
How an Infection of Sheep Revealed Prion Mechanisms in Alzheimer’s Disease and Other Neurodegenerative Disorders

Abstract
1. Introduction
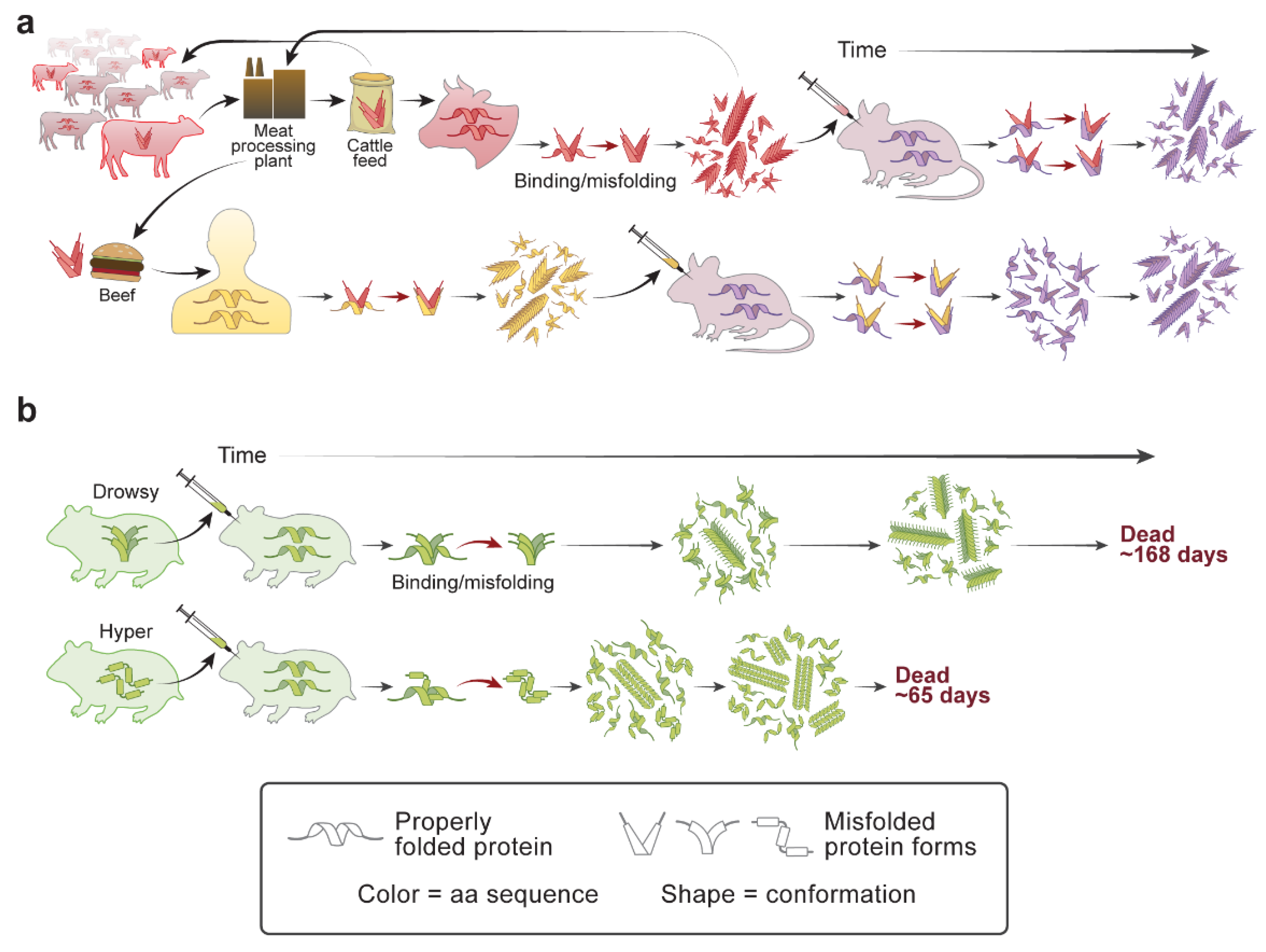
2. Scrapie and the Prion Protein
3. Prion Protein and Scrapie Incubation Time Genes Are Linked
4. Scrapie Strains
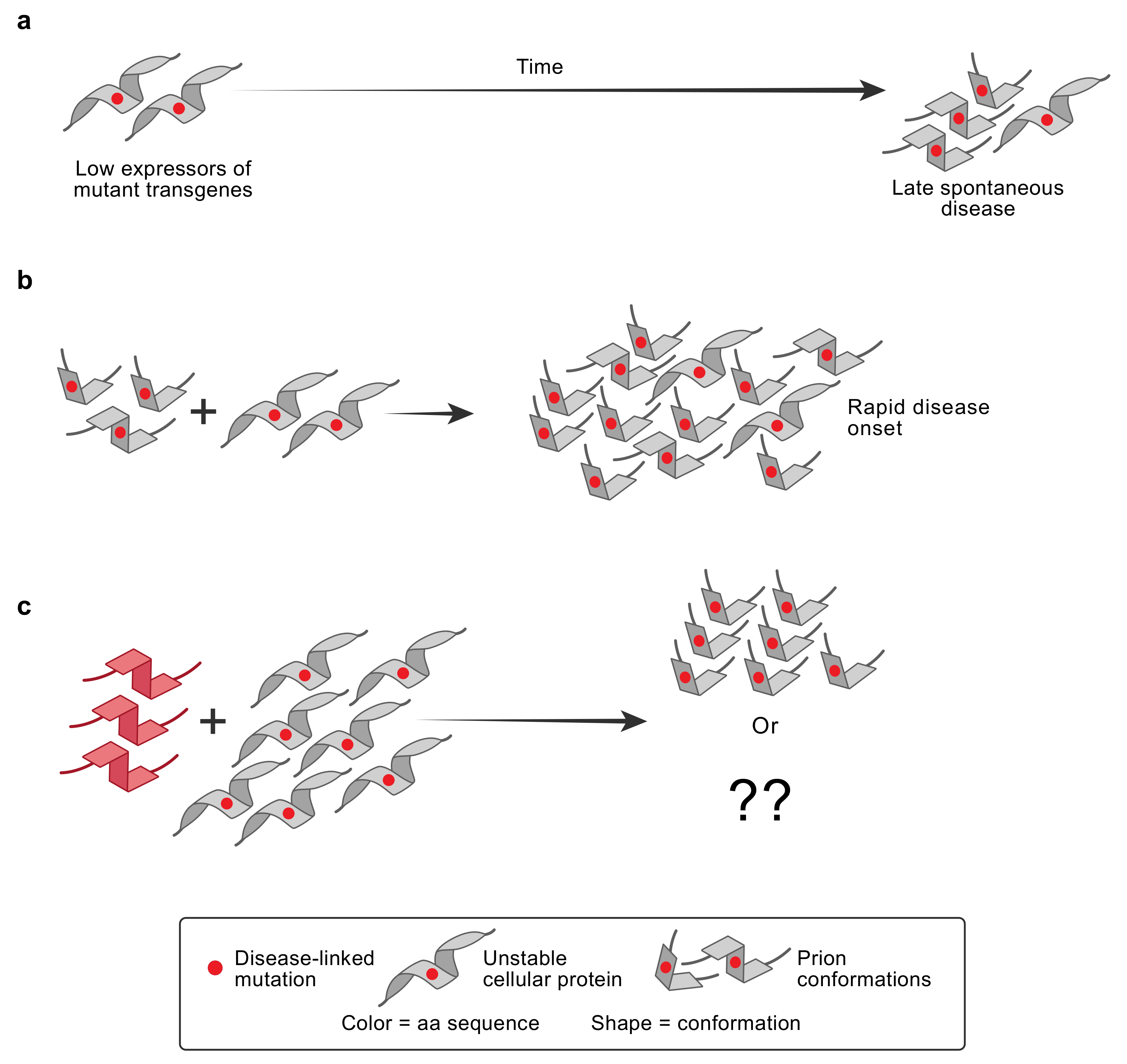
5. PRNP and Familial Prion Diseases
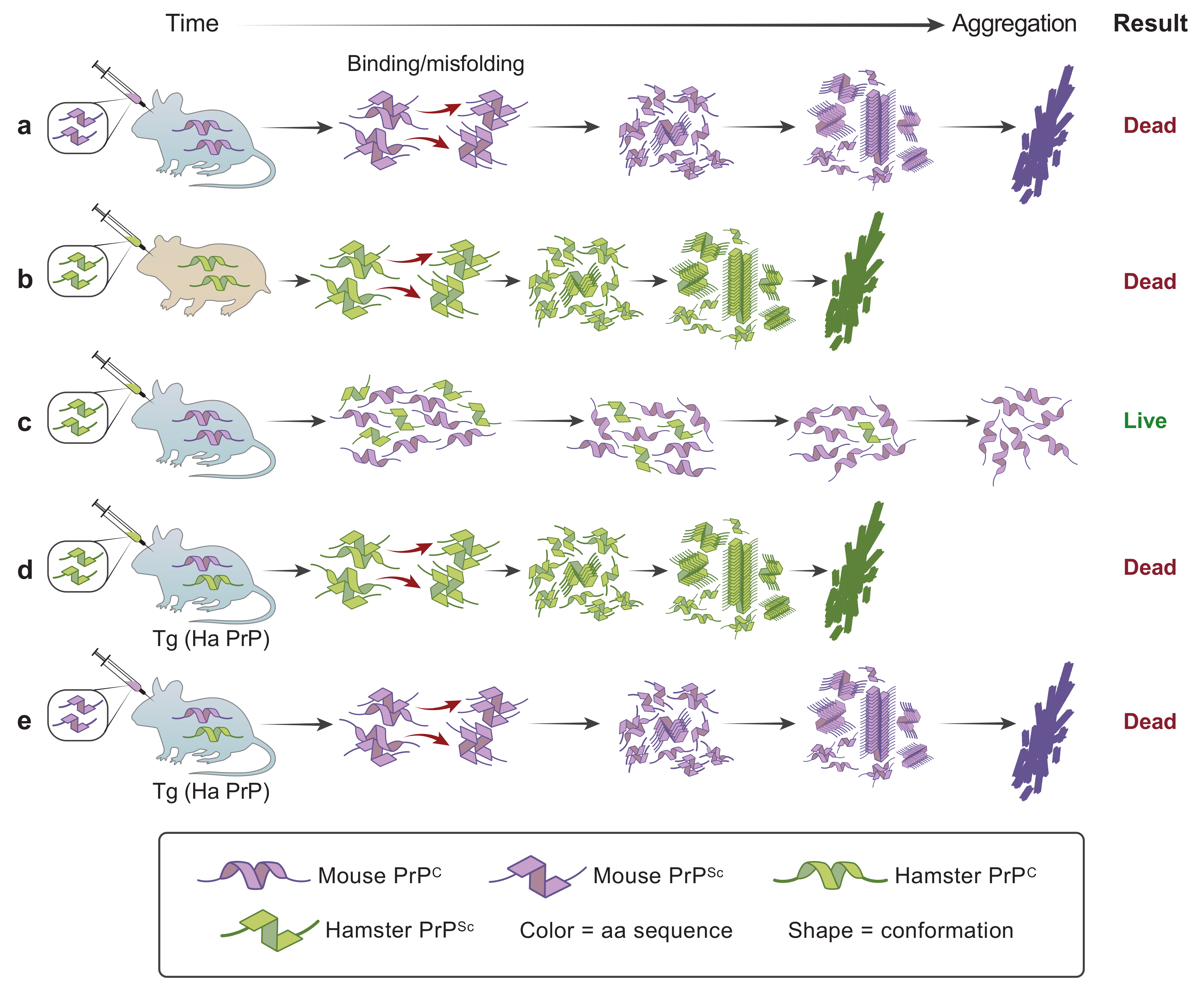
6. Protein Conformation Enciphers Heritable Information
7. Genetic Control of Susceptibility to Human PrP Prion Diseases
8. Prion Replication of Misfolded Proteins Causing AD, PD, and MSA
9. Aβ Prions
10. Tau Prions
11. Alzheimer’s Disease Is a Double Prion Disorder
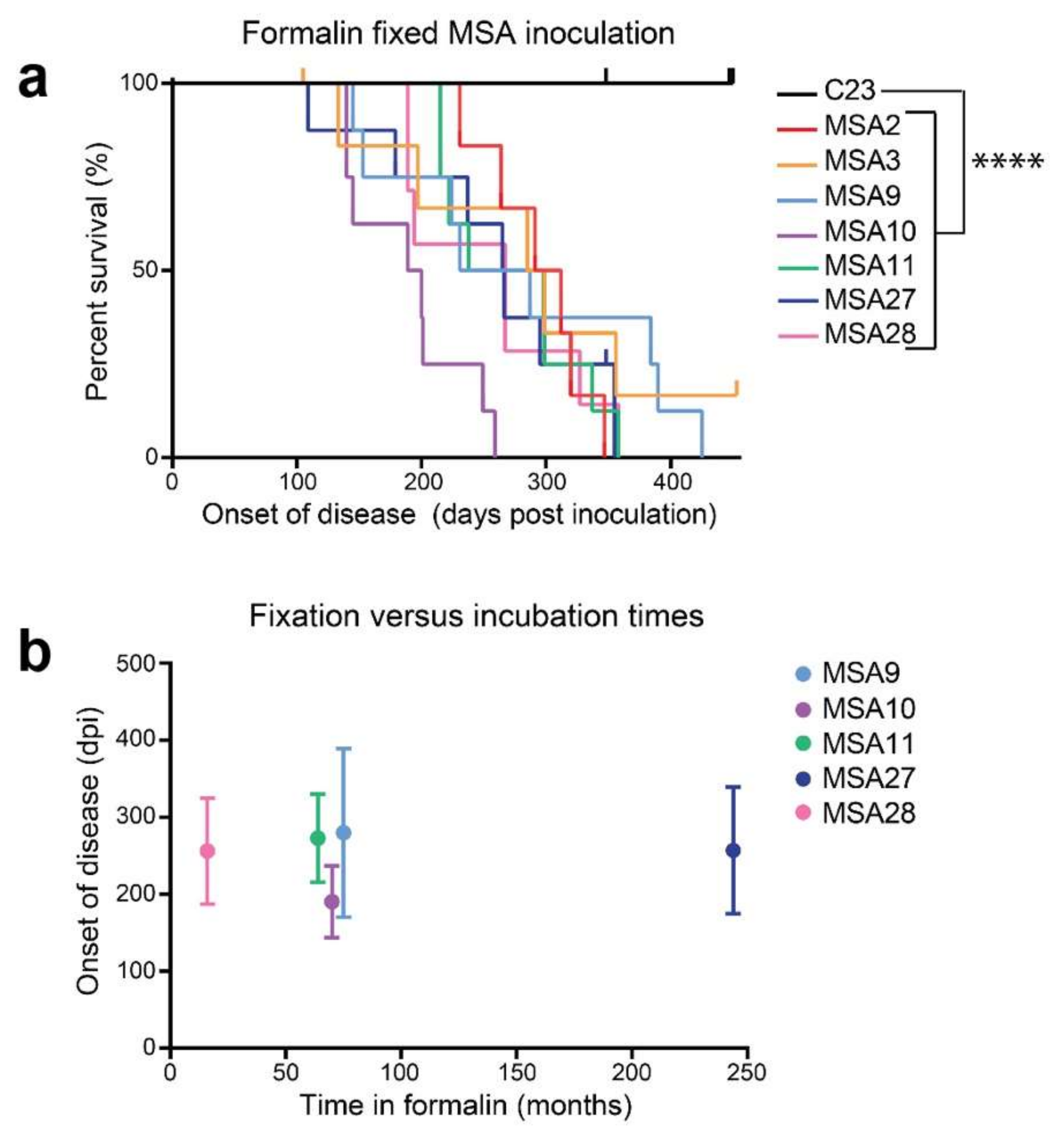
12. α-Synuclein Prions
13. The Quest for Human Prion Disease Therapeutics
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| BSE | Bovine spongiform encephalopathy |
| CAA | Cerebral amyloid angiopathy |
| CBD | Corticobasal degeneration |
| CJD | Creutzfeldt–Jakob disease |
| Cryo-EM | Cryo-electron microscopy |
| CTE | Chronic traumatic encephalopathy |
| DY | Drowsy |
| fAD | Familial Alzheimer’s disease |
| FFI | Fatal familial insomnia |
| FTD | Frontotemporal dementia |
| GCIs | Glial cytoplasmic inclusions |
| GSS | Gerstmann–Sträussler–Scheinker |
| HY | Hyper |
| iCJD | Iatrogenic Creutzfeldt–Jakob disease |
| MSA | Multiple system atrophy |
| NDs | Neurodegenerative diseases |
| NFTs | Neurofibrillary tangles |
| PD | Parkinson’s disease |
| PK | Proteinase K |
| PrP | Prion protein |
| RDs | Repeat domains |
| RML | Rocky Mountain Lab |
| sFI | Sporadic fatal insomnia |
| SHaPrP | Syrian hamster prion protein |
| TME | Transmissible mink encephalopathy |
| vCJD | Variant Creutzfeldt–Jakob disease |
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. (Ed.) An introduction to "Prion Biology". In Prion Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; pp. 1–15. [Google Scholar]
- Ayers, J.I.; Paras, N.A.; Prusiner, S.B. Expanding spectrum of prion diseases. Emerg. Top. Life Sci. 2020, 4, 155–167. [Google Scholar] [PubMed]
- Carlson, G.A. Prion protein and genetic susceptibility to diseases caused by its misfolding. Prog. Mol. Biol. Transl. Sci. 2017, 150, 123–145. [Google Scholar] [PubMed]
- Dugger, B.N.; Perl, D.P.; Carlson, G.A. Neurodegenerative disease transmission and transgenesis in mice. In Prion Biology; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; pp. 299–318. [Google Scholar]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.M.; Baker, H.F.; Windle, C.P.; Cummings, R.M. Very long term studies of the seeding of beta-amyloidosis in primates. J. Neural Transm. 2006, 113, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Tolnay, M.; Goedert, M. The prion-like behavior of assembled tau in transgenic mice. In Prion Diseases; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; pp. 137–148. [Google Scholar]
