
B Cells and Microbiota in Autoimmunity


{kind=link}
{kind=link}
Abstract
1. Introduction
2. B-Cell Lymphopoiesis and B-Cell Differentiation
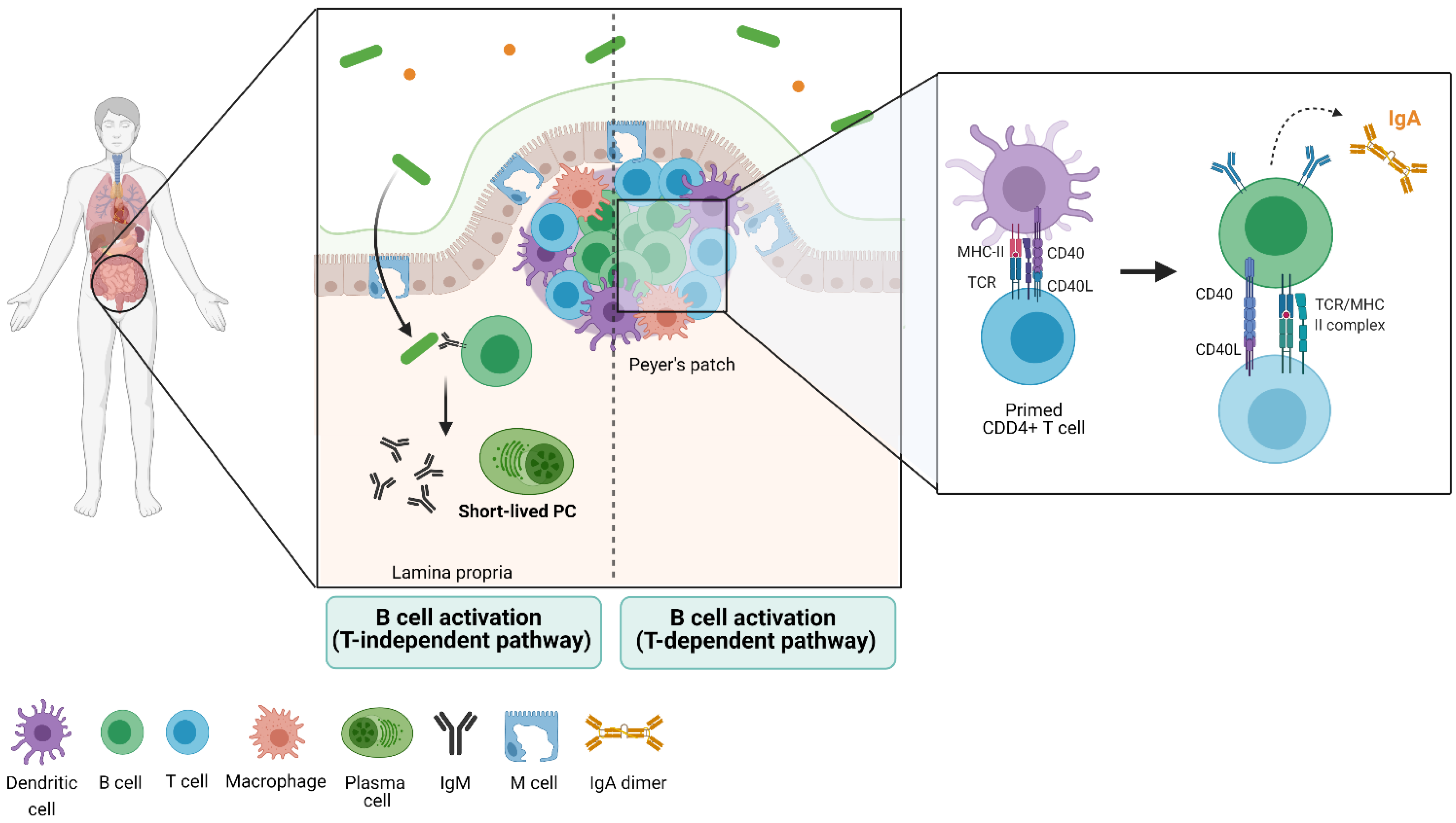
3. Gut Immune Structure and Immune Cell Distribution (the Gut-Associated Lymphoid Tissue)
4. The Microbiota
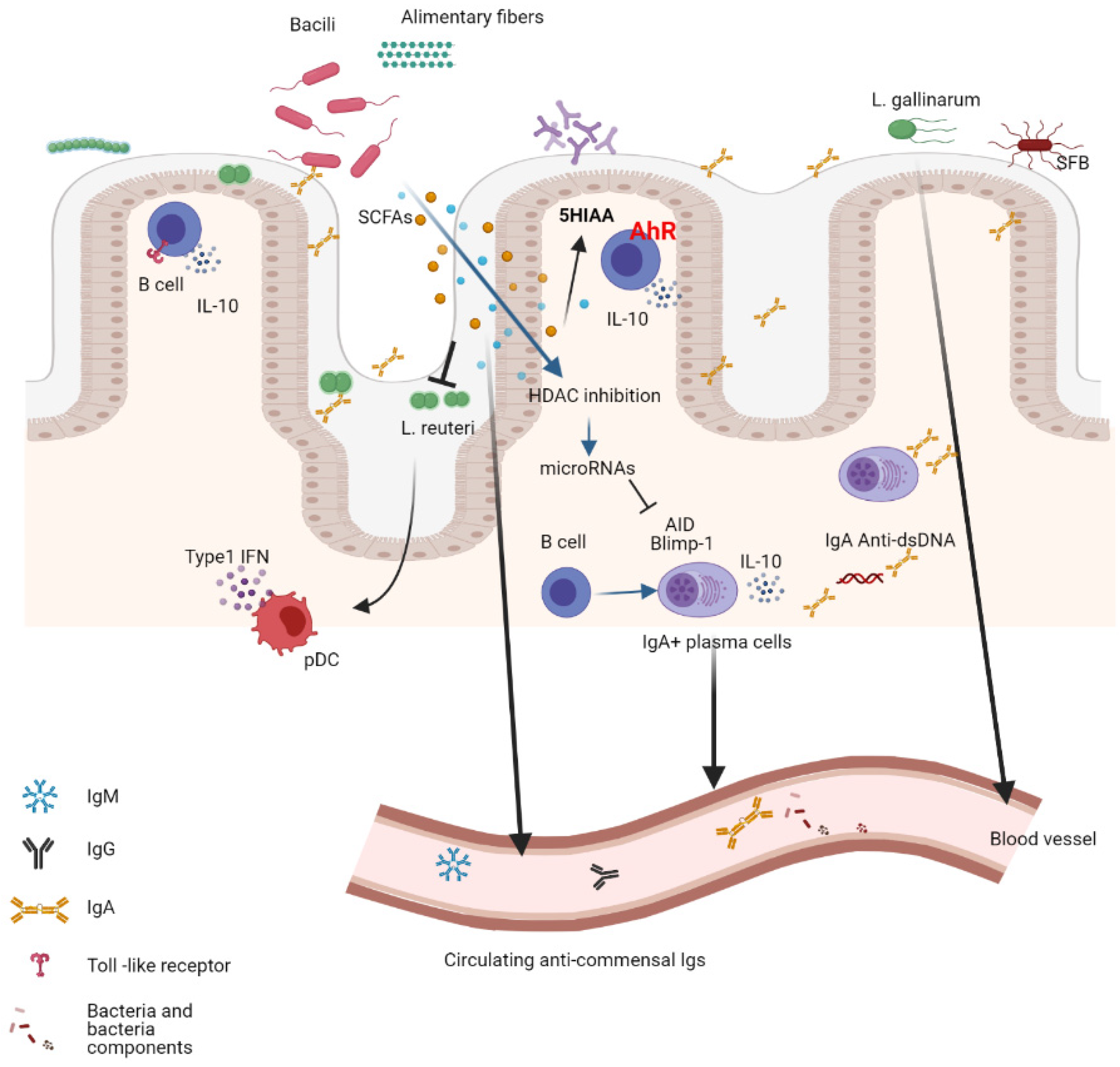
5. The B-Cell Response to the Microbiota
6. Shaping the Microbiota Composition by the B-Cell Response
7. The Interaction Between the Gut Microbiota and B Cells in Autoimmune Diseases
7.1. Systemic Lupus Erythematosus
7.2. Multiple Sclerosis
7.3. Rheumatoid Arthritis
7.4. Type I Diabetes
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pabst, O.; Slack, E. IgA and the intestinal microbiota: The importance of being specific. Mucosal Immunol. 2020, 13, 12–21. [Google Scholar] [CrossRef]
- Okai, S.; Usui, F.; Ohta, M.; Mori, H.; Kurokawa, K.; Matsumoto, S.; Kato, T.; Miyauchi, E.; Ohno, H.; Shinkura, R. Intestinal IgA as a modulator of the gut microbiota. Gut Microbes 2017, 8, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.J.; Flynn, T.M.; Koval, J.C.; Shaw, D.G.; Meisel, M.; McDonald, B.D.; Ishizuka, I.E.; Dent, A.L.; Wilson, P.C.; Jabri, B.; et al. Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 2015, 43, 541–553. [Google Scholar] [CrossRef]
- Greiling, T.M.; Dehner, C.; Chen, X.; Hughes, K.; Iniguez, A.J.; Boccitto, M.; Ruiz, D.Z.; Renfroe, S.C.; Vieira, S.M.; Ruff, W.E.; et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Ruff, W.E.; Dehner, C.; Kim, W.J.; Pagovich, O.; Aguiar, C.L.; Yu, A.T.; Roth, A.S.; Vieira, S.M.; Kriegel, C.; Adeniyi, O.; et al. Pathogenic Autoreactive T and B Cells Cross-React with Mimotopes Expressed by a Common Human Gut Commensal to Trigger Autoimmunity. Cell Host Microbe 2019, 26, 100–113.E8. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gudi, R.R.; Johnson, B.M.; Vasu, C. Abundance and nuclear antigen reactivity of intestinal and fecal Immunoglobulin A in lupus-prone mice at younger ages correlate with the onset of eventual systemic autoimmunity. Sci. Rep. 2020, 10, 14258. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.Y.; Cisalpino, D.; Varadarajan, S.; Hellman, J.; Warren, H.S.; Cascalho, M.; Inohara, N.; Nunez, G. Gut Microbiota-Induced Immunoglobulin G Controls Systemic Infection by Symbiotic Bacteria and Pathogens. Immunity 2016, 44, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Edwards, M.R.; Swartwout, B.K.; Cabana Puig, X.; Mao, J.; Zhu, J.; Grieco, J.; Cecere, T.E.; Prakash, M.; Reilly, C.M.; et al. Gut Microbiota and Bacterial DNA Suppress Autoimmunity by Stimulating Regulatory B Cells in a Murine Model of Lupus. Front. Immunol. 2020, 11, 593353. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Pearson, J.A.; Peng, J.; Hu, Y.; Sha, S.; Xing, Y.; Huang, G.; Li, X.; Hu, F.; Xie, Z.; et al. Gut microbial metabolites alter IgA immunity in type 1 diabetes. JCI Insight 2020, 5, e135718. [Google Scholar] [CrossRef]
