
Low Replicative Stress Triggers Cell-Type Specific Inheritable Advanced Replication Timing

 ,
,  , , , ,
, , , ,  and
and
Abstract
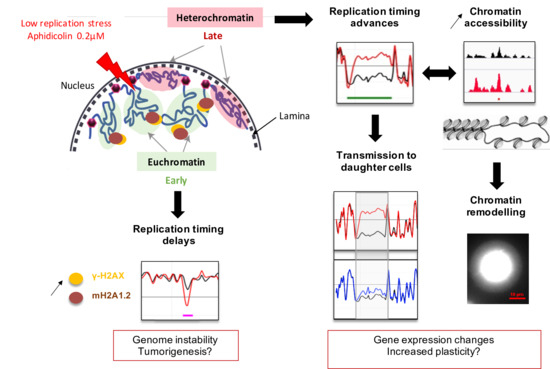
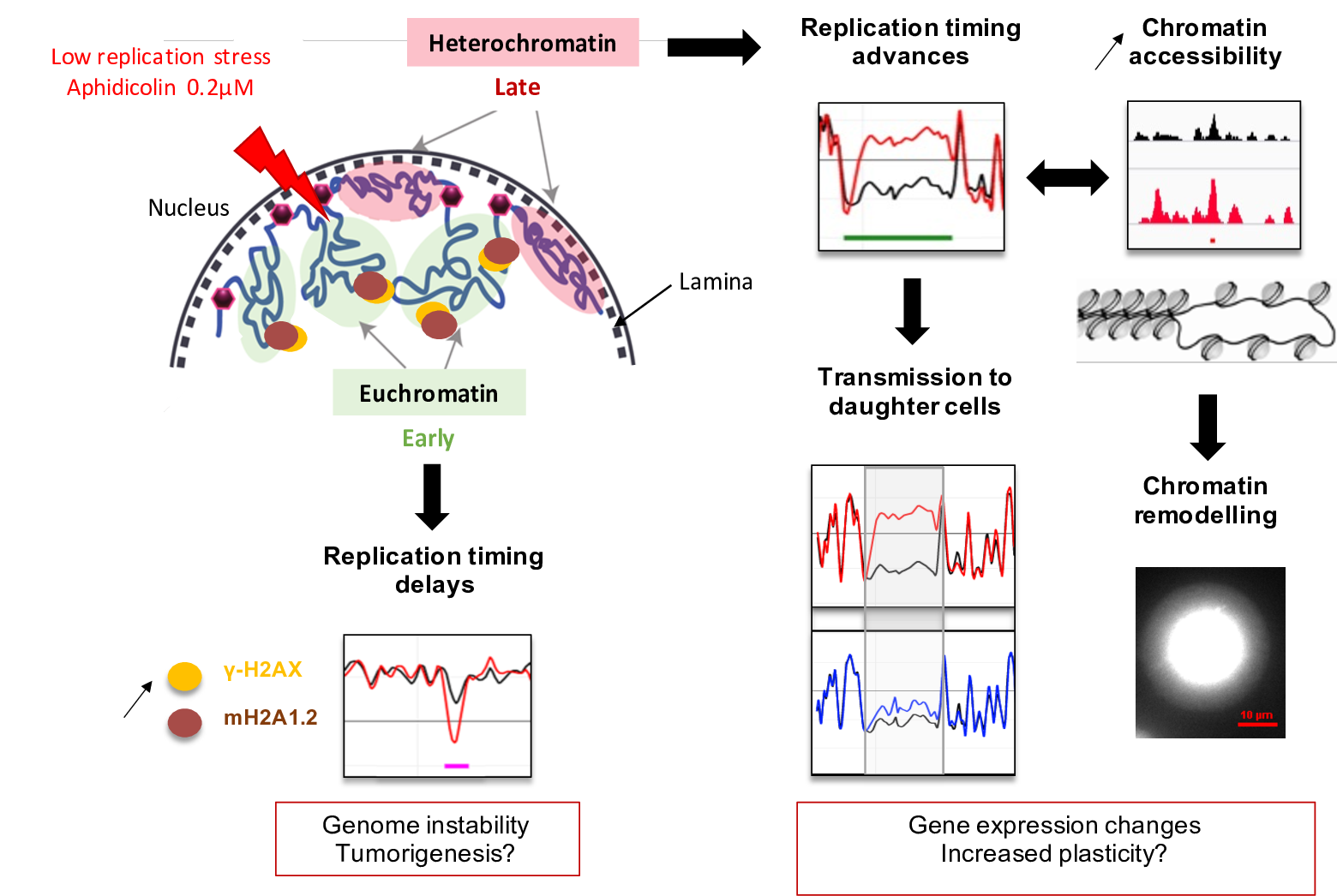{kind=link}
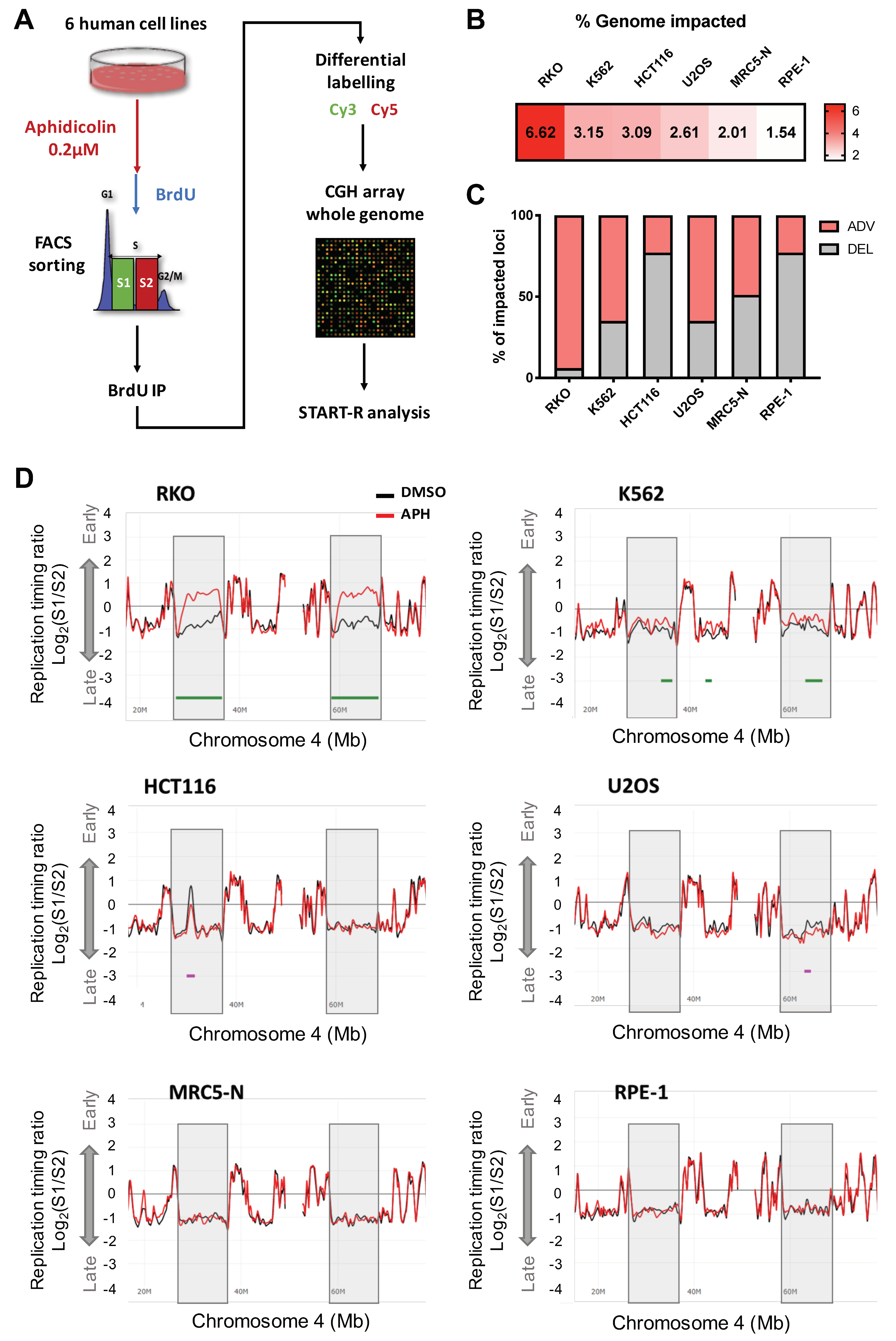{kind=link}
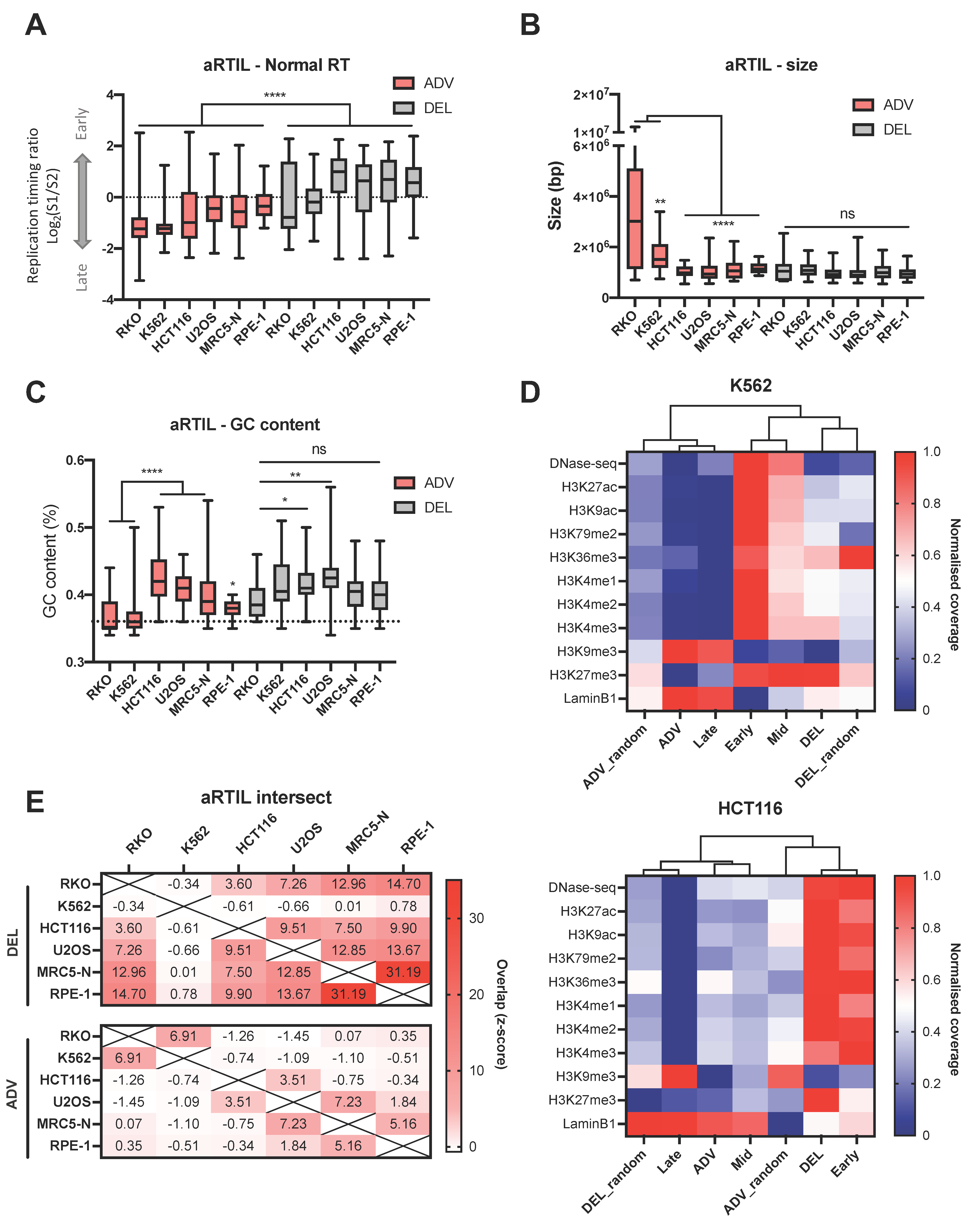{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Low Replication Stress Induces Cell-Type Replication Timing Changes
2.1.1. Genomic and Epigenomic Characterization of aRTIL
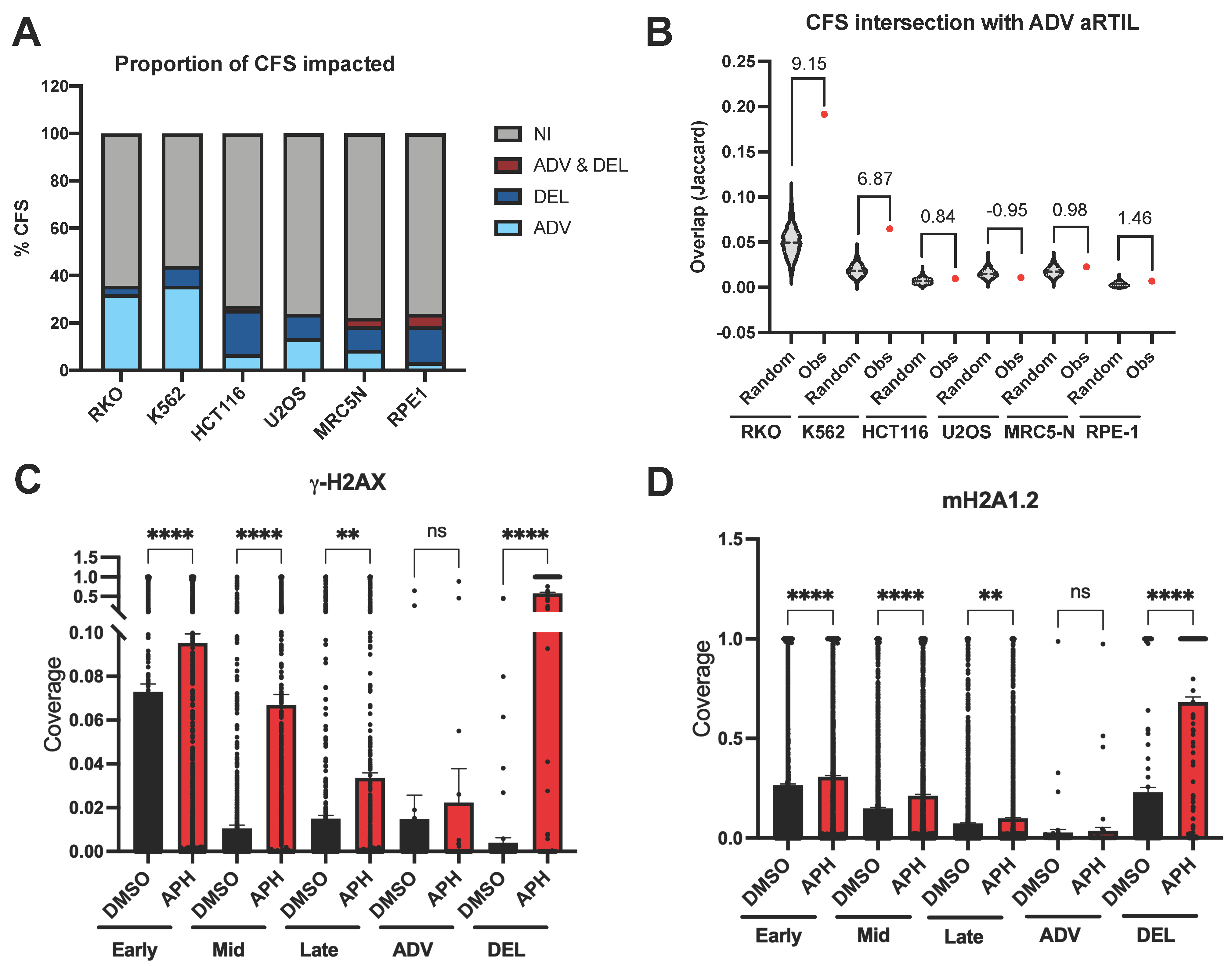
2.1.2. ADV aRTIL Are Related to CFS but Not Enriched in DNA Damage Signaling Histone Marks
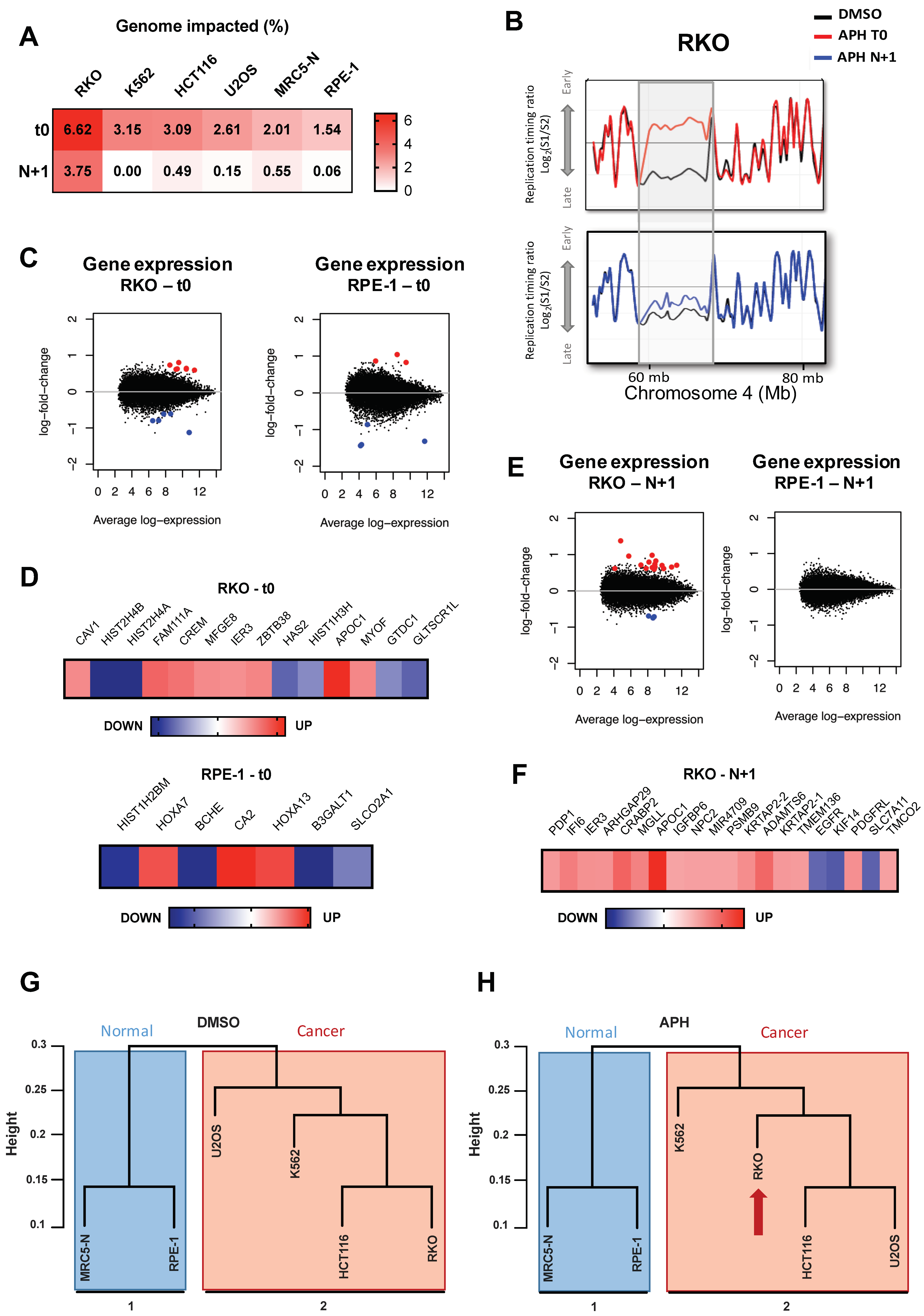
2.2. ADV aRTIL Can Be Transmitted to Daughter Cells
2.2.1. Aphidicolin Affects the RT-Related Identity of Cells
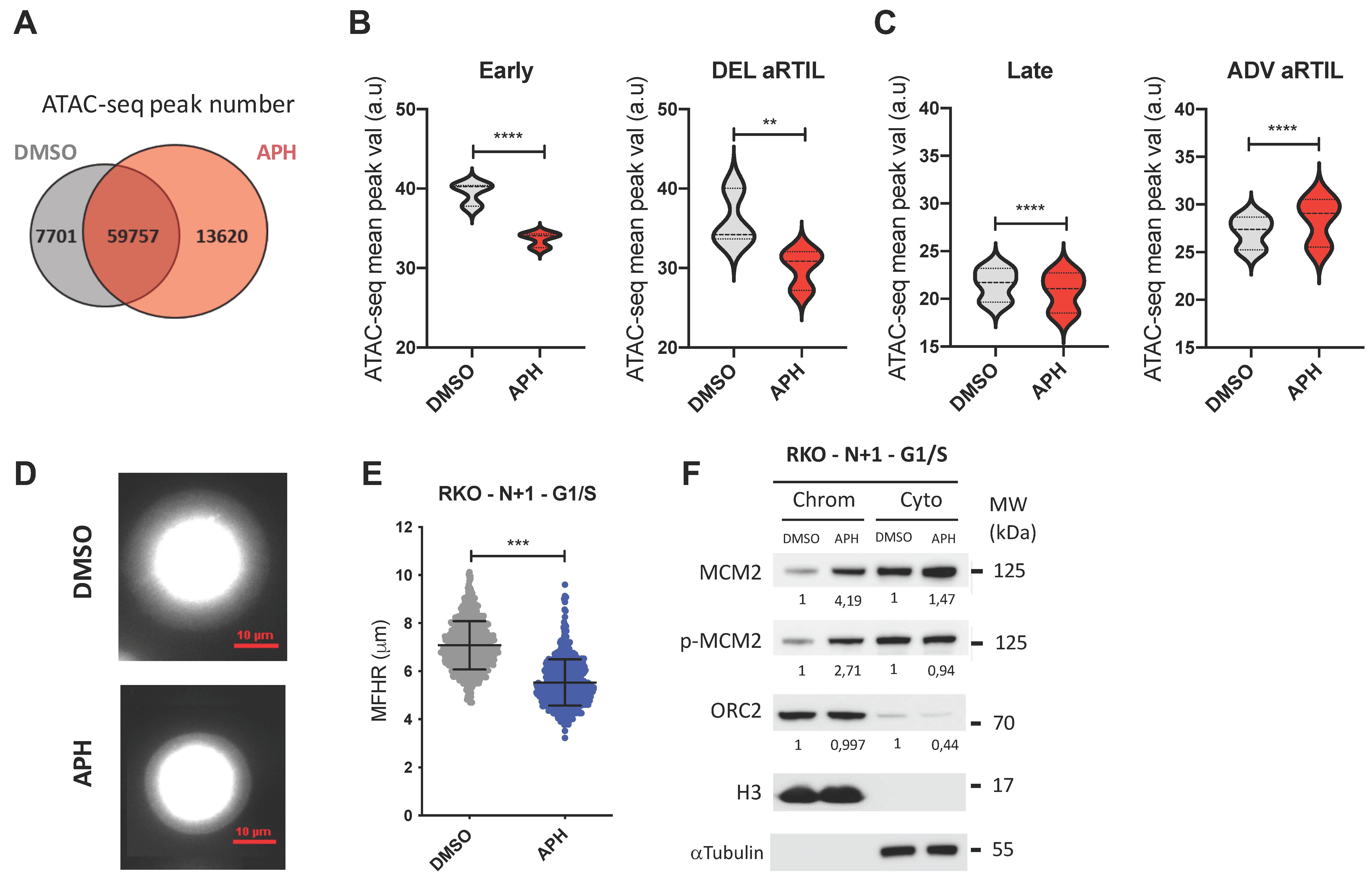
2.2.2. Aphidicolin Modulates Chromatin Accessibility within RKO ADV aRTIL
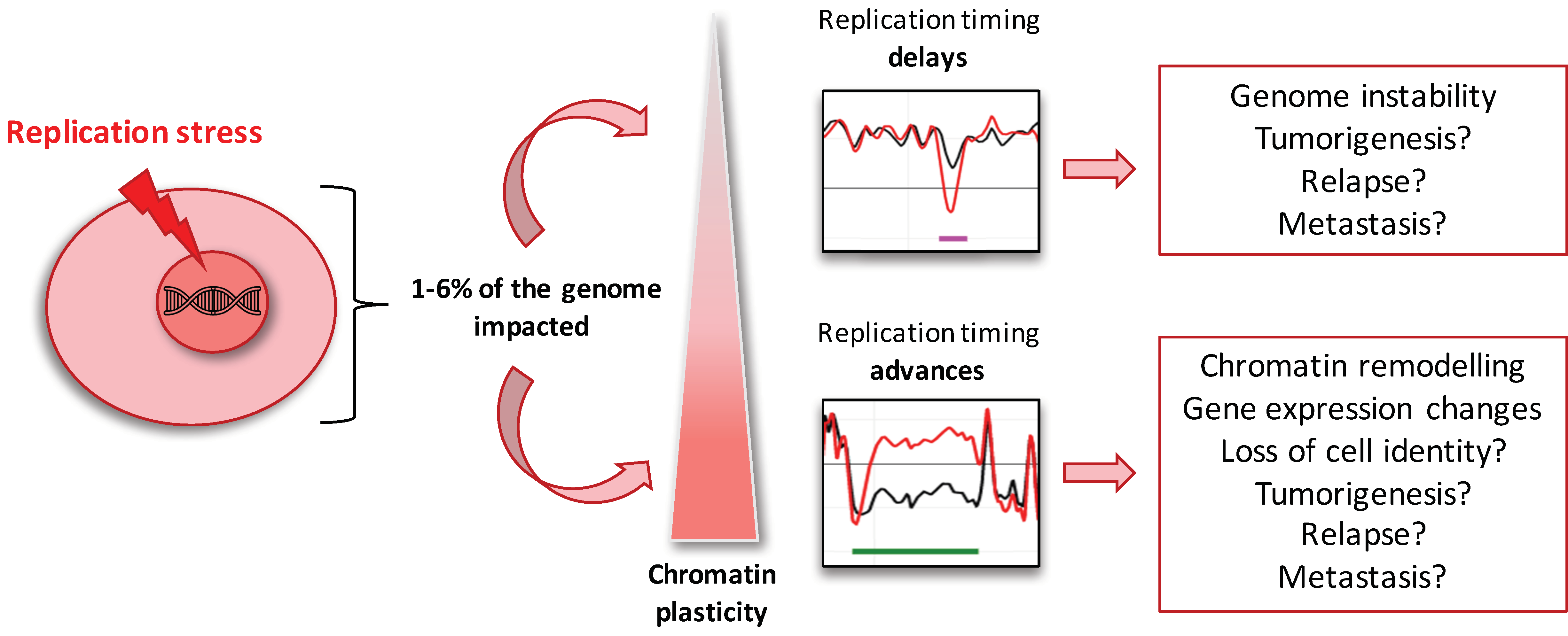
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture, and Drugs
4.2. Cell Lysis, Fractionation, and Western Blotting
4.3. Replication Timing Analysis
4.4. Hierachical Clustering Based on Replication Timing
4.5. Genomic Studies of Advanced and Delayed Replication Timing Domains
4.6. aRTIL Intersections and Randomization Procedure
4.7. Gene Expression Microarrays
4.8. ATAC-seq
4.9. Processing ChIP-seq Data from Public Databases
4.10. Fluorescent DNA Halo
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. Eukaryotic Chromosome DNA Replication: Where, When, and How? Annu. Rev. Biochem. 2010, 79, 89–130. [Google Scholar] [CrossRef]
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of Eukaryotic Replicons, Replicon Clusters, and Replication Foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.-W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global Reorganization of Replication Domains During Embryonic Stem Cell Differentiation. PLOS Biol. 2008, 6, e245. [Google Scholar] [CrossRef]
- Pope, B.D.; Hiratani, I.; Gilbert, D.M. Domain-Wide Regulation of DNA Replication Timing during Mammalian Development. Chromosom. Res. 2010, 18, 127–136. [Google Scholar] [CrossRef]
- Hiratani, I.; Ryba, T.; Itoh, M.; Rathjen, J.; Kulik, M.; Papp, B.; Fussner, E.; Bazett-Jones, D.P.; Plath, K.; Dalton, S.; et al. Genome-Wide Dynamics of Replication Timing Revealed by in Vitro Models of Mouse Embryogenesis. Genome Res. 2010, 20, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Julienne, H.; Audit, B.; Arneodo, A. Embryonic Stem Cell Specific “Master” Replication Origins at the Heart of the Loss of Pluripotency. PLoS Comput. Biol. 2015, 11, e1003969. [Google Scholar] [CrossRef] [PubMed]