- Holmes, B.B.; Diamond, M.I. Cellular models for the study of prions. In Prion Diseases; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; pp. 127–136. [Google Scholar]
- Woerman, A.L.; Aoyagi, A.; Patel, S.; Kazmi, S.A.; Lobach, I.; Grinberg, L.T.; McKee, A.C.; Seeley, W.W.; Olson, S.H.; Prusiner, S.B. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc. Natl. Acad. Sci. USA 2016, 113, E8187–E8196. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef]
- Woerman, A.L.; Stöhr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef]
- Schneider, K.; Fangerau, H.; Michaelsen, B.; Raab, W.H.-M. The early history of the transmissible spongiform encephalopathies exemplified by scrapie. Brain Res. Bull. 2008, 77, 343–355. [Google Scholar] [CrossRef]
- Cuillé, J.; Chelle, P.-L. La maladie dite tremblante du mouton est-elle inoculable? C. R. Acad. Sci. 1936, 203, 1552–1554. [Google Scholar]
- Cuillé, J.; Chelle, P.-L. Transmission experimentale de la tremblante a la chevre. C. R. Acad. Sci. 1939, 208, 1058–1060. [Google Scholar]
- Alper, T.; Haig, D.A.; Clarke, M.C. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun. 1966, 22, 278–284. [Google Scholar] [CrossRef]
- Alper, T.; Cramp, W.A.; Haig, D.A.; Clarke, M.C. Does the agent of scrapie replicate without nucleic acid? Nature 1967, 214, 764–766. [Google Scholar] [CrossRef]
- Latarjet, R.; Muel, B.; Haig, D.A.; Clarke, M.C.; Alper, T. Inactivation of the scrapie agent by near monochromatic ultraviolet light. Nature 1970, 227, 1341–1343. [Google Scholar] [CrossRef]
- Prusiner, S.B. Scrapie prions. Annu. Rev. Microbiol. 1989, 43, 345–374. [Google Scholar] [CrossRef]
- Prusiner, S.B. (Ed.) Prion Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; p. 456. [Google Scholar]
- Gajdusek, D.C.; Zigas, V. Degenerative disease of the central nervous system in New Guinea; The endemic occurrence of “kuru” in the native population. N. Engl. J. Med. 1957, 257, 974–978. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Zigas, V. Kuru—Clinical, pathological and epidemiological study of an acute progressive degenerative disease of the central nervous system among natives of the eastern highlands of New Guinea. Am. J. Med. 1959, 26, 442–469. [Google Scholar] [CrossRef]
- Hadlow, W.J. Scrapie and kuru. Lancet 1959, 274, 289–290. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Gibbs, C.J., Jr.; Alpers, M. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 1966, 209, 794–796. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- De Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Gordon, W.S. Advances in veterinary research. Vet. Rec. 1946, 58, 516–520. [Google Scholar]
- Pattison, I.H. Resistance of the scrapie agent to formalin. J. Comp. Pathol. 1965, 75, 159–164. [Google Scholar] [CrossRef]
- Chandler, R.L. Attempts to demonstrate antibodies in scrapie disease. Vet. Rec. 1959, 71, 58–59. [Google Scholar]
- Sigurdsson, B. Rida, a chronic encephalitis of sheep with general remarks on infections which develop slowly and some of their special characteristics. Br. Vet. J. 1954, 110, 341–354. [Google Scholar] [CrossRef]
