
Identification of miRNAs and Their Targets Involved in Flower and Fruit Development across Domesticated and Wild Capsicum Species

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
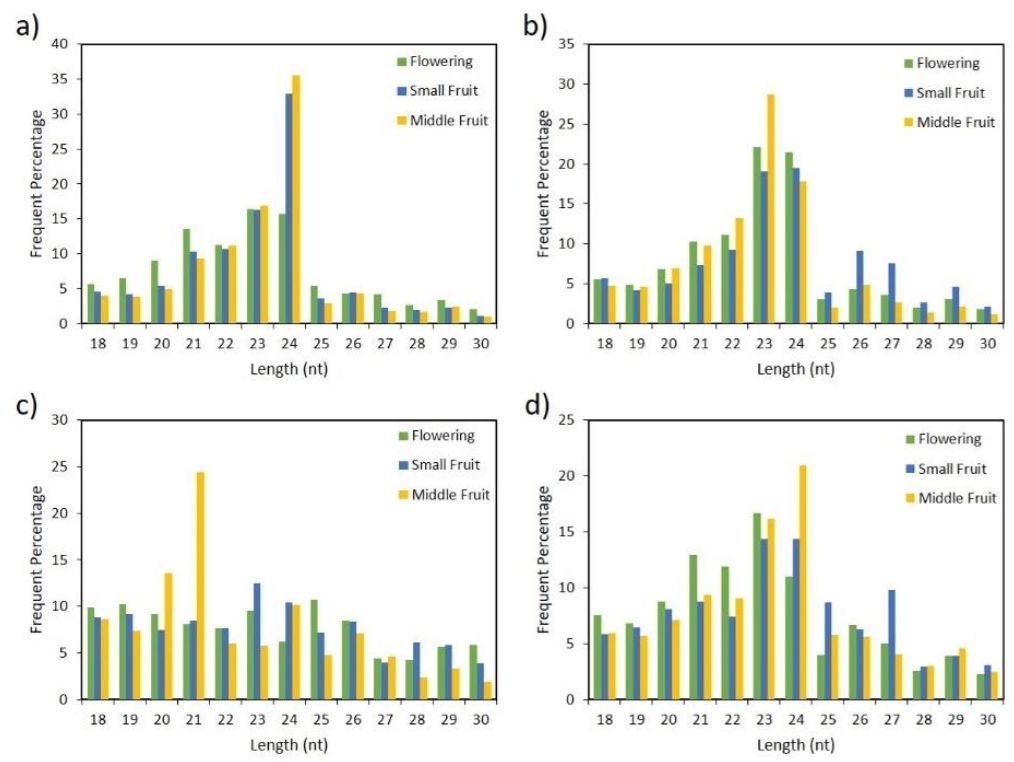
2.1. Analysis of Small RNAs in Pepper
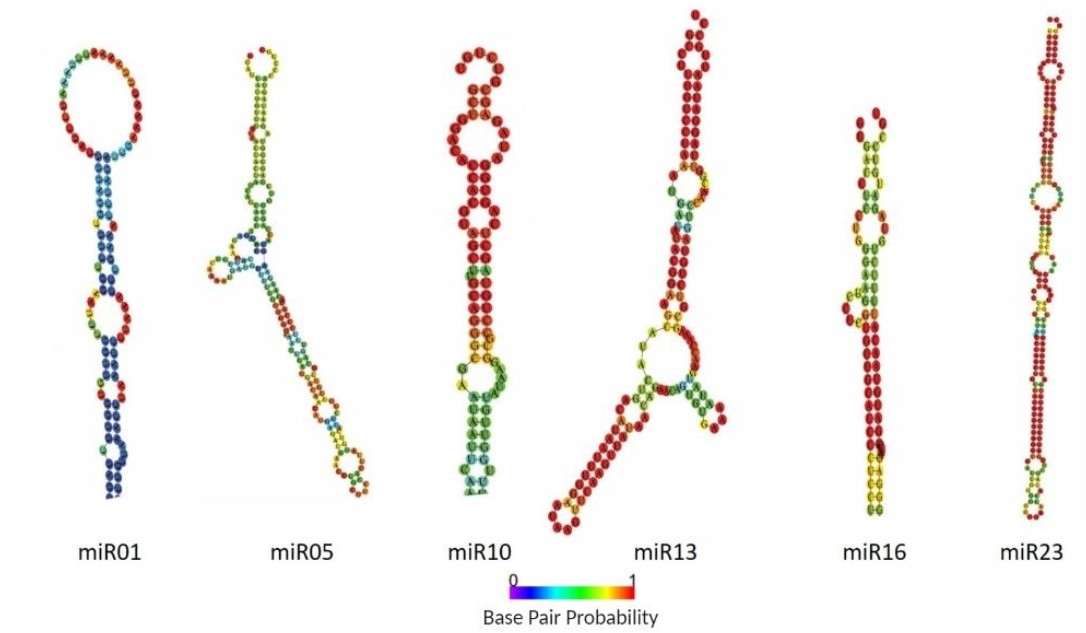
2.2. Identification of Known and Novel miRNAs in Pepper
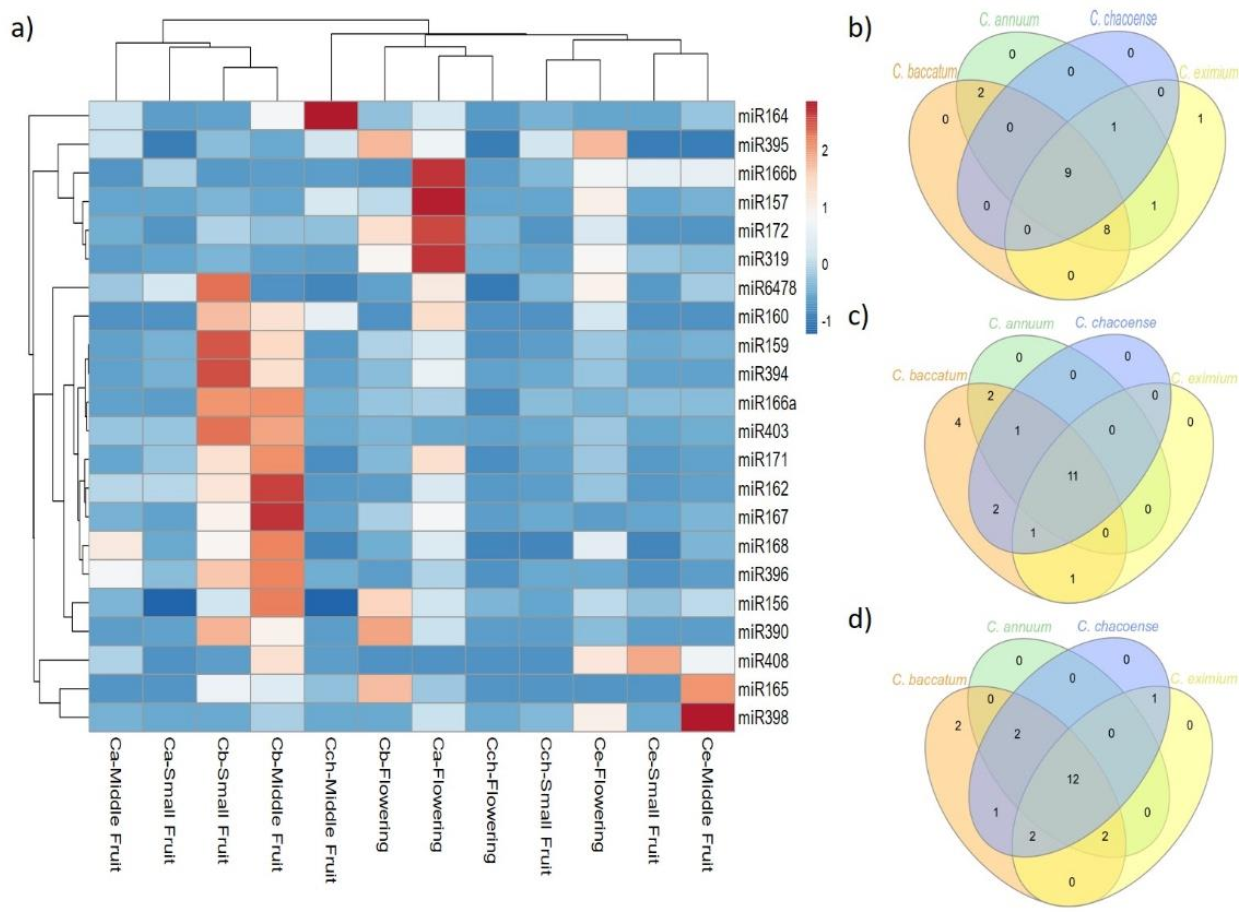
2.3. Expression Pattern of Known and Novel miRNAs in Pepper
2.4. Prediction of Putative Targets for Known and Novel miRNAs in Pepper
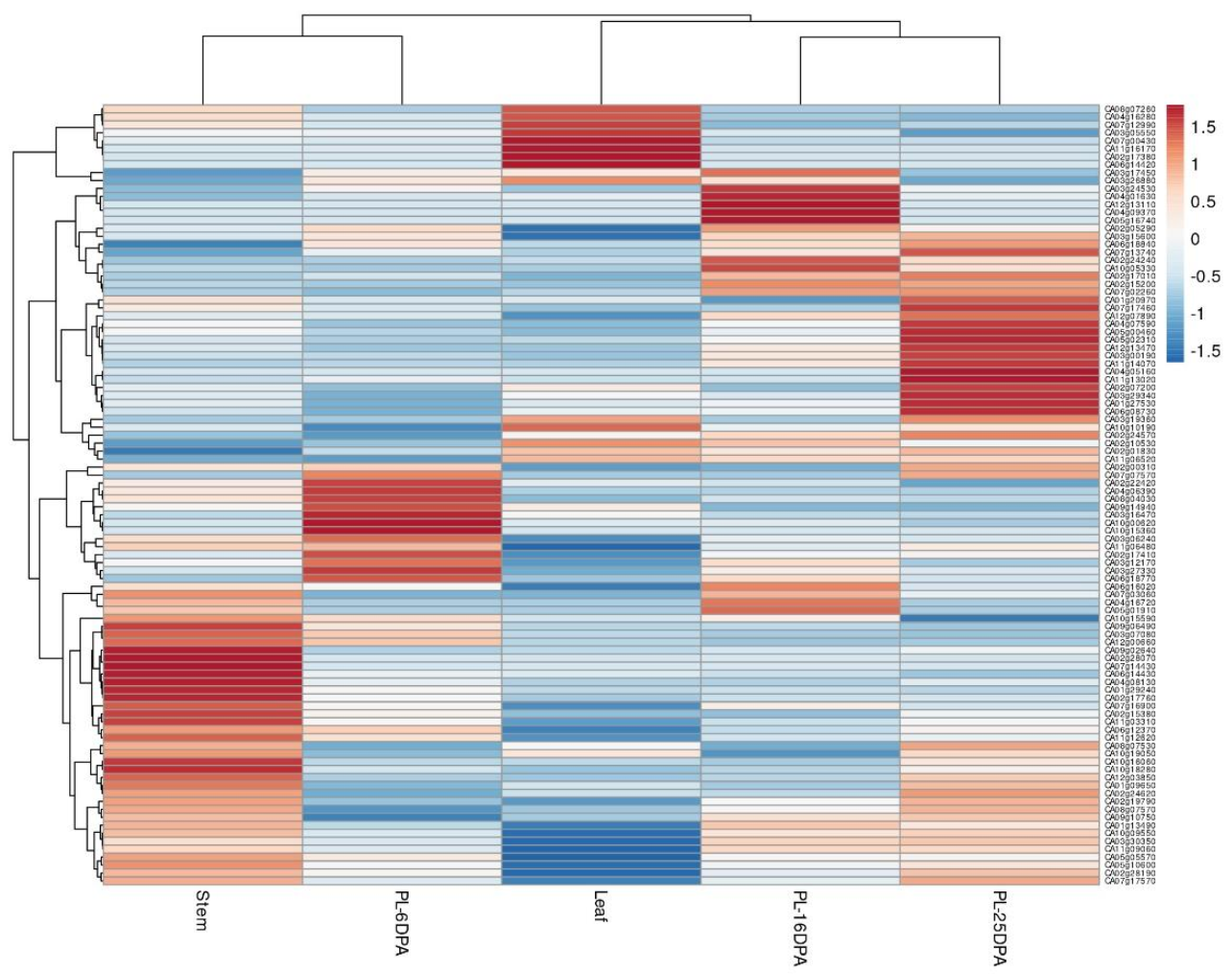
2.5. Expression Profile of miRNA Target Genes in C. annuum
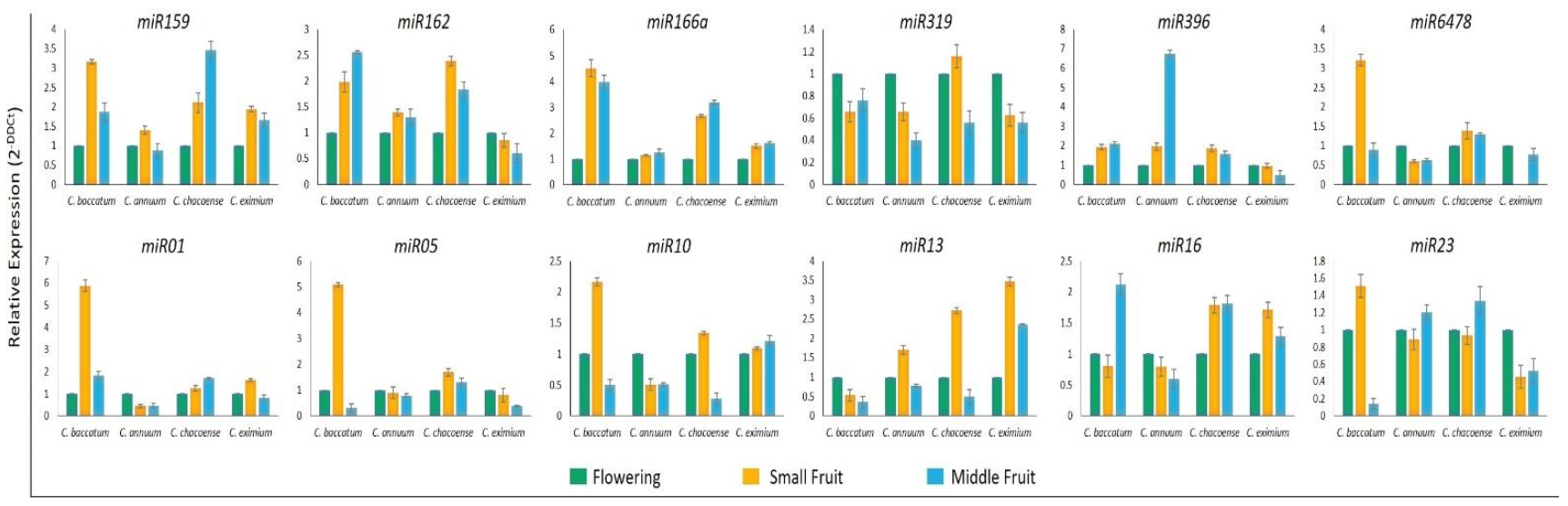
2.6. Validation of miRNA and Target Gene Expression
3. Discussion
3.1. Role of miRNAs and Their Regulators in Flowering across Capsicum Species
3.2. Role of miRNAs and Their Regulators in Pepper Fruit Development
4. Materials and Methods
4.1. Plant Materials
4.2. Construction and Sequencing of Small RNA Libraries
4.3. Bioinformatics Analysis of miRNAs
4.4. Analysis of Differentially Expressed miRNAs
4.5. Prediction of miRNA Targets and Enrichment Analyses
4.6. Quantitative RT-qPCR and Stem-Loop RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Song, Q.-X.; Liu, Y.-F.; Hu, X.-Y.; Zhang, W.-K.; Ma, B.; Chen, S.-Y.; Zhang, J.-S. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Murchison, E.P.; Hannon, G.J. miRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr. Opin. Cell Biol. 2004, 16, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Karlova, R.; van Haarst, J.C.; Maliepaard, C.; van de Geest, H.; Bovy, A.G.; Lammers, M.; Angenent, G.C.; de Maagd, R.A. Identification of microRNA targets in tomato fruit development using high-throughput sequencing and degradome analysis. J. Exp. Bot. 2013, 64, 1863–1878. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Reinhart, B.J.; Jones-Rhoades, M.W.; Tang, G.; Zamore, P.D.; Barton, M.K.; Bartel, D.P. MicroRNA control of PHABULOSA in leaf development: Importance of pairing to the microRNA 5′ region. EMBO J. 2004, 23, 3356–3364. [Google Scholar] [CrossRef]
- Bartel, B.; Bartel, D.P. MicroRNAs: At the root of plant development? Plant Physiol. 2003, 132, 709–717. [Google Scholar] [CrossRef]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef]
- Sunkar, R.; Kapoor, A.; Zhu, J.-K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef]
- Sunkar, R. MicroRNAs with macro-effects on plant stress responses. Semin. Cell Dev. Biol. 2010, 21, 805–811. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, F.; Cao, H.; Peng, H.; Ni, Z.; Sun, Q.; Yao, Y. TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS ONE 2012, 7, e48445. [Google Scholar] [CrossRef]
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.-B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef]
- Subramanian, S.; Fu, Y.; Sunkar, R.; Barbazuk, W.B.; Zhu, J.-K.; Yu, O. Novel and nodulation-regulated microRNAs in soybean roots. BMC Genom. 2008, 9, 160. [Google Scholar] [CrossRef]
- Pandey, S.P.; Shahi, P.; Gase, K.; Baldwin, I.T. Herbivory-induced changes in the small-RNA transcriptome and phytohormone signaling in Nicotiana attenuata. Proc. Natl. Acad. Sci. USA 2008, 105, 4559–4564. [Google Scholar] [CrossRef]
- Hewezi, T.; Howe, P.; Maier, T.R.; Baum, T.J. Arabidopsis small RNAs and their targets during cyst nematode parasitism. Mol. Plant Microbe Interact. 2008, 21, 1622–1634. [Google Scholar] [CrossRef]
- Liu, H.; Jin, T.; Liao, R.; Wan, L.; Xu, B.; Zhou, S.; Guan, J. miRFANs: An integrated database for Arabidopsis thaliana microRNA function annotations. BMC Plant Biol. 2012, 12, 1–8. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Knapp, S. Tobacco to tomatoes: A phylogenetic perspective on fruit diversity in the Solanaceae. J. Exp. Bot. 2002, 53, 2001–2022. [Google Scholar] [CrossRef]