- Dus-Szachniewicz, K.; Drobczynski, S.; Ziolkowski, P.; Kolodziej, P.; Walaszek, K.M.; Korzeniewska, A.K.; Agrawal, A.; Kupczyk, P.; Wozniak, M. Physiological Hypoxia (Physioxia) Impairs the Early Adhesion of Single Lymphoma Cell to Marrow Stromal Cell and Extracellular Matrix. Optical Tweezers Study. Int. J. Mol. Sci. 2018, 19, 1880. [Google Scholar] [CrossRef]
- Funk, P.E.; Kincade, P.W.; Witte, P.L. Native associations of early hematopoietic stem cells and stromal cells isolated in bone marrow cell aggregates. Blood 1994, 83, 361–369. [Google Scholar] [CrossRef]
- Jacobsen, K.; Kravitz, J.; Kincade, P.W.; Osmond, D.G. Adhesion receptors on bone marrow stromal cells: In vivo expression of vascular cell adhesion molecule-1 by reticular cells and sinusoidal endothelium in normal and gamma-irradiated mice. Blood 1996, 87, 73–82. [Google Scholar] [CrossRef]
- Addo, R.K.; Heinrich, F.; Heinz, G.A.; Schulz, D.; Sercan-Alp, O.; Lehmann, K.; Tran, C.L.; Bardua, M.; Matz, M.; Lohning, M.; et al. Single-cell transcriptomes of murine bone marrow stromal cells reveal niche-associated heterogeneity. Eur. J. Immunol. 2019, 49, 1372–1379. [Google Scholar] [CrossRef]
- Zehentmeier, S.; Pereira, J.P. Cell circuits and niches controlling B cell development. Immunol. Rev. 2019, 289, 142–157. [Google Scholar] [CrossRef]
- Azagra, A.; Marina-Zarate, E.; Ramiro, A.R.; Javierre, B.M.; Parra, M. From Loops to Looks: Transcription Factors and Chromatin Organization Shaping Terminal B Cell Differentiation. Trends Immunol. 2020, 41, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jain, S.; Li, P.; Lin, J.X.; Oh, J.; Qi, C.; Gao, Y.; Sun, J.; Sakai, T.; Naghashfar, Z.; et al. Transcription factors IRF8 and PU.1 are required for follicular B cell development and BCL6-driven germinal center responses. Proc. Natl. Acad. Sci. USA 2019, 116, 9511–9520. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Chen, G.; Zhao, Y.; Gao, X.M.; Wang, J. Regulation of the Development and Function of B Cells by ZBTB Transcription Factors. Front. Immunol. 2018, 9, 580. [Google Scholar] [CrossRef] [PubMed]
- Fistonich, C.; Zehentmeier, S.; Bednarski, J.J.; Miao, R.; Schjerven, H.; Sleckman, B.P.; Pereira, J.P. Cell circuits between B cell progenitors and IL-7(+) mesenchymal progenitor cells control B cell development. J. Exp. Med. 2018, 215, 2586–2599. [Google Scholar] [CrossRef]