- Gajdusek, D.C. Unconventional viruses and the origin and disappearance of kuru. Science 1977, 197, 943–960. [Google Scholar] [CrossRef]
- Griffith, J.S. Nature of the scrapie agent: Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef]
- Pattison, I.H.; Jones, K.M. The possible nature of the transmissible agent of scrapie. Vet. Rec. 1967, 80, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.H.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Carlson, G.A.; Kingsbury, D.T.; Goodman, P.A.; Coleman, S.; Marshall, S.T.; DeArmond, S.; Westaway, D.; Prusiner, S.B. Linkage of prion protein and scrapie incubation time genes. Cell 1986, 46, 503–511. [Google Scholar] [CrossRef]
- Carlson, G.A.; Goodman, P.A.; Lovett, M.; Taylor, B.A.; Marshall, S.T.; Peterson-Torchia, M.; Westaway, D.; Prusiner, S.B. Genetics and polymorphism of the mouse prion gene complex: Control of scrapie incubation time. Mol. Cell. Biol. 1988, 8, 5528–5540. [Google Scholar] [CrossRef]
- Westaway, D.; Goodman, P.A.; Mirenda, C.A.; McKinley, M.P.; Carlson, G.A.; Prusiner, S.B. Distinct prion proteins in short and long scrapie incubation period mice. Cell 1987, 51, 651–662. [Google Scholar] [CrossRef]
- Carlson, G.A.; Ebeling, C.; Torchia, M.; Westaway, D.; Prusiner, S.B. Delimiting the location of the scrapie prion incubation time gene on chromosome 2 of the mouse. Genetics 1993, 133, 979–988. [Google Scholar] [CrossRef]
- Moore, R.C.; Hope, J.; McBride, P.A.; McConnell, I.; Selfridge, J.; Melton, D.W.; Manson, J.C. Mice with gene targetted prion protein alterations show that Prnp, Sinc and Prni are congruent. Nat. Genet. 1998, 18, 118–125. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Mackay, J.M. Genetical control of the incubation period in mice of the neurological disease, scrapie. Heredity 1964, 19, 279–288. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Meikle, V.M.H. Host-genotype and agent effects in scrapie incubation: Change in allelic interaction with different strains of agent. Mol. Gen. Genet. 1971, 112, 73–79. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Meikle, V.M.H. A comparison of some biological characteristics of the mouse-passaged scrapie agents, 22A and ME7. Genet. Res. 1969, 13, 213–225. [Google Scholar] [CrossRef]
- Bruce, M.E.; Fraser, H. Scrapie strain variation and its implications. Curr. Top. Microbiol. Immunol. 1991, 172, 125–138. [Google Scholar]
- Carlson, G.A.; Westaway, D.; DeArmond, S.J.; Peterson-Torchia, M.; Prusiner, S.B. Primary structure of prion protein may modify scrapie isolate properties. Proc. Natl. Acad. Sci. USA 1989, 86, 7475–7479. [Google Scholar] [CrossRef]
- Westaway, D.; Mirenda, C.A.; Foster, D.; Zebarjadian, Y.; Scott, M.; Torchia, M.; Yang, S.-L.; Serban, H.; DeArmond, S.J.; Ebeling, C.; et al. Paradoxical shortening of scrapie incubation times by expression of prion protein transgenes derived from long incubation period mice. Neuron 1991, 7, 59–68. [Google Scholar] [CrossRef]