- Kim, S.; Park, M.; Yeom, S.-I.; Kim, Y.-M.; Lee, J.M.; Lee, H.-A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.-T. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014, 46, 270–278. [Google Scholar] [CrossRef]
- Qin, C.; Yu, C.; Shen, Y.; Fang, X.; Chen, L.; Min, J.; Cheng, J.; Zhao, S.; Xu, M.; Luo, Y. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. USA 2014, 111, 5135–5140. [Google Scholar] [CrossRef]
- Perla, V.; Nadimi, M.; Reddy, R.; Hankins, G.R.; Nimmakayala, P.; Harris, R.T.; Valluri, J.; Sirbu, C.; Reddy, U.K. Effect of ghost pepper on cell proliferation, apoptosis, senescence and global proteomic profile in human renal adenocarcinoma cells. PLoS ONE 2018, 13, e0206183. [Google Scholar] [CrossRef]
- Hwang, D.-G.; Park, J.H.; Lim, J.Y.; Kim, D.; Choi, Y.; Kim, S.; Reeves, G.; Yeom, S.-I.; Lee, J.-S.; Park, M. The hot pepper (Capsicum annuum) microRNA transcriptome reveals novel and conserved targets: A foundation for understanding microRNA functional roles in hot pepper. PLoS ONE 2013, 8, e64238. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Ou, L.; Kang, L.; Liu, Y.; Lv, J.; Wei, G.; Yang, B.; Yang, S.; Chen, W. Identification and characterization of novel microRNAs for fruit development and quality in hot pepper (Capsicum annuum L.). Gene 2017, 608, 66–72. [Google Scholar] [CrossRef]
- Czech, B.; Hannon, G.J. Small RNA sorting: Matchmaking for Argonautes. Nat. Rev. Genet. 2011, 12, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Bologna, N.G.; Voinnet, O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu. Rev. Plant Biol. 2014, 65, 473–503. [Google Scholar] [CrossRef]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Li, S.; Zhou, H.; Long, C. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef]
- Martinez, J.; Tuschl, T. RISC is a 5′ phosphomonoester-producing RNA endonuclease. Genes Dev. 2004, 18, 975–980. [Google Scholar] [CrossRef]
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. 2009, 25, 21–44. [Google Scholar] [CrossRef]
- Samad, A.F.; Sajad, M.; Nazaruddin, N.; Fauzi, I.A.; Murad, A.; Zainal, Z.; Ismail, I. MicroRNA and transcription factor: Key players in plant regulatory network. Front. Plant Sci. 2017, 8, 565. [Google Scholar] [CrossRef]
- Djami-Tchatchou, A.T.; Sanan-Mishra, N.; Ntushelo, K.; Dubery, I.A. Functional roles of microRNAs in agronomically important plants—potential as targets for crop improvement and protection. Front. Plant Sci. 2017, 8, 378. [Google Scholar] [CrossRef]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big impact on plant development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Q.; Allan, A.C.; Yin, X.-R. Small RNAs With a Big Impact on Horticultural Traits. Crit. Rev. Plant Sci. 2020, 39, 30–43. [Google Scholar] [CrossRef]
- Hong, Y.; Jackson, S. Floral induction and flower formation—the role and potential applications of mi RNA s. Plant Biotechnol. J. 2015, 13, 282–292. [Google Scholar] [CrossRef]
- Waheed, S.; Zeng, L. The Critical Role of miRNAs in Regulation of Flowering Time and Flower Development. Genes 2020, 11, 319. [Google Scholar] [CrossRef]
- Todesco, M.; Rubio-Somoza, I.; Paz-Ares, J.; Weigel, D. A collection of target mimics for comprehensive analysis of microRNA function in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1001031. [Google Scholar] [CrossRef]