- Madrid-Mencía, M.; Raineri, E.; Cao, T.B.N.; Pancaldi, V. Using GARDEN-NET and ChAseR to Explore Human Haematopoietic 3D Chromatin Interaction Networks. Nucleic Acids Res. 2020, 48, 4066–4080. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Buckley, Q.; Sasaki, T.; Zimmerman, J.; Didier, R.A.; Nazor, K.; Loring, J.F.; Lian, Z.; Weissman, S.; Robins, A.J.; et al. Dynamic Changes in Replication Timing and Gene Expression during Lineage Specification of Human Pluripotent Stem Cells. Genome Res. 2015, 25, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily Conserved Replication Timing Profiles Predict Long-Range Chromatin Interactions and Distinguish Closely Related Cell Types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef]
- Ryba, T.; Hiratani, I.; Sasaki, T.; Battaglia, D.; Kulik, M.; Zhang, J.; Dalton, S.; Gilbert, D.M. Replication Timing: A Fingerprint for Cell Identity and Pluripotency. PLoS Comput. Biol. 2011, 7, e1002225. [Google Scholar] [CrossRef]
- Takahashi, S.; Miura, H.; Shibata, T.; Nagao, K.; Okumura, K.; Ogata, M.; Obuse, C.; Takebayashi, S.-I.; Hiratani, I. Genome-Wide Stability of the DNA Replication Program in Single Mammalian Cells. Nat. Genet. 2019, 51, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Goren, A.; Cedar, H. Replicating by the Clock. Nat. Rev. Mol. Cell Biol. 2003, 4, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Donley, N.; Thayer, M.J. DNA Replication Timing, Genome Stability and Cancer. Semin. Cancer Biol. 2013, 23, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased Replication Initiation and Conflicts with Transcription Underlie Cyclin E-Induced Replication Stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Miron, K.; Golan-Lev, T.; Dvir, R.; Ben-David, E.; Kerem, B. Oncogenes Create a Unique Landscape of Fragile Sites. Nat. Commun. 2015, 6, 7094. [Google Scholar] [CrossRef]
- Gómez-González, B.; Aguilera, A. Transcription-Mediated Replication Hindrance: A Major Driver of Genome Instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [PubMed]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A. The Causes of Replication Stress and Their Consequences on Genome Stability and Cell Fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication Stress Induces Sister-Chromatid Bridging at Fragile Site Loci in Mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 Nuclear Bodies Form around DNA Lesions Generated by Mitotic Transmission of Chromosomes under Replication Stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC Pathway and BLM Collaborate during Mitosis to Prevent Micro-Nucleation and Chromosome Abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication Stress Induces 53BP1-Containing OPT Domains in G1 Cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef]