- Carlson, G.A.; Ebeling, C.; Yang, S.-L.; Telling, G.; Torchia, M.; Groth, D.; Westaway, D.; DeArmond, S.J.; Prusiner, S.B. Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 5690–5694. [Google Scholar] [CrossRef]
- Zlotnik, I.; Rennie, J.C. Experimental transmission of mouse passaged scrapie to goats, sheep, rats and hamsters. J. Comp. Pathol. 1965, 75, 147–157. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of scrapie: Agent multiplication in brain at the first and second passage of hamster scrapie in mice. J. Gen. Virol. 1979, 42, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.; Foster, D.; Mirenda, C.; Serban, D.; Coufal, F.; Wälchli, M.; Torchia, M.; Groth, D.; Carlson, G.; DeArmond, S.J.; et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 1989, 59, 847–857. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.-M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.-L.; Serban, D.; Carlson, G.A.; et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Hsiao, K.; Baker, H.F.; Crow, T.J.; Poulter, M.; Owen, F.; Terwilliger, J.D.; Westaway, D.; Ott, J.; Prusiner, S.B. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature 1989, 338, 342–345. [Google Scholar] [CrossRef]
- Hsiao, K.K.; Scott, M.; Foster, D.; Groth, D.F.; DeArmond, S.J.; Prusiner, S.B. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 1990, 250, 1587–1590. [Google Scholar] [CrossRef]
- Hsiao, K.K.; Groth, D.; Scott, M.; Yang, S.-L.; Serban, A.; Rapp, D.; Foster, D.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B. Genetic and transgenic studies of prion proteins in Gerstmann-Straussler–Scheinker disease. In Prion Diseases of Humans and Animals; Prusiner, S.B., Collinge, J., Powell, J., Anderton, B., Eds.; Ellis Horwood: London, UK, 1992; pp. 120–128. [Google Scholar]
- Brown, P.; Gajdusek, D.C.; Gibbs, C.J., Jr.; Asher, D.M. Potential epidemic of Creutzfeldt-Jakob disease from human growth hormone therapy. N. Engl. J. Med. 1985, 313, 728–731. [Google Scholar] [CrossRef]
- Hsiao, K.; Meiner, Z.; Kahana, E.; Cass, C.; Kahana, I.; Avrahami, D.; Scarlato, G.; Abramsky, O.; Prusiner, S.B.; Gabizon, R. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N. Engl. J. Med. 1991, 324, 1091–1097. [Google Scholar] [CrossRef]
- Medori, R.; Tritschler, H.-J.; LeBlanc, A.; Villare, F.; Manetto, V.; Chen, H.Y.; Xue, R.; Leal, S.; Montagna, P.; Cortelli, P.; et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N. Engl. J. Med. 1992, 326, 444–449. [Google Scholar] [CrossRef]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.C.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef]
- Bessen, R.A.; Marsh, R.F. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 1992, 73, 329–334. [Google Scholar] [CrossRef]
- Bessen, R.A.; Marsh, R.F. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 1994, 68, 7859–7868. [Google Scholar] [CrossRef]