- Karlova, R.; Rosin, F.M.; Busscher-Lange, J.; Parapunova, V.; Do, P.T.; Fernie, A.R.; Fraser, P.D.; Baxter, C.; Angenent, G.C.; de Maagd, R.A. Transcriptome and metabolite profiling show that APETALA2a is a major regulator of tomato fruit ripening. Plant Cell 2011, 23, 923–941. [Google Scholar] [CrossRef]
- Porto, D.D.; Bruneau, M.; Perini, P.; Anzanello, R.; Renou, J.-P.; Santos, H.P.d.; Fialho, F.B.; Revers, L.F. Transcription profiling of the chilling requirement for bud break in apples: A putative role for FLC-like genes. J. Exp. Bot. 2015, 66, 2659–2672. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Xu, M.; Hu, T.; Zhao, J.; Park, M.-Y.; Earley, K.W.; Wu, G.; Yang, L.; Poethig, R.S. Developmental functions of miR156-regulated SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) genes in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006263. [Google Scholar] [CrossRef]
- Nag, A.; King, S.; Jack, T. miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 22534–22539. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.; Mou, M.; Chen, Y.; Xiang, S.; Chen, L.; Yu, D. Arabidopsis class II TCP transcription factors integrate with the FT–FD module to control flowering. Plant Physiol. 2019, 181, 97–111. [Google Scholar] [CrossRef]
- Wang, X.; Xu, X.; Mo, X.; Zhong, L.; Zhang, J.; Mo, B.; Kuai, B. Overexpression of TCP8 delays Arabidopsis flowering through a FLOWERING LOCUS C-dependent pathway. BMC Plant Biol. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Wang, W.; Shi, T.; Ni, X.; Xu, Y.; Qu, S.; Gao, Z. The role of miR319a and its target gene TCP4 in the regulation of pistil development in Prunus mume. Genome 2018, 61, 43–48. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, P.; Liu, S.; Yang, Q.; Guo, H. The Control of Developmental Phase Transitions by microRNAs and Their Targets in Seed Plants. Int. J. Mol. Sci. 2020, 21, 1971. [Google Scholar] [CrossRef]
- Yanhui, C.; Xiaoyuan, Y.; Kun, H.; Meihua, L.; Jigang, L.; Zhaofeng, G.; Zhiqiang, L.; Yunfei, Z.; Xiaoxiao, W.; Xiaoming, Q. The MYB transcription factor superfamily of Arabidopsis: Expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 2006, 60, 107–124. [Google Scholar] [CrossRef]
- Millar, A.A.; Lohe, A.; Wong, G. Biology and Function of miR159 in Plants. Plants 2019, 8, 255. [Google Scholar] [CrossRef]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef]
- Osakabe, Y.; Maruyama, K.; Seki, M.; Satou, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Leucine-rich repeat receptor-like kinase1 is a key membrane-bound regulator of abscisic acid early signaling in Arabidopsis. Plant Cell 2005, 17, 1105–1119. [Google Scholar] [CrossRef]
- Rayson, S.; Arciga-Reyes, L.; Wootton, L.; Zabala, M.D.T.; Truman, W.; Graham, N.; Grant, M.; Davies, B. A role for nonsense-mediated mRNA decay in plants: Pathogen responses are induced in Arabidopsis thaliana NMD mutants. PLoS ONE 2012, 7, e31917. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zhou, X. SGS3 cooperates with RDR6 in triggering geminivirus-induced gene silencing and in suppressing geminivirus infection in Nicotiana benthamiana. Viruses 2017, 9, 247. [Google Scholar] [CrossRef]
- Tripodi, P.; Greco, B. Large scale phenotyping provides insight into the diversity of vegetative and reproductive organs in a wide collection of wild and domesticated peppers (Capsicum spp.). Plants 2018, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Paran, I.; Van Der Knaap, E. Genetic and molecular regulation of fruit and plant domestication traits in tomato and pepper. J. Exp. Bot. 2007, 58, 3841–3852. [Google Scholar] [CrossRef] [PubMed]
- Paran, I.; Fallik, E. Breeding for fruit quality in pepper (Capsicum spp.). Breed. Fruit Qual. 2011, 307–322. [Google Scholar] [CrossRef]