- Hecht, F.; Glover, T.W. Cancer Chromosome Breakpoints and Common Fragile Sites Induced by Aphidicolin. Cancer Genet. Cytogenet. 1984, 13, 185–188. [Google Scholar] [CrossRef]
- Ozeri-Galai, E.; Bester, A.C.; Kerem, B. The Complex Basis Underlying Common Fragile Site Instability in Cancer. Trends Genet. 2012, 28, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Casper, A.M.; Glover, T.W. Common Fragile Sites. Cytogenet. Genome Res. 2003, 100, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA Damage Checkpoint and Genomic Instability in Human Precancerous Lesions. Nat. Cell Biol. 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Brison, O.; El-Hilali, S.; Azar, D.; Koundrioukoff, S.; Schmidt, M.; Nähse, V.; Jaszczyszyn, Y.; Lachages, A.-M.; Dutrillaux, B.; Thermes, C.; et al. Transcription-Mediated Organization of the Replication Initiation Program across Large Genes Sets Common Fragile Sites Genome-Wide. Nat. Commun. 2019, 10, 5693. [Google Scholar] [CrossRef]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common Fragile Site Profiling in Epithelial and Erythroid Cells Reveals That Most Recurrent Cancer Deletions Lie in Fragile Sites Hosting Large Genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef]
- Sarni, D.; Sasaki, T.; Tur-Sinai, M.I.; Miron, K.; Rivera-Mulia, J.C.; Magnuson, B.; Ljungman, M.; Gilbert, D.M.; Kerem, B. 3D Genome Organization Contributes to Genome Instability at Fragile Sites. Nat. Commun. 2020, 11, 3613. [Google Scholar] [CrossRef]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-Type-Specific Replication Initiation Programs Set Fragility of the FRA3B Fragile Site. Nat. Cell Biol. 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large Transcription Units Unify Copy Number Variants and Common Fragile Sites Arising under Replication Stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef]
- Ryba, T.; Battaglia, D.; Chang, B.H.; Shirley, J.W.; Buckley, Q.; Pope, B.D.; Devidas, M.; Druker, B.J.; Gilbert, D.M. Abnormal Developmental Control of Replication-Timing Domains in Pediatric Acute Lymphoblastic Leukemia. Genome Res. 2012, 22, 1833–1844. [Google Scholar] [CrossRef]