- Bessen, R.A.; Kocisko, D.A.; Raymond, G.J.; Nandan, S.; Lansbury, P.T.; Caughey, B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995, 375, 698–700. [Google Scholar] [CrossRef]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef]
- Bruce, M.E.; McConnell, I.; Fraser, H.; Dickinson, A.G. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: Implications for the nature of the agent and host control of pathogenesis. J. Gen. Virol. 1991, 72, 595–603. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prion diseases and the BSE crisis. Science 1997, 278, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Barash, J.A. Clinical features of sporadic fatal insomnia. Rev. Neurol. Dis. 2009, 6, E87–E93. [Google Scholar] [PubMed]
- Collinge, J.; Whitfield, J.; McKintosh, E.; Beck, J.; Mead, S.; Thomas, D.J.; Alpers, M.P. Kuru in the 21st century—An acquired human prion disease with very long incubation periods. Lancet 2006, 367, 2068–2074. [Google Scholar] [CrossRef]
- Mead, S.; Whitfield, J.; Poulter, M.; Shah, P.; Uphill, J.; Campbell, T.; Al-Dujaily, H.; Hummerich, H.; Beck, J.; Mein, C.A.; et al. A novel protective prion protein variant that colocalizes with kuru exposure. N. Engl. J. Med. 2009, 361, 2056–2065. [Google Scholar] [CrossRef]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian evolution of prions in cell culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef]
- Berry, D.B.; Lu, D.; Geva, M.; Watts, J.C.; Bhardwaj, S.; Oehler, A.; Renslo, A.R.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Drug resistance confounding prion therapeutics. Proc. Natl. Acad. Sci. USA 2013, 110, E4160–E4169. [Google Scholar] [CrossRef] [PubMed]
- Oelschlegel, A.M.; Weissmann, C. Acquisition of drug resistance and dependence by prions. PLoS Pathog. 2013, 9, e1003158. [Google Scholar] [CrossRef]
- Olanow, C.W.; Prusiner, S.B. Is Parkinson’s disease a prion disorder? Proc. Natl. Acad. Sci. USA 2009, 106, 12571–12572. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.Y. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef]
- Woerman, A.L.; Watts, J.C.; Aoyagi, A.; Giles, K.; Middleton, L.T.; Prusiner, S.B. α-Synuclein: Multiple system atrophy prions. In Prion Diseases; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; pp. 319–330. [Google Scholar]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef]
- Alzheimer, A. Über eigenartige Krankheitsfälle des späteren Alters. Zentralbl. Gesamte Neurol. Psychiatr. 1911, 4, 356–385. [Google Scholar] [CrossRef]
- Van Duinen, S.G.; Castano, E.M.; Prelli, F.; Bots, G.T.A.B.; Luyendij, K.W.; Frangione, B. Hereditary cerebral haemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1987, 84, 5991–5994. [Google Scholar] [CrossRef]
- Maat-Schieman, M.L.C.; Yamaguchi, H.; Hegeman-Kleinn, I.M.; Welling-Graafland, C.; Natté, R.; Roos, R.A.C.; van Duinen, S.G. Glial reactions and the clearance of amyloid β protein in the brains of patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type. Acta Neuropathol. 2004, 107, 389–398. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef]