- FAN, S.-s.; LI, Q.-n.; GUO, G.-j.; GAO, J.-c.; WANG, X.-x.; GUO, Y.-m.; Snyder, J.C.; DU, Y.-c. Identification of microRNAs in two species of tomato, Solanum lycopersicum and Solanum habrochaites, by deep sequencing. J. Integr. Agric. 2015, 14, 42–49. [Google Scholar] [CrossRef]
- Chen, W.; Kong, J.; Lai, T.; Manning, K.; Wu, C.; Wang, Y.; Qin, C.; Li, B.; Yu, Z.; Zhang, X. Tuning LeSPL-CNR expression by SlymiR157 affects tomato fruit ripening. Sci. Rep. 2015, 5, 1–6. [Google Scholar] [CrossRef]
- Swetha, C.; Basu, D.; Pachamuthu, K.; Tirumalai, V.; Nair, A.; Prasad, M.; Shivaprasad, P.V. Major domestication-related phenotypes in indica rice are due to loss of miRNA-mediated laccase silencing. Plant Cell 2018, 30, 2649–2662. [Google Scholar] [CrossRef]
- Chen, X.; Xia, J.; Xia, Z.; Zhang, H.; Zeng, C.; Lu, C.; Zhang, W.; Wang, W. Potential functions of microRNAs in starch metabolism and development revealed by miRNA transcriptome profiling of cassava cultivars and their wild progenitor. BMC Plant Biol. 2015, 15, 1–11. [Google Scholar] [CrossRef]
- Zeng, S.; Liu, Y.; Pan, L.; Hayward, A.; Wang, Y. Identification and characterization of miRNAs in ripening fruit of Lycium barbarum L. using high-throughput sequencing. Front. Plant Sci. 2015, 6, 778. [Google Scholar] [CrossRef]
- Saminathan, T.; Alvarado, A.; Lopez, C.; Shinde, S.; Gajanayake, B.; Abburi, V.L.; Vajja, V.G.; Jagadeeswaran, G.; Reddy, K.R.; Nimmakayala, P. Elevated carbon dioxide and drought modulate physiology and storage-root development in sweet potato by regulating microRNAs. Funct. Integr. Genom. 2019, 19, 171–190. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, Y.; Zhu, A.; Wu, X.; Ye, J.; Yu, K.; Guo, W.; Deng, X. Discovery and comparative profiling of microRNAs in a sweet orange red-flesh mutant and its wild type. BMC Genom. 2010, 11, 1–17. [Google Scholar] [CrossRef]
- Hou, Y.; Zhai, L.; Li, X.; Xue, Y.; Wang, J.; Yang, P.; Cao, C.; Li, H.; Cui, Y.; Bian, S. Comparative analysis of fruit ripening-related miRNAs and their targets in blueberry using small RNA and degradome sequencing. Int. J. Mol. Sci. 2017, 18, 2767. [Google Scholar] [CrossRef]
- Manohar, S.; Jagadeeswaran, G.; Nimmakayala, P.; Tomason, Y.; Almeida, A.; Sunkar, R.; Levi, A.; Reddy, U.K. Dynamic regulation of novel and conserved miRNAs across various tissues of diverse cucurbit species. Plant Mol. Biol. Report. 2013, 31, 335–343. [Google Scholar] [CrossRef]
- Chung, M.Y.; Vrebalov, J.; Alba, R.; Lee, J.; McQuinn, R.; Chung, J.D.; Klein, P.; Giovannoni, J. A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J. 2010, 64, 936–947. [Google Scholar] [CrossRef]
- Yao, J.-L.; Tomes, S.; Xu, J.; Gleave, A.P. How microRNA172 affects fruit growth in different species is dependent on fruit type. Plant Signal. Behav. 2016, 11, 417–427. [Google Scholar] [CrossRef]
- Zhang, X.; Zou, Z.; Zhang, J.; Zhang, Y.; Han, Q.; Hu, T.; Xu, X.; Liu, H.; Li, H.; Ye, Z. Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant. FEBS Lett. 2011, 585, 435–439. [Google Scholar] [CrossRef]
- Yao, J.L.; Xu, J.; Cornille, A.; Tomes, S.; Karunairetnam, S.; Luo, Z.; Bassett, H.; Whitworth, C.; Rees-George, J.; Ranatunga, C. A micro RNA allele that emerged prior to apple domestication may underlie fruit size evolution. Plant J. 2015, 84, 417–427. [Google Scholar] [CrossRef]
- da Silva, E.M.; Silva, G.F.F.e.; Bidoia, D.B.; da Silva Azevedo, M.; de Jesus, F.A.; Pino, L.E.; Peres, L.E.P.; Carrera, E.; López-Díaz, I.; Nogueira, F.T.S. micro RNA 159-targeted Sl GAMYB transcription factors are required for fruit set in tomato. Plant J. 2017, 92, 95–109. [Google Scholar] [CrossRef]