- Rivera-Mulia, J.C.; Desprat, R.; Trevilla-Garcia, C.; Cornacchia, D.; Schwerer, H.; Sasaki, T.; Sima, J.; Fells, T.; Studer, L.; Lemaitre, J.-M.; et al. DNA Replication Timing Alterations Identify Common Markers between Distinct Progeroid Diseases. Proc. Natl. Acad. Sci. USA 2017, 114, E10972–E10980. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Sasaki, T.; Trevilla-Garcia, C.; Nakamichi, N.; Knapp, D.J.H.F.; Hammond, C.A.; Chang, B.H.; Tyner, J.W.; Devidas, M.; Zimmerman, J.; et al. Replication Timing Alterations in Leukemia Affect Clinically Relevant Chromosome Domains. Blood Adv. 2019, 3, 3201–3213. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.-C.; Drac, M.; Schwob, E.; Baldacci, G.; Cazaux, C.; Hoffmann, J.-S. A Role for DNA Polymerase θ in the Timing of DNA Replication. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Almeida, R.; Fernández-Justel, J.M.; Santa-María, C.; Cadoret, J.-C.; Cano-Aroca, L.; Lombraña, R.; Herranz, G.; Agresti, A.; Gómez, M. Chromatin Conformation Regulates the Coordination between DNA Replication and Transcription. Nat. Commun. 2018, 9, 1590. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, D.; Denecker, T.; Maric, C.; Fauchereau, F.; Baldacci, G.; Cadoret, J.-C. Characterization of the Replication Timing Program of 6 Human Model Cell Lines. Genom. Data 2016, 9, 113–117. [Google Scholar] [CrossRef]
- Hadjadj, D.; Denecker, T.; Guérin, E.; Kim, S.-J.; Fauchereau, F.; Baldacci, G.; Maric, C.; Cadoret, J.-C. Efficient, Quick and Easy-to-Use DNA Replication Timing Analysis with START-R Suite. NAR Genom. Bioinform. 2020, 2, lqaa045. [Google Scholar] [CrossRef] [PubMed]
- Maccaroni, K.; Balzano, E.; Mirimao, F.; Giunta, S.; Pelliccia, F. Impaired Replication Timing Promotes Tissue-Specific Expression of Common Fragile Sites. Genes 2020, 11, 326. [Google Scholar] [CrossRef]
- Pehrson, J.R.; Fried, V.A. MacroH2A, a Core Histone Containing a Large Nonhistone Region. Science 1992, 257, 1398–1400. [Google Scholar] [CrossRef]
- Barrero, M.J.; Sese, B.; Kuebler, B.; Bilic, J.; Boue, S.; Martí, M.; Belmonte, J.C.I. Macrohistone Variants Preserve Cell Identity by Preventing the Gain of H3K4me2 during Reprogramming to Pluripotency. Cell Rep. 2013, 3, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Janich, P.; Cantariño, N.; Noguera, M.; Valero, V.; Musulén, E.; Douet, J.; Posavec, M.; Martín-Caballero, J.; Sumoy, L.; et al. MacroH2A1 Regulates the Balance between Self-Renewal and Differentiation Commitment in Embryonic and Adult Stem Cells. Mol. Cell. Biol. 2012, 32, 1442–1452. [Google Scholar] [CrossRef]
- Kim, J.; Oberdoerffer, P.; Khurana, S. The Histone Variant MacroH2A1 Is a Splicing-Modulated Caretaker of Genome Integrity and Tumor Growth. Mol. Cell Oncol. 2018, 5, e1441629. [Google Scholar] [CrossRef] [PubMed]