- Fisher, M.R.; Rivera-Reyes, A.; Bloch, N.B.; Schatz, D.G.; Bassing, C.H. Immature Lymphocytes Inhibit Rag1 and Rag2 Transcription and V(D)J Recombination in Response to DNA Double-Strand Breaks. J. Immunol. 2017, 198, 2943–2956. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Carmack, C.E.; Shinton, S.A.; Kemp, J.D.; Hayakawa, K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991, 173, 1213–1225. [Google Scholar] [CrossRef]
- Phares, T.W.; DiSano, K.D.; Stohlman, S.A.; Bergmann, C.C. Progression from IgD+ IgM+ to isotype-switched B cells is site specific during coronavirus-induced encephalomyelitis. J. Virol. 2014, 88, 8853–8867. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, D.; Roes, J.; Kuhn, R.; Rajewsky, K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 1991, 350, 423–426. [Google Scholar] [CrossRef]
- Kallies, A.; Hasbold, J.; Fairfax, K.; Pridans, C.; Emslie, D.; McKenzie, B.S.; Lew, A.M.; Corcoran, L.M.; Hodgkin, P.D.; Tarlinton, D.M.; et al. Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity 2007, 26, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Taubenheim, N.; Hasbold, J.; Corcoran, L.M.; Hodgkin, P.D. The genetic network controlling plasma cell differentiation. Semin. Immunol. 2011, 23, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.; Li, B.E.; Mishra, B.P.; Jones, K.L.; Ernst, P. MLL1 Promotes IL-7 Responsiveness and Survival during B Cell Differentiation. J. Immunol. 2018, 200, 1682–1691. [Google Scholar] [CrossRef]
- Yu, M.; Chen, Y.; Zeng, H.; Zheng, Y.; Fu, G.; Zhu, W.; Broeckel, U.; Aggarwal, P.; Turner, A.; Neale, G.; et al. PLCgamma-dependent mTOR signalling controls IL-7-mediated early B cell development. Nat. Commun. 2017, 8, 1457. [Google Scholar] [CrossRef]
- Melamed, D.; Benschop, R.J.; Cambier, J.C.; Nemazee, D. Developmental regulation of B lymphocyte immune tolerance compartmentalizes clonal selection from receptor selection. Cell 1998, 92, 173–182. [Google Scholar] [CrossRef]
- Gu, H.; Tarlinton, D.; Muller, W.; Rajewsky, K.; Forster, I. Most peripheral B cells in mice are ligand selected. J. Exp. Med. 1991, 173, 1357–1371. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.K. Innate response activator B cells: Origins and functions. Int. Immunol. 2015, 27, 537–541. [Google Scholar] [CrossRef]
- Godin, I.E.; Garcia-Porrero, J.A.; Coutinho, A.; Dieterlen-Lievre, F.; Marcos, M.A. Para-aortic splanchnopleura from early mouse embryos contains B1a cell progenitors. Nature 1993, 364, 67–70. [Google Scholar] [CrossRef]