- Vallarino, J.G.; Osorio, S.; Bombarely, A.; Casañal, A.; Cruz-Rus, E.; Sánchez-Sevilla, J.F.; Amaya, I.; Giavalisco, P.; Fernie, A.R.; Botella, M.A. Central role of Fa GAMYB in the transition of the strawberry receptacle from development to ripening. New Phytol. 2015, 208, 482–496. [Google Scholar] [CrossRef]
- Nimmakayala, P.; Abburi, V.L.; Saminathan, T.; Alaparthi, S.B.; Almeida, A.; Davenport, B.; Nadimi, M.; Davidson, J.; Tonapi, K.; Yadav, L. Genome-wide diversity and association mapping for capsaicinoids and fruit weight in Capsicum annuum L. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Durbak, A.R.; Tax, F.E. CLAVATA signaling pathway receptors of Arabidopsis regulate cell proliferation in fruit organ formation as well as in meristems. Genetics 2011, 189, 177–194. [Google Scholar] [CrossRef]
- Muños, S.; Ranc, N.; Botton, E.; Bérard, A.; Rolland, S.; Duffé, P.; Carretero, Y.; Le Paslier, M.-C.; Delalande, C.; Bouzayen, M. Increase in tomato locule number is controlled by two single-nucleotide polymorphisms located near WUSCHEL. Plant Physiol. 2011, 156, 2244–2254. [Google Scholar] [CrossRef]
- Zhang, N.; Brewer, M.T.; van der Knaap, E. Fine mapping of fw3. 2 controlling fruit weight in tomato. Theor. Appl. Genet. 2012, 125, 273–284. [Google Scholar] [CrossRef]
- Lopez-Ortiz, C.; Dutta, S.K.; Natarajan, P.; Peña-Garcia, Y.; Abburi, V.; Saminathan, T.; Nimmakayala, P.; Reddy, U.K. Genome-wide identification and gene expression pattern of ABC transporter gene family in Capsicum spp. PLoS ONE 2019, 14, e0215901. [Google Scholar] [CrossRef]
- Chen, C.; Zeng, Z.; Liu, Z.; Xia, R. Small RNAs, emerging regulators critical for the development of horticultural traits. Hortic. Res. 2018, 5, 1–14. [Google Scholar] [CrossRef]
- De Jong, M.; Wolters-Arts, M.; Feron, R.; Mariani, C.; Vriezen, W.H. The Solanum lycopersicum auxin response factor 7 (SlARF7) regulates auxin signaling during tomato fruit set and development. Plant J. 2009, 57, 160–170. [Google Scholar] [CrossRef]
- Yang, T.; Wang, Y.; Teotia, S.; Wang, Z.; Shi, C.; Sun, H.; Gu, Y.; Zhang, Z.; Tang, G. The interaction between miR160 and miR165/166 in the control of leaf development and drought tolerance in Arabidopsis. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Liu, J.; Pei, W.; Li, X.; Wang, N.; Ma, J.; Zang, X.; Zhang, J.; Yu, S.; Wu, M. Analysis of the MIR160 gene family and the role of MIR160a_A05 in regulating fiber length in cotton. Planta 2019, 250, 2147–2158. [Google Scholar] [CrossRef]
- Damodharan, S.; Zhao, D.; Arazi, T. A common mi RNA 160-based mechanism regulates ovary patterning, floral organ abscission and lamina outgrowth in tomato. Plant J. 2016, 86, 458–471. [Google Scholar] [CrossRef]
- Hendelman, A.; Buxdorf, K.; Stav, R.; Kravchik, M.; Arazi, T. Inhibition of lamina outgrowth following Solanum lycopersicum AUXIN RESPONSE FACTOR 10 (SlARF10) derepression. Plant Mol. Biol. 2012, 78, 561–576. [Google Scholar] [CrossRef]
- Houben, M.; Van de Poel, B. 1-Aminocyclopropane-1-carboxylic acid oxidase (ACO): The enzyme that makes the plant hormone ethylene. Front. Plant Sci. 2019, 10, 695. [Google Scholar] [CrossRef]
- Grierson, D. 10 Ethylene Biosynthesis. In Fruit Ripening: Physiology, Signalling and Genomics; CABI: London, UK, 2014; p. 178. [Google Scholar]