- Pasque, V.; Radzisheuskaya, A.; Gillich, A.; Halley-Stott, R.P.; Panamarova, M.; Zernicka-Goetz, M.; Surani, M.A.; Silva, J.C.R. Histone Variant MacroH2A Marks Embryonic Differentiation in Vivo and Acts as an Epigenetic Barrier to Induced Pluripotency. J. Cell Sci. 2012, 125, 6094–6104. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sturgill, D.; Sebastian, R.; Khurana, S.; Tran, A.D.; Edwards, G.B.; Kruswick, A.; Burkett, S.; Hosogane, E.K.; Hannon, W.W.; et al. Replication Stress Shapes a Protective Chromatin Environment across Fragile Genomic Regions. Mol. Cell 2018, 69, 36–47.e7. [Google Scholar] [CrossRef]
- Seo, J.; Kim, K.; Chang, D.-Y.; Kang, H.-B.; Shin, E.-C.; Kwon, J.; Choi, J.K. Genome-Wide Reorganization of Histone H2AX toward Particular Fragile Sites on Cell Activation. Nucleic Acids Res. 2014, 42, 1016–1025. [Google Scholar] [CrossRef]
- Hiratani, I.; Leskovar, A.; Gilbert, D.M. Differentiation-Induced Replication-Timing Changes Are Restricted to AT-Rich/Long Interspersed Nuclear Element (LINE)-Rich Isochores. Proc. Natl. Acad. Sci. USA 2004, 101, 16861–16866. [Google Scholar] [CrossRef]
- Perry, P.; Sauer, S.; Billon, N.; Richardson, W.D.; Spivakov, M.; Warnes, G.; Livesey, F.J.; Merkenschlager, M.; Fisher, A.G.; Azuara, V. A Dynamic Switch in the Replication Timing of Key Regulator Genes in Embryonic Stem Cells upon Neural Induction. Cell Cycle 2004, 3, 1645–1650. [Google Scholar] [CrossRef]
- Azuara, V.; Brown, K.E.; Williams, R.R.E.; Webb, N.; Dillon, N.; Festenstein, R.; Buckle, V.; Merkenschlager, M.; Fisher, A.G. Heritable Gene Silencing in Lymphocytes Delays Chromatid Resolution without Affecting the Timing of DNA Replication. Nat. Cell Biol. 2003, 5, 668–674. [Google Scholar] [CrossRef]
- Gartler, S.M.; Goldstein, L.; Tyler-Freer, S.E.; Hansen, R.S. The Timing of XIST Replication: Dominance of the Domain. Hum. Mol. Genet. 1999, 8, 1085–1089. [Google Scholar] [CrossRef]
- White, E.J.; Emanuelsson, O.; Scalzo, D.; Royce, T.; Kosak, S.; Oakeley, E.J.; Weissman, S.; Gerstein, M.; Groudine, M.; Snyder, M.; et al. DNA Replication-Timing Analysis of Human Chromosome 22 at High Resolution and Different Developmental States. Proc. Natl. Acad. Sci. USA 2004, 101, 17771–17776. [Google Scholar] [CrossRef]
- Shaikh, N.; Mazzagatti, A.; Bakker, B.; Spierings, D.C.J.; Wardenaar, R.; Muliaditan, D.; Maniati, E.; Larsson, P.; Wang, J.; Foijer, F.; et al. Replication Stress Generates Multiple Distinct Classes of Copy Number Alterations. bioRxiv 2019, 743658. [Google Scholar] [CrossRef]