- Ansel, K.M.; Harris, R.B.; Cyster, J.G. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity 2002, 16, 67–76. [Google Scholar] [CrossRef]
- Wardemann, H.; Boehm, T.; Dear, N.; Carsetti, R. B-1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J. Exp. Med. 2002, 195, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Klinman, N.R. The “clonal selection hypothesis” and current concepts of B cell tolerance. Immunity 1996, 5, 189–195. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Burrows, P.D.; Wang, J.Y. B Cell Development and Maturation. In B Cells in Immunity and Tolerance. Advances in Experimental Medicine and Biology; Springer: Singapore, 2020; Volume 1254, pp. 1–22. [Google Scholar] [CrossRef]
- Wesemann, D.R.; Portuguese, A.J.; Meyers, R.M.; Gallagher, M.P.; Cluff-Jones, K.; Magee, J.M.; Panchakshari, R.A.; Rodig, S.J.; Kepler, T.B.; Alt, F.W. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 2013, 501, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Melnick, A. Mechanisms of action of BCL6 during germinal center B cell development. Sci. China Life Sci. 2015, 58, 1226–1232. [Google Scholar] [CrossRef]
- McHeyzer-Williams, M.; Okitsu, S.; Wang, N.; McHeyzer-Williams, L. Molecular programming of B cell memory. Nat. Rev. Immunol. 2011, 12, 24–34. [Google Scholar] [CrossRef]
- Shlomchik, M.J.; Luo, W.; Weisel, F. Linking signaling and selection in the germinal center. Immunol. Rev. 2019, 288, 49–63. [Google Scholar] [CrossRef]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef]
- MacLennan, I.C. Germinal centers. Annu. Rev. Immunol. 1994, 12, 117–139. [Google Scholar] [CrossRef]
- Dent, A.L.; Shaffer, A.L.; Yu, X.; Allman, D.; Staudt, L.M. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 1997, 276, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.H.; Cattoretti, G.; Shen, Q.; Zhang, J.; Hawe, N.; de Waard, R.; Leung, C.; Nouri-Shirazi, M.; Orazi, A.; Chaganti, R.S.; et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat. Genet. 1997, 16, 161–170. [Google Scholar] [CrossRef]
- Bergqvist, P.; Stensson, A.; Hazanov, L.; Holmberg, A.; Mattsson, J.; Mehr, R.; Bemark, M.; Lycke, N.Y. Re-utilization of germinal centers in multiple Peyer’s patches results in highly synchronized, oligoclonal, and affinity-matured gut IgA responses. Mucosal Immunol. 2013, 6, 122–135. [Google Scholar] [CrossRef]
