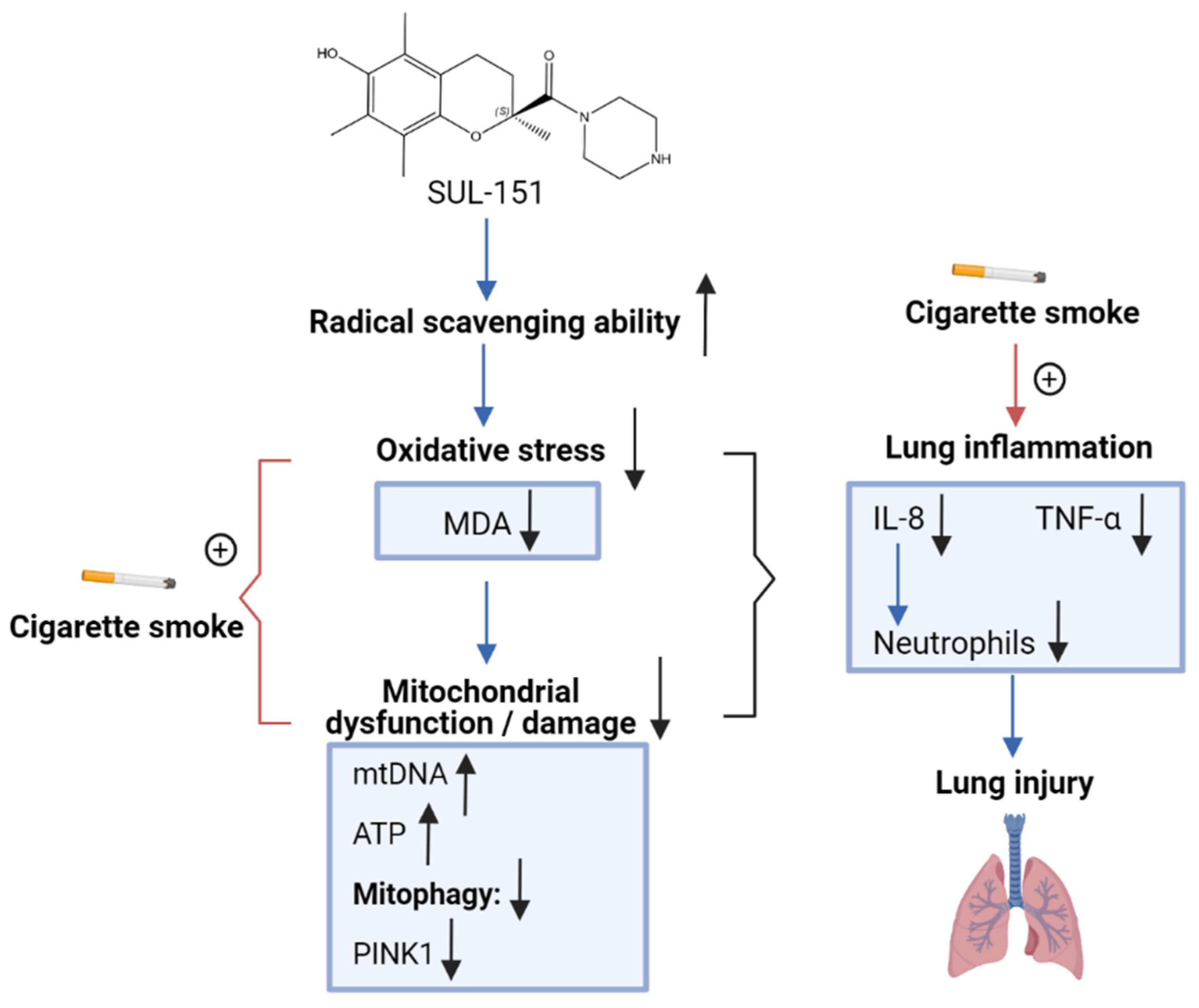
3. Discussion
4. Materials and Methods
4.1. Animals
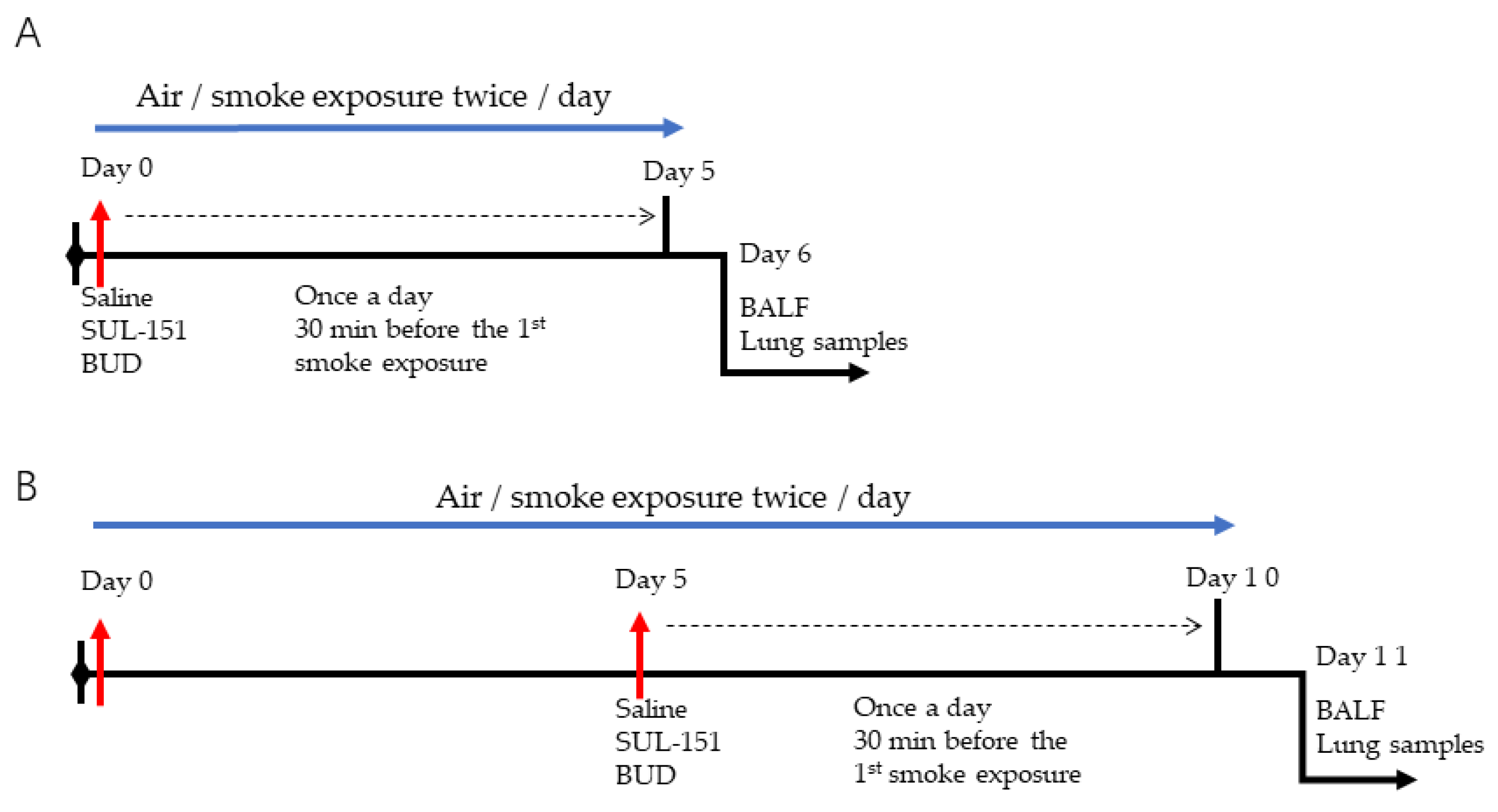
4.2. Experimental In Vivo Procedures
4.3. Bronchoalveolar Lavage Fluid (BALF)
4.4. Radical Scavenging Activity, Lipid Peroxidation Product Malondialdehyde (MDA), and ATP Measurements
4.5. mtDNA Copy Number
4.6. Immunoblotting
4.7. Assessment of SUL-151 Levels in Serum and Lung
4.8. KC Measurement in the Lung Homogenates
4.9. CSE Preparation and IL-8 Measurement in the Human Bronchial Epithelial Cells
4.10. Statistical Analysis
4.11. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eapen, M.S.; Myers, S.; Walters, E.H.; Sohal, S.S. Airway inflammation in chronic obstructive pulmonary disease (COPD): A true paradox. Expert Rev. Respir. Med. 2017, 11, 827–839. [Google Scholar] [CrossRef]
- Lundbäck, B.; Lindberg, A.; Lindström, M.; Rönmark, E.; Jonsson, A.; Jönsson, E.; Larsson, L.-G.; Andersson, S.; Sandström, T.; Larsson, K. Not 15 but 50% of smokers develop COPD?—Report from the Obstructive Lung Disease in Northern Sweden Studies. Respir. Med. 2003, 97, 115–122. [Google Scholar] [CrossRef]
- Tsubouchi, K.; Araya, J.; Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018, 38, 1–9. [Google Scholar] [CrossRef]
- Dunn, J.D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Kuwano, K.; Araya, J. Mitochondrial Quality Control in COPD and IPF. Cells 2018, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, W.; Zhu, B.; Wang, X. Epithelial Mitochondrial Dysfunction in Lung Disease. Chem. Biol. Pteridines Folates 2017, 1038, 201–217. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, X.; Hu, D. Mitochondrial alterations during oxidative stress in chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1153–1162. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; De Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and pharmacological targets for chronic lung diseases. Br. J. Pharmacol. 2016, 173, 2305–2318. [Google Scholar] [CrossRef]
- Liu, S.-F.; Kuo, H.-C.; Tseng, C.-W.; Huang, H.-T.; Chen, Y.-C.; Tseng, C.-C.; Lin, M.-C. Leukocyte Mitochondrial DNA Copy Number Is Associated with Chronic Obstructive Pulmonary Disease. PLoS ONE 2015, 10, e0138716. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef]
- Ernst, P.; Saad, N.; Suissa, S. Inhaled corticosteroids in COPD: The clinical evidence. Eur. Respir. J. 2014, 45, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. COPD 2020: New directions needed. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L884–L886. [Google Scholar] [CrossRef]