- Du, Q.; Bert, S.A.; Armstrong, N.J.; Caldon, C.E.; Song, J.Z.; Nair, S.S.; Gould, C.M.; Luu, P.-L.; Peters, T.; Khoury, A.; et al. Replication Timing and Epigenome Remodelling Are Associated with the Nature of Chromosomal Rearrangements in Cancer. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.; Wu, B.; Chang, H.; Greenleaf, W. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Miao, N.; Sun, T. Detect Accessible Chromatin Using ATAC-Sequencing, from Principle to Applications. Hereditas 2019, 156, 29. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S.; Gilbert, D.M. The Spatial Position and Replication Timing of Chromosomal Domains Are Both Established in Early G1 Phase. Mol. Cell 1999, 4, 983–993. [Google Scholar] [CrossRef]
- Lu, J.; Li, F.; Murphy, C.S.; Davidson, M.W.; Gilbert, D.M. G2 Phase Chromatin Lacks Determinants of Replication Timing. J. Cell Biol. 2010, 189, 967–980. [Google Scholar] [CrossRef]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef]
- Vogelstein, B.; Pardoll, D.M.; Coffey, D.S. Supercoiled Loops and Eucaryotic DNA Replication. Cell 1980, 22, 79–85. [Google Scholar] [CrossRef]
- Lemaitre, J.-M.; Danis, E.; Pasero, P.; Vassetzky, Y.; Méchali, M. Mitotic Remodeling of the Replicon and Chromosome Structure. Cell 2005, 123, 787–801. [Google Scholar] [CrossRef]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, J.I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin Organizes Chromatin Loops at DNA Replication Factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Hernández-Muñoz, R.; Martínez, F.; Aranda-Anzaldo, A. DNA Moves Sequentially towards the Nuclear Matrix during DNA Replication in Vivo. BMC Cell Biol. 2011, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Moindrot, B.; Audit, B.; Klous, P.; Baker, A.; Thermes, C.; de Laat, W.; Bouvet, P.; Mongelard, F.; Arneodo, A. 3D Chromatin Conformation Correlates with Replication Timing and Is Conserved in Resting Cells. Nucleic Acids Res. 2012, 40, 9470–9481. [Google Scholar] [CrossRef]
- Yaffe, E.; Farkash-Amar, S.; Polten, A.; Yakhini, Z.; Tanay, A.; Simon, I. Comparative Analysis of DNA Replication Timing Reveals Conserved Large-Scale Chromosomal Architecture. PLoS Genet 2010, 6, e1001011. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain Organization of Human Chromosomes Revealed by Mapping of Nuclear Lamina Interactions. Nat. Cell Biol. 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Duriez, B.; Chilaka, S.; Bercher, J.-F.; Hercul, E.; Prioleau, M.-N. Replication Dynamics of Individual Loci in Single Living Cells Reveal Changes in the Degree of Replication Stochasticity through S Phase. Nucleic Acids Res. 2019, 47, 5155–5169. [Google Scholar] [CrossRef]
- Brueckner, L.; Zhao, P.A.; van Schaik, T.; Leemans, C.; Sima, J.; Peric-Hupkes, D.; Gilbert, D.M.; van Steensel, B. Local Rewiring of Genome–Nuclear Lamina Interactions by Transcription. EMBO J. 2020, 39, e103159. [Google Scholar] [CrossRef]