- Goldgaber, D.; Lerman, M.I.; McBride, O.W.; Saffiotti, U.; Gajdusek, D.C. Charaterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 1987, 235, 877–880. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Beyreuther, K.; Masters, C.L. Serpents on the road to dementia and death. Accumulating evidence from several studies points to the normal function of presenilin 1 and suggests how the mutant protein contributes to deposition of amyloid plaques in Alzheimer’s disease. Nat. Med. 1997, 3, 723–725. [Google Scholar] [CrossRef]
- Goate, A.; Hardy, J. Twenty years of Alzheimer’s disease-causing mutations. J. Neurochem. 2012, 120 (Suppl. 1), 3–8. [Google Scholar] [CrossRef]
- Goudsmit, J.; Morrow, C.H.; Asher, D.M.; Yanagihara, R.T.; Masters, C.L.; Gibbs, C.J., Jr.; Gajdusek, D.C. Evidence for and against the transmissibility of Alzheimer’s disease. Neurology 1980, 30, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Godec, M.S.; Asher, D.M.; Masters, C.L.; Kozachuk, W.E.; Friedland, R.P.; Gibbs, C.J., Jr.; Gajdusek, D.C.; Rapoport, S.I.; Schapiro, M.B. Evidence against the transmissibility of Alzheimer’s disease. Neurology 1991, 41, 1320. [Google Scholar] [CrossRef] [PubMed]
- Baker, H.F.; Ridley, R.M.; Duchen, L.W.; Crow, T.J.; Bruton, C.J. Induction of ß(A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Mol. Neurobiol. 1994, 8, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Bolmont, T.; Heikenwalder, M.; Langer, F.; Jacobson, L.H.; Yan, Z.X.; Roth, K.; Aguzzi, A.; Staufenbiel, M.; Walker, L.C.; et al. Induction of cerebral β-amyloidosis: Intracerebral versus systemic Aβ inoculation. Proc. Natl. Acad. Sci. USA 2009, 106, 12926–12931. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef]
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef]
- Hsiao, K.K.; Groth, D.; Scott, M.; Yang, S.-L.; Serban, H.; Rapp, D.; Foster, D.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc. Natl. Acad. Sci. USA 1994, 91, 9126–9130. [Google Scholar] [CrossRef]
- Shimizu, S.; Hoshi, K.; Muramoto, T.; Homma, M.; Ironside, J.W.; Kuzuhara, S.; Sato, T.; Yamamoto, T.; Kitamoto, T. Creutzfeldt-Jakob disease with florid-type plaques after cadaveric dura mater grafting. Arch. Neurol. 1999, 56, 357–362. [Google Scholar] [CrossRef][Green Version]
- Mills, J.L.; Schonberger, L.B.; Wysowski, D.K.; Brown, P.; Durako, S.J.; Cox, C.; Kong, F.; Fradkin, J.E. Long-term mortality in the United States cohort of pituitary-derived growth hormone recipients. J. Pediatr. 2004, 144, 430–436. [Google Scholar] [CrossRef][Green Version]
- Aoyagi, A.; Condello, C.; Stöhr, J.; Yue, W.; Lee, J.C.; Rivera, B.M.; Woerman, A.L.; Halliday, G.; van Duinen, S.; Ingelsson, M.; et al. Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci. Transl. Med. 2019, 11, eaat8462. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 75, 1858–1862. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef]