- Wang, M.T.; Liou, J.T.; Lin, C.W.; Tsai, C.L.; Wang, Y.H.; Hsu, Y.J.; Lai, J.H. Association of Cardiovascular Risk with Inhaled Long-Acting Bronchodilators in Patients with Chronic Obstructive Pulmonary Disease: A Nested Case-Control Study. JAMA Intern. Med. 2018, 178, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhu, L. Update on molecular mechanisms of corticosteroid resistance in chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2016, 37, 1–8. [Google Scholar] [CrossRef]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Marwick, J.A.; Ito, K.; Adcock, I.M.; A Kirkham, P. Oxidative stress and steroid resistance in asthma and COPD: Pharmacological manipulation of HDAC-2 as a therapeutic strategy. Expert Opin. Ther. Targets 2007, 11, 745–755. [Google Scholar] [CrossRef]
- Hajmousa, G.; Vogelaar, P.; Brouwer, L.A.; Van Der Graaf, A.C.; Henning, R.H.; Krenning, G. The 6-chromanol derivate SUL-109 enables prolonged hypothermic storage of adipose tissue-derived stem cells. Biomaterials 2017, 119, 43–52. [Google Scholar] [CrossRef]
- Han, B.; Poppinga, W.J.; Zuo, H.; Zuidhof, A.B.; Bos, I.S.T.; Smit, M.; Vogelaar, P.; Krenning, G.; Henning, R.H.; Maarsingh, H.; et al. The novel compound Sul-121 inhibits airway inflammation and hyperresponsiveness in experimental models of chronic obstructive pulmonary disease. Sci. Rep. 2016, 6, 26928. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- O’Donnell, R.A.; Peebles, C.; A Ward, J.; Daraker, A.; Angco, G.; Broberg, P.; Pierrou, S.; Lund, J.; Holgate, S.T.; Davies, D.E.; et al. Relationship between peripheral airway dysfunction, airway obstruction, and neutrophilic inflammation in COPD. Thorax 2004, 59, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Hanania, N.A.; Lareau, S.C.; Yawn, B.P. Safety of inhaled long-acting anti-muscarinic agents in COPD. Postgrad. Med. 2017, 129, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhu, J.; Liu, Y.; Li, Y.; Liu, W.; Zhang, M.; Chen, B.; Zhu, S. Optimization of Nebulized Budesonide in the Treatment of Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Lambooy, S.P.H.; Bidadkosh, A.; Nakladal, D.; Van Buiten, A.; Girgis, R.A.T.; Van Der Graaf, A.C.; Wiedenmann, T.J.; Koster, R.A.; Vogelaar, P.; Buikema, H.; et al. The Novel Compound Sul-121 Preserves Endothelial Function and Inhibits Progression of Kidney Damage in Type 2 Diabetes Mellitus in Mice. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Butler, A.; Walton, G.M.; Sapey, E. Neutrophilic Inflammation in the Pathogenesis of Chronic Obstructive Pulmonary Disease. COPD: J. Chronic Obstr. Pulm. Dis. 2018, 15, 392–404. [Google Scholar] [CrossRef]