- Bronshtein, I.; Kepten, E.; Kanter, I.; Berezin, S.; Lindner, M.; Redwood, A.B.; Mai, S.; Gonzalo, S.; Foisner, R.; Shav-Tal, Y.; et al. Loss of Lamin A Function Increases Chromatin Dynamics in the Nuclear Interior. Nat. Commun. 2015, 6, 8044. [Google Scholar] [CrossRef]
- Ranade, D.; Pradhan, R.; Jayakrishnan, M.; Hegde, S.; Sengupta, K. Lamin A/C and Emerin Depletion Impacts Chromatin Organization and Dynamics in the Interphase Nucleus. BMC Mol. Cell Biol. 2019, 20, 11. [Google Scholar] [CrossRef]
- Desprat, R.; Thierry-Mieg, D.; Lailler, N.; Lajugie, J.; Schildkraut, C.; Thierry-Mieg, J.; Bouhassira, E.E. Predictable Dynamic Program of Timing of DNA Replication in Human Cells. Genome Res. 2009, 19, 2288–2299. [Google Scholar] [CrossRef]
- Karnani, N.; Taylor, C.; Malhotra, A.; Dutta, A. Pan-S Replication Patterns and Chromosomal Domains Defined by Genome-Tiling Arrays of ENCODE Genomic Areas. Genome Res. 2007, 17, 865–876. [Google Scholar] [CrossRef]
- MacAlpine, D.M.; Rodríguez, H.K.; Bell, S.P. Coordination of Replication and Transcription along a Drosophila Chromosome. Genes Dev. 2004, 18, 3094–3105. [Google Scholar] [CrossRef]
- Müller, C.A.; Nieduszynski, C.A. DNA Replication Timing Influences Gene Expression Level. J. Cell Biol. 2017, 216, 1907–1914. [Google Scholar] [CrossRef]
- Schübeler, D.; Scalzo, D.; Kooperberg, C.; van Steensel, B.; Delrow, J.; Groudine, M. Genome-Wide DNA Replication Profile for Drosophila Melanogaster: A Link between Transcription and Replication Timing. Nat. Genet. 2002, 32, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Simon, I.; Tenzen, T.; Mostoslavsky, R.; Fibach, E.; Lande, L.; Milot, E.; Gribnau, J.; Grosveld, F.; Fraser, P.; Cedar, H. Developmental Regulation of DNA Replication Timing at the Human β Globin Locus. EMBO J. 2001, 20, 6150–6157. [Google Scholar] [CrossRef]
- Woodfine, K.; Fiegler, H.; Beare, D.M.; Collins, J.E.; McCann, O.T.; Young, B.D.; Debernardi, S.; Mott, R.; Dunham, I.; Carter, N.P. Replication Timing of the Human Genome. Hum. Mol. Genet. 2003, 13, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Alver, R.C.; Chadha, G.S.; Blow, J.J. The Contribution of Dormant Origins to Genome Stability: From Cell Biology to Human Genetics. DNA Repair 2014, 19, 182–189. [Google Scholar] [CrossRef]