- Nowosad, C.R.; Mesin, L.; Castro, T.B.R.; Wichmann, C.; Donaldson, G.P.; Araki, T.; Schiepers, A.; Lockhart, A.A.K.; Bilate, A.M.; Mucida, D.; et al. Tunable dynamics of B cell selection in gut germinal centres. Nature 2020, 588, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Defrance, T.; Taillardet, M.; Genestier, L. T cell-independent B cell memory. Curr. Opin. Immunol. 2011, 23, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, A.; Anderson, S.M.; Khalil, A.; Shlomchik, M.J. Maintenance of the plasma cell pool is independent of memory B cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4802–4807. [Google Scholar] [CrossRef] [PubMed]
- Hiepe, F.; Dorner, T.; Hauser, A.E.; Hoyer, B.F.; Mei, H.; Radbruch, A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat. Rev. Rheumatol. 2011, 7, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Slifka, M.K.; Ahmed, R. Long-term humoral immunity against viruses: Revisiting the issue of plasma cell longevity. Trends Microbiol. 1996, 4, 394–400. [Google Scholar] [CrossRef]
- Tunyaplin, C.; Shaffer, A.L.; Angelin-Duclos, C.D.; Yu, X.; Staudt, L.M.; Calame, K.L. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J. Immunol. 2004, 173, 1158–1165. [Google Scholar] [CrossRef]
- Wang, Y.H.; Tsai, D.Y.; Ko, Y.A.; Yang, T.T.; Lin, I.Y.; Hung, K.H.; Lin, K.I. Blimp-1 Contributes to the Development and Function of Regulatory B Cells. Front. Immunol. 2019, 10, 1909. [Google Scholar] [CrossRef]
- Haaijman, J.J.; Schuit, H.R.; Hijmans, W. Immunoglobulin-containing cells in different lymphoid organs of the CBA mouse during its life-span. Immunology 1977, 32, 427–434. [Google Scholar] [PubMed]
- Benner, R.; Hijmans, W.; Haaijman, J.J. The bone marrow: The major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin. Exp. Immunol. 1981, 46, 1–8. [Google Scholar]
- Benet, Z.; Jing, Z.; Fooksman, D.R. Plasma cell dynamics in the bone marrow niche. Cell Rep. 2021, 34, 108733. [Google Scholar] [CrossRef] [PubMed]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.; Dorner, T.; Hiepe, F. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; McKee, T.; Bosshard, C.; Durual, S.; Matthes, T.; Myit, S.; Donze, O.; Frossard, C.; Chizzolini, C.; Favre, C.; et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J. Clin. Investig. 2008, 118, 2887–2895. [Google Scholar] [CrossRef]
- Odendahl, M.; Mei, H.; Hoyer, B.F.; Jacobi, A.M.; Hansen, A.; Muehlinghaus, G.; Berek, C.; Hiepe, F.; Manz, R.; Radbruch, A.; et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood 2005, 105, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Verbrugghe, P.; Kujala, P.; Waelput, W.; Peters, P.J.; Cuvelier, C.A. Clusterin in human gut-associated lymphoid tissue, tonsils, and adenoids: Localization to M cells and follicular dendritic cells. Histochem. Cell Biol. 2008, 129, 311–320. [Google Scholar] [CrossRef]