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and novel exons of the human τ gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Bird, T.D.; Ghetti, B. Frontotemporal dementia and parkinsonism linked to chromosome 17: A new group of tauopathies. Brain Pathol. 1998, 8, 387–402. [Google Scholar] [CrossRef]
- Arrasate, M.; Pérez, M.; Armas-Portela, R.; Ávila, J. Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 1999, 446, 199–202. [Google Scholar] [CrossRef]
- Nacharaju, P.; Lewis, J.; Easson, C.; Yen, S.; Hackett, J.; Hutton, M.; Yen, S.-H. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999, 447, 195–199. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Iaccarino, L.; Schonhaut, D.R.; Brown, J.A.; La Joie, R.; O’Neil, J.P.; Janabi, M.; Baker, S.L.; Kramer, J.H.; Gorno-Tempini, M.L.; et al. Tau covariance patterns in Alzheimer’s disease patients match intrinsic connectivity networks in the healthy brain. Neuroimage Clin. 2019, 23, 101848. [Google Scholar] [CrossRef]
- Franzmeier, N.; Neitzel, J.; Rubinski, A.; Smith, R.; Strandberg, O.; Ossenkoppele, R.; Hansson, O.; Ewers, M.; Alzheimer’s Disease Neuroimaging Initiative. Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat. Commun. 2020, 11, 347. [Google Scholar] [CrossRef]
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative; Swedish BioFinder Study. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef]
- He, Z.; McBride, J.D.; Xu, H.; Changolkar, L.; Kim, S.J.; Zhang, B.; Narasimhan, S.; Gibbons, G.S.; Guo, J.L.; Kozak, M.; et al. Transmission of tauopathy strains is independent of their isoform composition. Nat. Commun. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Gotz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Bennett, R.E.; DeVos, S.L.; Dujardin, S.; Corjuc, B.; Gor, R.; Gonzalez, J.; Roe, A.D.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Enhanced tau aggregation in the presence of amyloid β. Am. J. Pathol. 2017, 187, 1601–1612. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef]
- Crowther, R.A.; Jakes, R.; Spillantini, M.G.; Goedert, M. Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 1998, 436, 309–312. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef]
- Mougenot, A.-L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchère, J.; Lakhdar, L.; Legastelois, S.; Baron, T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [Google Scholar] [CrossRef]
- Ayers, J.I.; Brooks, M.M.; Rutherford, N.J.; Howard, J.K.; Sorrentino, Z.A.; Riffe, C.J.; Giasson, B.I. Robust central nervous system pathology in transgenic mice following peripheral injection of α-synuclein fibrils. J. Virol. 2017, 91, e02095-16. [Google Scholar] [CrossRef]