- Van de Poel, B.; Vandenzavel, N.; Smet, C.; Nicolay, T.; Bulens, I.; Mellidou, I.; Vandoninck, S.; Hertog, M.L.; Derua, R.; Spaepen, S. Tissue specific analysis reveals a differential organization and regulation of both ethylene biosynthesis and E8 during climacteric ripening of tomato. BMC Plant Biol. 2014, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Wang, M.; Zhang, H.-Y.; Liu, J.-H. The miR396b of Poncirus trifoliata functions in cold tolerance by regulating ACC oxidase gene expression and modulating ethylene–polyamine homeostasis. Plant Cell Physiol. 2016, 57, 1865–1878. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chung, E.-J.; Joung, Y.-H.; Choi, D. Non-climacteric fruit ripening in pepper: Increased transcription of EIL-like genes normally regulated by ethylene. Funct. Integr. Genom. 2010, 10, 135–146. [Google Scholar] [CrossRef]
- Osorio, S.; Alba, R.; Nikoloski, Z.; Kochevenko, A.; Fernie, A.R.; Giovannoni, J.J. Integrative comparative analyses of transcript and metabolite profiles from pepper and tomato ripening and development stages uncovers species-specific patterns of network regulatory behavior. Plant Physiol. 2012, 159, 1713–1729. [Google Scholar] [CrossRef]
- Hou, B.-Z.; Li, C.-L.; Han, Y.-Y.; Shen, Y.-Y. Characterization of the hot pepper (Capsicum frutescens) fruit ripening regulated by ethylene and ABA. BMC Plant Biol. 2018, 18, 1–12. [Google Scholar] [CrossRef]
- Villavicencio, L.E.; Blankenship, S.M.; Sanders, D.C.; Swallow, W.H. Ethylene and carbon dioxide concentrations in attached fruits of pepper cultivars during ripening. Sci. Hortic. 2001, 91, 17–24. [Google Scholar] [CrossRef]
- Zhu, M.; Chen, G.; Zhou, S.; Tu, Y.; Wang, Y.; Dong, T.; Hu, Z. A new tomato NAC (N AM/A TAF1/2/C UC2) transcription factor, SlNAC4, functions as a positive regulator of fruit ripening and carotenoid accumulation. Plant Cell Physiol. 2014, 55, 119–135. [Google Scholar] [CrossRef]
- Jagadeesh, B.H.; Prabha, T.N.; Srinivasan, K. Activities of glycosidases during fruit development and ripening of tomato (Lycopersicum esculantum L.): Implication in fruit ripening. Plant Sci. 2004, 166, 1451–1459. [Google Scholar] [CrossRef]
- Ghosh, S.; Meli, V.S.; Kumar, A.; Thakur, A.; Chakraborty, N.; Chakraborty, S.; Datta, A. The N-glycan processing enzymes α-mannosidase and β-D-N-acetylhexosaminidase are involved in ripening-associated softening in the non-climacteric fruits of capsicum. J. Exp. Bot. 2011, 62, 571–582. [Google Scholar] [CrossRef]
- Mammadov, J.; Buyyarapu, R.; Guttikonda, S.K.; Parliament, K.; Abdurakhmonov, I.Y.; Kumpatla, S.P. Wild relatives of maize, rice, cotton, and soybean: Treasure troves for tolerance to biotic and abiotic stresses. Front. Plant Sci. 2018, 9, 886. [Google Scholar] [CrossRef]
- Bae, C.; Kim, S.-m.; Lee, D.J.; Choi, D. Multiple classes of immune-related proteases associated with the cell death response in pepper plants. PLoS ONE 2013, 8, e63533. [Google Scholar] [CrossRef]
- Chen, R.; Guo, W.; Yin, Y.; Gong, Z.-H. A novel F-box protein CaF-box is involved in responses to plant hormones and abiotic stress in pepper (Capsicum annuum L.). Int. J. Mol. Sci. 2014, 15, 2413–2430. [Google Scholar] [CrossRef]
- Lim, J.; Lim, C.W.; Lee, S.C. Functional Analysis of Pepper F-box Protein CaDIF1 and Its Interacting Partner CaDIS1: Modulation of ABA Signaling and Drought Stress Response. Front. Plant Sci. 2019, 10, 1365. [Google Scholar] [CrossRef]
- Harvey, J.J.; Lewsey, M.G.; Patel, K.; Westwood, J.; Heimstädt, S.; Carr, J.P.; Baulcombe, D.C. An antiviral defense role of AGO2 in plants. PLoS ONE 2011, 6, e14639. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J. Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi-Gasic, E.; Hellens, R.P. Quantitative stem-loop RT-PCR for detection of microRNAs. In RNAi and Plant Gene Function Analysis; Springer: Berlin/Heidelberg, Germany, 2011; pp. 145–157. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Capsicum baccatum | Capsicum annuum | Capsicum chacoense | Capsicum eximium | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Flowering | Small Fruit | Middle Fruit | Flowering | Small Fruit | Middle Fruit | Flowering | Small Fruit | Middle Fruit | Flowering | Small Fruit | Middle Fruit | miRNA Sequence |