- Delecluse, S.; Tsai, M.H.; Shumilov, A.; Bencun, M.; Arrow, S.; Beshirova, A.; Cottignies-Calamarte, A.; Lasitschka, F.; Bulut, O.C.; Munz, C.; et al. Epstein-Barr Virus Induces Expression of the LPAM-1 Integrin in B Cells In Vitro and In Vivo. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Beller, A.; Kruglov, A.; Durek, P.; von Goetze, V.; Werner, K.; Heinz, G.A.; Ninnemann, J.; Lehmann, K.; Maier, R.; Hoffmann, U.; et al. Specific microbiota enhances intestinal IgA levels by inducing TGF-beta in T follicular helper cells of Peyer’s patches in mice. Eur. J. Immunol. 2020, 50, 783–794. [Google Scholar] [CrossRef]
- Gribonika, I.; Eliasson, D.G.; Chandode, R.K.; Schon, K.; Stromberg, A.; Bemark, M.; Lycke, N.Y. Class-switch recombination to IgA in the Peyer’s patches requires natural thymus-derived Tregs and appears to be antigen independent. Mucosal Immunol. 2019, 12, 1268–1279. [Google Scholar] [CrossRef]
- Girard-Madoux, M.J.H.; Gomez de Aguero, M.; Ganal-Vonarburg, S.C.; Mooser, C.; Belz, G.T.; Macpherson, A.J.; Vivier, E. The immunological functions of the Appendix: An example of redundancy? Semin. Immunol. 2018, 36, 31–44. [Google Scholar] [CrossRef]
- Fawkner-Corbett, D.; Antanaviciute, A.; Parikh, K.; Jagielowicz, M.; Geros, A.S.; Gupta, T.; Ashley, N.; Khamis, D.; Fowler, D.; Morrissey, E.; et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell 2021, 184, 810–826.E23. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.M.; Jorgensen, P.B.; Niss, K.; Rubin, S.J.S.; Morbe, U.M.; Riis, L.B.; Da Silva, C.; Plumb, A.; Vandamme, J.; Jakobsen, H.L.; et al. Immune Profiling of Human Gut-Associated Lymphoid Tissue Identifies a Role for Isolated Lymphoid Follicles in Priming of Region-Specific Immunity. Immunity 2020, 52, 557–570.e6. [Google Scholar] [CrossRef]
- Nagashima, K.; Sawa, S.; Nitta, T.; Prados, A.; Koliaraki, V.; Kollias, G.; Nakashima, T.; Takayanagi, H. Targeted deletion of RANKL in M cell inducer cells by the Col6a1-Cre driver. Biochem. Biophys. Res. Commun. 2017, 493, 437–443. [Google Scholar] [CrossRef]
- Vazquez-Torres, A.; Jones-Carson, J.; Baumler, A.J.; Falkow, S.; Valdivia, R.; Brown, W.; Le, M.; Berggren, R.; Parks, W.T.; Fang, F.C. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature 1999, 401, 804–808. [Google Scholar] [CrossRef]
- Martinoli, C.; Chiavelli, A.; Rescigno, M. Entry route of Salmonella typhimurium directs the type of induced immune response. Immunity 2007, 27, 975–984. [Google Scholar] [CrossRef]