- Woerman, A.L.; Kazmi, S.A.; Patel, S.; Freyman, Y.; Oehler, A.; Aoyagi, A.; Mordes, D.A.; Halliday, G.M.; Middleton, L.T.; Gentleman, S.M.; et al. MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol. 2018, 135, 49–63. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- van Dyck, C.H. Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: Pitfalls and promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Misfolded Protein in Prions | Familial Mutations | Strains | Aggregate/Amyloid Location | Spread within the Brain | Human-to-Human Transmission | Environmental Cause |
|---|---|---|---|---|---|---|---|
| Kuru | PrP | No | Probably | Extracellular | Interstitial fluid/cerebrospinal fluid and neural pathways | Cannibalism | No evidence |
| Creutzfeldt–Jakob disease (CJD) | PrP | Yes; <10% familial CJD, Gerstmann–Sträussler–Scheinker (GSS), fatal familial insomnia (FFI) | Yes | Extracellular | Interstitial fluid/cerebrospinal fluid and neural pathways | Iatrogenic: Dura mater grafts, contaminated cadaver-derived human growth hormone | No evidence |
| Alzheimer’s disease | Aβ1-40, Aβ1-42(43) amyloid plaques | APP, PSEN1, and PSEN2 mutations elevate Aβ levels | Aβ40/42 ratio | Extracellular | Interstitial fluid/CSF | Contaminated cadaver-derived human growth hormone | Head trauma increases risk |
| Alzheimer’s disease | 3R and 4R tau | No; wild-type tau only | Yes | Intracellular neurofibrillary tangles | Transsynaptic along neural pathways | Unknown | Unknown |
| Chronic traumatic encephalopathy | 3R and 4R tau | No; wild-type tau only | Unknown | Extracellular and intracellular; perivascular | Interstitial fluid/CSF | Unknown | Repeated head trauma |
| Frontotemporal dementia-tau | 4R tau | Yes, in ~20% of patients | Yes | Intracellular neurofibrillary tangles | Transsynaptic along neural pathways | Unknown | Unknown |
| Parkinson’s disease | α-synuclein | Yes, in ~10% of patients | Yes | Intracellular Lewy bodies | Transsynaptic along neural pathways | Unknown | Herbicide/pesticide exposure increases risk |
| Multiple system atrophy | α-synuclein | No | Yes | Intracellular | Along neural pathways | Unknown | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlson, G.A.; Prusiner, S.B. How an Infection of Sheep Revealed Prion Mechanisms in Alzheimer’s Disease and Other Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4861. https://doi.org/10.3390/ijms22094861
Carlson GA, Prusiner SB. How an Infection of Sheep Revealed Prion Mechanisms in Alzheimer’s Disease and Other Neurodegenerative Disorders. International Journal of Molecular Sciences. 2021; 22(9):4861. https://doi.org/10.3390/ijms22094861
Chicago/Turabian StyleCarlson, George A., and Stanley B. Prusiner. 2021. "How an Infection of Sheep Revealed Prion Mechanisms in Alzheimer’s Disease and Other Neurodegenerative Disorders" International Journal of Molecular Sciences 22, no. 9: 4861. https://doi.org/10.3390/ijms22094861
APA StyleCarlson, G. A., & Prusiner, S. B. (2021). How an Infection of Sheep Revealed Prion Mechanisms in Alzheimer’s Disease and Other Neurodegenerative Disorders. International Journal of Molecular Sciences, 22(9), 4861. https://doi.org/10.3390/ijms22094861