- Verheijden, K.A.T.; Willemsen, L.E.M.; Braber, S.; Leusink-Muis, T.; Delsing, D.J.M.; Garssen, J.; Kraneveld, A.D.; Folkerts, G. Dietary galacto-oligosaccharides prevent airway eosinophilia and hyperresponsiveness in a murine house dust mite-induced asthma model. Respir. Res. 2015, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zhu, Y.-H.; Liu, D.-X.; Li, J.; Zhao, P.-C.; Zhong, Y.-P.; Chen, Y.-Q.; Xu, W.; Zhu, Z.-Q. Intranasal Application of Budesonide Attenuates Lipopolysaccharide-Induced Acute Lung Injury by Suppressing Nucleotide-Binding Oligomerization Domain-Like Receptor Family, Pyrin Domain-Containing 3 Inflammasome Activation in Mice. J. Immunol. Res. 2019, 2019, 7264383. [Google Scholar] [CrossRef]
- Van Eeden, S.F.; Sin, D.D. Oxidative stress in chronic obstructive pulmonary disease: A lung and systemic process. Can. Respir. J. 2013, 20, 27–29. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative Stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Ceylan, E.; Kocyigit, A.; Gencer, M.; Aksoy, N.; Selek, S. Increased DNA damage in patients with chronic obstructive pulmonary disease who had once smoked or been exposed to biomass. Respir. Med. 2006, 100, 1270–1276. [Google Scholar] [CrossRef]
- Ciencewicki, J.; Trivedi, S.; Kleeberger, S.R. Oxidants and the pathogenesis of lung diseases. J. Allergy Clin. Immunol. 2008, 122, 456–468. [Google Scholar] [CrossRef]
- Kluchová, Z.; Petrásová, D.; Joppa, P.; Dorková, Z.; Tkácová, R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol. Res. 2006, 56, 51–56. [Google Scholar] [PubMed]
- Montaño, M.; Cisneros, J.; Ramírez-Venegas, A.; Pedraza-Chaverri, J.; Mercado, D.; Ramos, C.; Sansores, R.H. Malondialdehyde and superoxide dismutase correlate with FEV 1 in patients with COPD associated with wood smoke exposure and tobacco smoking. Inhal. Toxicol. 2010, 22, 868–874. [Google Scholar] [CrossRef]
- Paredi, P.; Kharitonov, S.A.; Leak, D.; Ward, S.; Cramer, D.; Barnes, P.J. Exhaled Ethane, a Marker of Lipid Peroxidation, Is Elevated in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2000, 162, 369–373. [Google Scholar] [CrossRef]
- Kant, S.; Bajpai, J.; Prakash, V.; Verma, A.K.; Srivastava, A.; Bajaj, D.K.; Ahmad, M.; Agarwal, A. Study of oxidative stress biomarkers in chronic obstructive pulmonary disease and their correlation with disease severity in north Indian population cohort. Lung India 2017, 34, 324–329. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, X.; Ge, D.; Wang, S.; Qi, B. Protective Effects of Astragaloside IV Combined with Budesonide in Bronchitis in Rats by Regulation of Nrf2/Keap1 Pathway. Med. Sci. Monit. 2018, 24, 8481–8488. [Google Scholar] [CrossRef] [PubMed]