- Cremonesi, E.; Governa, V.; Garzon, J.F.G.; Mele, V.; Amicarella, F.; Muraro, M.G.; Trella, E.; Galati-Fournier, V.; Oertli, D.; Daster, S.R.; et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut 2018, 67, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.; Sawa, S.; Nitta, T.; Tsutsumi, M.; Okamura, T.; Penninger, J.M.; Nakashima, T.; Takayanagi, H. Identification of subepithelial mesenchymal cells that induce IgA and diversify gut microbiota. Nat. Immunol. 2017, 18, 675–682. [Google Scholar] [CrossRef]
- Mantis, N.J.; Cheung, M.C.; Chintalacharuvu, K.R.; Rey, J.; Corthesy, B.; Neutra, M.R. Selective adherence of IgA to murine Peyer’s patch M cells: Evidence for a novel IgA receptor. J. Immunol. 2002, 169, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Boullier, S.; Tanguy, M.; Kadaoui, K.A.; Caubet, C.; Sansonetti, P.; Corthesy, B.; Phalipon, A. Secretory IgA-mediated neutralization of Shigella flexneri prevents intestinal tissue destruction by down-regulating inflammatory circuits. J. Immunol. 2009, 183, 5879–5885. [Google Scholar] [CrossRef]
- Fransen, F.; Zagato, E.; Mazzini, E.; Fosso, B.; Manzari, C.; El Aidy, S.; Chiavelli, A.; D’Erchia, A.M.; Sethi, M.K.; Pabst, O.; et al. BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity 2015, 43, 527–540. [Google Scholar] [CrossRef]
- Vely, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F.; et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, Q.; Zheng, M.; Hao, S.; Lum, J.S.; Chen, X.; Huang, X.F.; Yu, Y.; Zheng, K. Supplement of microbiota-accessible carbohydrates prevents neuroinflammation and cognitive decline by improving the gut microbiota-brain axis in diet-induced obese mice. J. Neuroinflamm. 2020, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Limenitakis, J.P.; Greiff, V.; Yilmaz, B.; Scharen, O.; Urbaniak, C.; Zund, M.; Lawson, M.A.E.; Young, I.D.; Rupp, S.; et al. Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature 2020, 584, 274–278. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Y.; Ye, A.Y.; Du, Z.; Xu, M.; Lee, C.S.; Hwang, J.K.; Kyritsis, N.; Ba, Z.; Neuberg, D.; et al. BCR selection and affinity maturation in Peyer’s patch germinal centres. Nature 2020, 582, 421–425. [Google Scholar] [CrossRef]
- Bunker, J.J.; Erickson, S.A.; Flynn, T.M.; Henry, C.; Koval, J.C.; Meisel, M.; Jabri, B.; Antonopoulos, D.A.; Wilson, P.C.; Bendelac, A. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 2017, 358. [Google Scholar] [CrossRef] [PubMed]