- Courbet, S.; Gay, S.; Arnoult, N.; Wronka, G.; Anglana, M.; Brison, O.; Debatisse, M. Replication Fork Movement Sets Chromatin Loop Size and Origin Choice in Mammalian Cells. Nat. Cell Biol. 2008, 455, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Courtot, L.; Hoffmann, J.-S.; Bergoglio, V. The Protective Role of Dormant Origins in Response to Replicative Stress. Int. J. Mol. Sci. 2018, 19, 3569. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM Proteins Protect Human Cells from Replicative Stress by Licensing Backup Origins of Replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, D.; Blow, J.J. Dormant Origins, the Licensing Checkpoint, and the Response to Replicative Stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, a012955. [Google Scholar] [CrossRef]
- Shima, N.; Pederson, K.D. Dormant Origins as a Built-in Safeguard in Eukaryotic DNA Replication against Genome Instability and Disease Development. DNA Repair 2017, 56, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Battaglia, D.; Pope, B.D.; Hiratani, I.; Gilbert, D.M. Genome-Scale Analysis of Replication Timing: From Bench to Bioinformatics. Nat. Protoc. 2011, 6, 870–895. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Cadoret, J.-C.; Audit, B.; Arneodo, A.; Alberti, A.; Battail, C.; Duret, L.; Prioleau, M.-N. The Spatiotemporal Program of DNA Replication Is Associated with Specific Combinations of Chromatin Marks in Human Cells. PLOS Genet. 2014, 10, e1004282. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An Improved ATAC-Seq Protocol Reduces Background and Enables Interrogation of Frozen Tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Courtot, L.; Bournique, E.; Maric, C.; Guitton-Sert, L.; Madrid-Mencía, M.; Pancaldi, V.; Cadoret, J.-C.; Hoffmann, J.-S.; Bergoglio, V. Low Replicative Stress Triggers Cell-Type Specific Inheritable Advanced Replication Timing. Int. J. Mol. Sci. 2021, 22, 4959. https://doi.org/10.3390/ijms22094959
Courtot L, Bournique E, Maric C, Guitton-Sert L, Madrid-Mencía M, Pancaldi V, Cadoret J-C, Hoffmann J-S, Bergoglio V. Low Replicative Stress Triggers Cell-Type Specific Inheritable Advanced Replication Timing. International Journal of Molecular Sciences. 2021; 22(9):4959. https://doi.org/10.3390/ijms22094959
Chicago/Turabian StyleCourtot, Lilas, Elodie Bournique, Chrystelle Maric, Laure Guitton-Sert, Miguel Madrid-Mencía, Vera Pancaldi, Jean-Charles Cadoret, Jean-Sébastien Hoffmann, and Valérie Bergoglio. 2021. "Low Replicative Stress Triggers Cell-Type Specific Inheritable Advanced Replication Timing" International Journal of Molecular Sciences 22, no. 9: 4959. https://doi.org/10.3390/ijms22094959
APA StyleCourtot, L., Bournique, E., Maric, C., Guitton-Sert, L., Madrid-Mencía, M., Pancaldi, V., Cadoret, J.-C., Hoffmann, J.-S., & Bergoglio, V. (2021). Low Replicative Stress Triggers Cell-Type Specific Inheritable Advanced Replication Timing. International Journal of Molecular Sciences, 22(9), 4959. https://doi.org/10.3390/ijms22094959