- Van der Toorn, M.; Slebos, D.J.; de Bruin, H.G.; Leuvenink, H.G.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; van Oosterhout, A.J.; Kauffman, H.F. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am. J. Physiol. Lung. Cell Mol. Physiol. 2007, 292, L1211–L1218. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Harrison, C.M.; Chuang, G.C.; Ballinger, S.W. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutat. Res. Mol. Mech. Mutagen. 2007, 621, 61–74. [Google Scholar] [CrossRef]
- Wu, K.; Luan, G.; Xu, Y.; Shen, S.; Qian, S.; Zhu, Z.; Zhang, X.; Yin, S.; Ye, J. Cigarette smoke extract increases mitochondrial membrane permeability through activation of adenine nucleotide translocator (ANT) in lung epithelial cells. Biochem. Biophys. Res. Commun. 2020, 525, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Marcatti, M.; Ahmad, A.; Montalbano, M.; Brunyánszki, A.; Bibli, S.-I.; Papapetropoulos, A.; Szabo, C. Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.-S.; Izumchenko, E.; Dasgupta, S.; O Hoque, M. Mitochondria in chronic obstructive pulmonary disease and lung cancer: Where are we now? Biomark. Med. 2017, 11, 475–489. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Thomas, P.S. Oxidative Stress in COPD and Its Measurement through Exhaled Breath Condensate. Clin. Transl. Sci. 2009, 2, 150–155. [Google Scholar] [CrossRef]
- Fischer, B.M.; A Voynow, J.; Ghio, A.J. COPD: Balancing oxidants and antioxidants. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 261–276. [Google Scholar] [CrossRef]
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2010, 163, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Mechanisms and resistance in glucocorticoid control of inflammation. J. Steroid Biochem. Mol. Biol. 2010, 120, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Rogliani, P.; Stolz, D.; Matera, M.G. Pharmacological treatment and current controversies in COPD. F1000Research 2019, 8, 1533. [Google Scholar] [CrossRef]
- Kumawat, K.; Geerdink, R.J.; Hennus, M.P.; Roda, M.A.; Van Ark, I.; Leusink-Muis, T.; Folkerts, G.; Van Oort-Jansen, A.; Mazharian, A.; Watson, S.P.; et al. LAIR-1 Limits Neutrophilic Airway Inflammation. Front. Immunol. 2019, 10, 842. [Google Scholar] [CrossRef]
- Braber, S.; Koelink, P.J.; Henricks, P.A.J.; Jackson, P.L.; Nijkamp, F.P.; Garssen, J.; Kraneveld, A.D.; Blalock, J.E.; Folkerts, G. Cigarette smoke-induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen breakdown. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L255–L265. [Google Scholar] [CrossRef] [PubMed]