| miR156 | 20 | 10 | 25 | 10 | 0 | 6 | 6 | 5 | 0 | 9 | 7 | 9 | UGACAGAAGAGAGUGAGCAC |
| miR157 | 47 | 12 | 0 | 281 | 0 | 0 | 0 | 0 | 68 | 132 | 0 | 9 | UUGACAGAAGAUAGAGAGCAC |
| miR159 | 7417 | 32107 | 22478 | 10667 | 3720 | 2206 | 578 | 1893 | 1419 | 6182 | 3176 | 3912 | UUUGGAUUGAAGGGAGCUCUA |
| miR160 | 0 | 36 | 30 | 31 | 0 | 0 | 0 | 0 | 19 | 14 | 0 | 0 | UGCCUGGCUCCCUGUAUGCCA |
| miR162 | 26 | 733 | 1252 | 385 | 260 | 262 | 6 | 30 | 19 | 187 | 7 | 45 | UCGAUAAACCUCUGCAUCCAG |
| miR164 | 194 | 55 | 598 | 377 | 52 | 334 | 25 | 123 | 1422 | 83 | 86 | 204 | UGGAGAAGCAGGGCACGUGC |
| miR165 | 31 | 17 | 14 | 7 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 35 | GUUGAGGGGAAUGUUGUCUGG |
| miR166a | 1699 | 7225 | 7342 | 2025 | 729 | 815 | 383 | 1498 | 1167 | 1234 | 1523 | 1477 | UCGGACCAGGCUUCAUUCCCC |
| miR166 | 0 | 16 | 25 | 1718 | 313 | 0 | 29 | 183 | 24 | 713 | 624 | 631 | GGGGAAUGUUGUCUGGCUCG |
| miR167 | 116 | 354 | 725 | 302 | 8 | 44 | 0 | 29 | 7 | 0 | 16 | 51 | UGAAGCUGCCAGCAUGAUCUA |
| miR168 | 11 | 46 | 79 | 32 | 10 | 52 | 0 | 0 | 0 | 35 | 0 | 13 | CCCGCCUUGCAUCAACUGAAU |
| miR171 | 77 | 363 | 495 | 372 | 102 | 42 | 0 | 38 | 6 | 106 | 26 | 41 | UAUUGGCCUGGUUCACUCAGA |
| miR172 | 68 | 21 | 15 | 108 | 0 | 8 | 11 | 0 | 15 | 34 | 0 | 0 | AGAAUCUUGAUGAUGCUGCAU |
| miR319 | 2098 | 403 | 121 | 4461 | 154 | 90 | 290 | 126 | 54 | 2027 | 604 | 484 | UUGGACUGAAGGGAGCUCCC |
| miR390 | 320 | 305 | 197 | 92 | 8 | 0 | 0 | 0 | 0 | 45 | 0 | 0 | AAGCUCAGGAGGGAUAGCGC |
| miR394 | 29 | 303 | 193 | 118 | 19 | 0 | 0 | 8 | 0 | 38 | 0 | 0 | UUGGCAUUCUGUCCACCUCC |
| miR395 | 106 | 26 | 17 | 63 | 0 | 41 | 0 | 44 | 45 | 107 | 0 | 0 | CUGAAGUGUUUGGGGGAACUC |
| miR396 | 144 | 2668 | 3232 | 778 | 507 | 1681 | 9 | 282 | 351 | 292 | 27 | 144 | UUCCACAGCUUUCUUGAACUG |
| miR398 | 25 | 20 | 428 | 641 | 0 | 79 | 0 | 196 | 20 | 1618 | 0 | 3520 | UGUGUUCUCAGGUCACCCCUU |
| miR403 | 123 | 1819 | 1584 | 35 | 235 | 235 | 0 | 55 | 53 | 254 | 14 | 86 | UUAGAUUCACGCACAAACUCG |
| miR408 | 0 | 7 | 117 | 0 | 0 | 38 | 0 | 0 | 8 | 110 | 147 | 79 | UGCACUGCCUCUUCCCUGGCU |
| miR6478 | 2750 | 15436 | 2122 | 10213 | 6236 | 4524 | 1001 | 3789 | 1508 | 9530 | 2404 | 4669 | CCGACCUUAGCUCAGUUGGUAGA |
| Capsicum baccatum | Capsicum annuum | Capsicum chacoense | Capsicum eximium | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | MFE | Flowering | Small Fruit | Middle Fruit | Flowering | Small Fruit | Middle Fruit | Flowering | Small Fruit | Middle Fruit | Flowering | Small Fruit | Middle Fruit | miRNA Sequence |
| miR01 | −18.8 | 57 | 11265 | 465 | 9 | 0 | 0 | 0 | 11 | 0 | 231 | 236 | 89 | AACCCUGAACCCUGAACCCU |
| miR02 | −30.7 | 11 | 85 | 11380 | 0 | 0 | 15 | 0 | 0 | 0 | 0 | 61 | 253 | AGGGAUGGCCUUGGCUCAGC |
| miR03 | −49.9 | 158 | 120 | 1468 | 54 | 30 | 29 | 126 | 126 | 172 | 102 | 214 | 86 | GAAGUCCUCGUGUUGCAUCCCU |
| miR04 | −47.9 | 0 | 252 | 23 | 26 | 36 | 19 | 0 | 0 | 0 | 25 | 0 | 0 | GACUAGGACGGUCUGAGGCUU |
| miR05 | −46.3 | 1252 | 12908 | 32 | 640 | 568 | 432 | 92 | 214 | 60 | 402 | 137 | 130 | GCACCAGUGGUCUAGUGGUAGAAU |
| miR06 | −18.3 | 75 | 715 | 584 | 359 | 183 | 131 | 8 | 122 | 38 | 381 | 36 | 136 | GCCCGUCUAGCUCAGUUGGUAGA |
| miR07 | −62.6 | 7487 | 0 | 10012 | 2965 | 0 | 2156 | 4328 | 0 | 14605 | 6921 | 4728 | 5898 | GCCGGCCGGGGGACGGACUG |
| miR08 | −56.1 | 0 | 55 | 592 | 17 | 0 | 12 | 0 | 22 | 66 | 49 | 13 | 35 | GCCGUCUUAGCUCAGCGGUA |