- Verheijden, K.A.T.; Braber, S.; Leusink-Muis, T.; Jeurink, P.V.; Thijssen, S.; Kraneveld, A.D.; Garssen, J.; Folkerts, G.; Willemsen, L.E.M. The Combination Therapy of Dietary Galacto-Oligosaccharides with Budesonide Reduces Pulmonary Th2 Driving Mediators and Mast Cell Degranulation in a Murine Model of House Dust Mite Induced Asthma. Front. Immunol. 2018, 9, 2419. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP Decolorization Assay of Antioxidant Capacity Reaction Pathways. Int. J. Mol. Sci. 2020, 21, 1131. [Google Scholar] [CrossRef]
- Vogelaar, P.C.; Roorda, M.; De Vrij, E.L.; Houwertjes, M.C.; Goris, M.; Bouma, H.; Van Der Graaf, A.C.; Krenning, G.; Henning, R.H. The 6-hydroxychromanol derivative SUL-109 ameliorates renal injury after deep hypothermia and rewarming in rats. Nephrol. Dial. Transplant. 2018, 33, 2128–2138. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Pelgrim, C.E.; Swart, D.H.; Krenning, G.; van der Graaf, A.C.; Kraneveld, A.D.; Leusink-Muis, T.; van Ark, I.; Garssen, J.; Folkerts, G.; et al. SUL-151 Decreases Airway Neutrophilia as a Prophylactic and Therapeutic Treatment in Mice after Cigarette Smoke Exposure. Int. J. Mol. Sci. 2021, 22, 4991. https://doi.org/10.3390/ijms22094991
Wang L, Pelgrim CE, Swart DH, Krenning G, van der Graaf AC, Kraneveld AD, Leusink-Muis T, van Ark I, Garssen J, Folkerts G, et al. SUL-151 Decreases Airway Neutrophilia as a Prophylactic and Therapeutic Treatment in Mice after Cigarette Smoke Exposure. International Journal of Molecular Sciences. 2021; 22(9):4991. https://doi.org/10.3390/ijms22094991
Chicago/Turabian StyleWang, Lei, Charlotte E. Pelgrim, Daniël H. Swart, Guido Krenning, Adrianus C. van der Graaf, Aletta D. Kraneveld, Thea Leusink-Muis, Ingrid van Ark, Johan Garssen, Gert Folkerts, and et al. 2021. "SUL-151 Decreases Airway Neutrophilia as a Prophylactic and Therapeutic Treatment in Mice after Cigarette Smoke Exposure" International Journal of Molecular Sciences 22, no. 9: 4991. https://doi.org/10.3390/ijms22094991
APA StyleWang, L., Pelgrim, C. E., Swart, D. H., Krenning, G., van der Graaf, A. C., Kraneveld, A. D., Leusink-Muis, T., van Ark, I., Garssen, J., Folkerts, G., & Braber, S. (2021). SUL-151 Decreases Airway Neutrophilia as a Prophylactic and Therapeutic Treatment in Mice after Cigarette Smoke Exposure. International Journal of Molecular Sciences, 22(9), 4991. https://doi.org/10.3390/ijms22094991