| miR09 | −30.3 | 0 | 89 | 1532 | 68 | 90 | 48 | 0 | 0 | 320 | 165 | 0 | 0 | GCCGUCUUAGCUCAGUGGUAGAGC |
| miR10 | −30.8 | 865 | 3358 | 64 | 416 | 0 | 0 | 62 | 452 | 0 | 654 | 389 | 309 | GCGCCUGUAGCUCAGUGGAUA |
| miR11 | −44.8 | 0 | 42 | 1827 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | GCGGAAGAUCCUGAAUUUGAGACU |
| miR12 | −23.7 | 239 | 530 | 8656 | 194 | 215 | 173 | 268 | 148 | 0 | 274 | 217 | 150 | GCGGGGAUAGCUCAGUUGGGAGA |
| miR13 | −27.6 | 7777 | 18 | 140 | 6089 | 4959 | 2735 | 4746 | 9380 | 705 | 7712 | 8554 | 8715 | GCGUCUGUAGUCCAACGGUUAGG |
| miR14 | −26.5 | 1317 | 8787 | 118 | 7068 | 18987 | 16637 | 1832 | 4800 | 9219 | 11268 | 421 | 2526 | GCUCAGUGGUAGAGCAUUUGACU |
| miR15 | −37 | 1854 | 2927 | 265 | 1155 | 549 | 314 | 1232 | 2325 | 2893 | 2456 | 1162 | 1624 | GGAUGCGAUCAUACCAGCACU |
| miR16 | −34.3 | 697 | 543 | 3629 | 224 | 125 | 0 | 754 | 750 | 550 | 340 | 742 | 371 | GGGAAGUCCUCGUGUUGCAUCCCU |
| miR17 | −29.7 | 539 | 468 | 21 | 609 | 131 | 148 | 121 | 383 | 302 | 753 | 128 | 288 | GGGAUUGUAGUUCAAUCGGUCAGA |
| miR18 | −15.9 | 0 | 0 | 4668 | 0 | 493 | 772 | 0 | 0 | 1690 | 0 | 0 | 0 | GGGGAUGUAGCUCAAAUGGU |
| miR19 | −18.1 | 8955 | 3485 | 0 | 1592 | 0 | 0 | 7949 | 8286 | 0 | 3517 | 10077 | 7444 | GGGGAUGUAGCUCAAAUGGUAGA |
| miR20 | −23 | 0 | 0 | 0 | 0 | 1815 | 1882 | 0 | 0 | 6380 | 5022 | 0 | 0 | GGGGAUGUAGCUCAGAUGGUA |
| miR21 | −24.9 | 11206 | 4473 | 0 | 3305 | 0 | 0 | 14304 | 13268 | 0 | 0 | 16839 | 12566 | GGGGAUGUAGCUCAGAUGGUAGA |
| miR22 | −55.1 | 210 | 501 | 456 | 455 | 312 | 0 | 190 | 321 | 413 | 1032 | 118 | 372 | GUCGAUAUGUCCGAGUGGUUAAGG |
| miR23 | −50.5 | 1781 | 2755 | 12 | 1676 | 811 | 726 | 771 | 1077 | 1094 | 2750 | 1114 | 1937 | GUGGACGUGCCGGAGUGGUUAUC |
| miR24 | −40.6 | 50 | 0 | 0 | 56 | 11 | 0 | 31 | 33 | 32 | 89 | 68 | 60 | GUGGGCGUGCCGGAGUGGUUAUC |
| miR25 | −36.6 | 0 | 0 | 0 | 0 | 0 | 0 | 285 | 103 | 115 | 0 | 143 | 0 | UAGUGGUAUGAUUCUCGCUU |
| miR26 | −37.8 | 98 | 0 | 0 | 0 | 238 | 112 | 438 | 235 | 0 | 429 | 668 | 159 | UAGUGGUCAGGACAUUGGACU |
| miR27 | −30 | 0 | 270 | 62 | 33 | 0 | 0 | 0 | 0 | 0 | 20 | 0 | 0 | UCACCAUCUUUCGGCUGAGAUU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Ortiz, C.; Peña-Garcia, Y.; Bhandari, M.; Abburi, V.L.; Natarajan, P.; Stommel, J.; Nimmakayala, P.; Reddy, U.K. Identification of miRNAs and Their Targets Involved in Flower and Fruit Development across Domesticated and Wild Capsicum Species. Int. J. Mol. Sci. 2021, 22, 4866. https://doi.org/10.3390/ijms22094866
Lopez-Ortiz C, Peña-Garcia Y, Bhandari M, Abburi VL, Natarajan P, Stommel J, Nimmakayala P, Reddy UK. Identification of miRNAs and Their Targets Involved in Flower and Fruit Development across Domesticated and Wild Capsicum Species. International Journal of Molecular Sciences. 2021; 22(9):4866. https://doi.org/10.3390/ijms22094866
Chicago/Turabian StyleLopez-Ortiz, Carlos, Yadira Peña-Garcia, Menuka Bhandari, Venkata Lakshmi Abburi, Purushothaman Natarajan, John Stommel, Padma Nimmakayala, and Umesh K. Reddy. 2021. "Identification of miRNAs and Their Targets Involved in Flower and Fruit Development across Domesticated and Wild Capsicum Species" International Journal of Molecular Sciences 22, no. 9: 4866. https://doi.org/10.3390/ijms22094866
APA StyleLopez-Ortiz, C., Peña-Garcia, Y., Bhandari, M., Abburi, V. L., Natarajan, P., Stommel, J., Nimmakayala, P., & Reddy, U. K. (2021). Identification of miRNAs and Their Targets Involved in Flower and Fruit Development across Domesticated and Wild Capsicum Species. International Journal of Molecular Sciences, 22(9), 4866. https://doi.org/10.3390/ijms22094866