- Wilmore, J.R.; Gaudette, B.T.; Gomez Atria, D.; Hashemi, T.; Jones, D.D.; Gardner, C.A.; Cole, S.D.; Misic, A.M.; Beiting, D.P.; Allman, D. Commensal Microbes Induce Serum IgA Responses that Protect against Polymicrobial Sepsis. Cell Host Microbe 2018, 23, 302–311.e3. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, Z.; Frazer, G.; Ferro, A.; Clare, S.; Bouladoux, N.; Ferdinand, J.; Tuong, Z.K.; Negro-Demontel, M.L.; Kumar, N.; Suchanek, O.; et al. Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature 2020, 587, 472–476. [Google Scholar] [CrossRef]
- Rosser, E.C.; Oleinika, K.; Tonon, S.; Doyle, R.; Bosma, A.; Carter, N.A.; Harris, K.A.; Jones, S.A.; Klein, N.; Mauri, C. Regulatory B cells are induced by gut microbiota-driven interleukin-1beta and interleukin-6 production. Nat. Med. 2014, 20, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Oka, A.; Liu, B.; Herzog, J.W.; Eun, C.S.; Fan, T.J.; Bulik-Sullivan, E.; Carroll, I.M.; Hansen, J.J.; Chen, L.; et al. Microbiota maintain colonic homeostasis by activating TLR2/MyD88/PI3K signaling in IL-10-producing regulatory B cells. J. Clin. Investig. 2019, 129, 3702–3716. [Google Scholar] [CrossRef] [PubMed]
- Daien, C.I.; Tan, J.; Audo, R.; Mielle, J.; Quek, L.E.; Krycer, J.R.; Angelatos, A.S.; Duares, M.; Pinget, G.V.; Ni, D.; et al. Gut-derived acetate promotes B10 cells with anti-inflammatory effects. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Matsumoto, M.; Baba, A.; Yokota, T.; Nishikawa, H.; Ohkawa, Y.; Kayama, H.; Kallies, A.; Nutt, S.L.; Sakaguchi, S.; Takeda, K.; et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 2014, 41, 1040–1051. [Google Scholar] [CrossRef]
- Rojas, O.L.; Probstel, A.K.; Porfilio, E.A.; Wang, A.A.; Charabati, M.; Sun, T.; Lee, D.S.W.; Galicia, G.; Ramaglia, V.; Ward, L.A.; et al. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell 2019, 176, 610–624.e18. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Shinkura, R.; Doi, Y.; Maruya, M.; Fagarasan, S.; Honjo, T. Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat. Immunol. 2011, 12, 264–270. [Google Scholar] [CrossRef]
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 13574. [Google Scholar] [CrossRef]
- Fadlallah, J.; El Kafsi, H.; Sterlin, D.; Juste, C.; Parizot, C.; Dorgham, K.; Autaa, G.; Gouas, D.; Almeida, M.; Lepage, P.; et al. Microbial ecology perturbation in human IgA deficiency. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Zegarra-Ruiz, D.F.; El Beidaq, A.; Iniguez, A.J.; Lubrano Di Ricco, M.; Manfredo Vieira, S.; Ruff, W.E.; Mubiru, D.; Fine, R.L.; Sterpka, J.; Greiling, T.M.; et al. A Diet-Sensitive Commensal Lactobacillus Strain Mediates TLR7-Dependent Systemic Autoimmunity. Cell Host Microbe 2019, 25, 113–127.E6. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, H.N.; Moroney, J.B.; Gan, H.; Shen, T.; Im, J.L.; Li, T.; Taylor, J.R.; Zan, H.; Casali, P. B cell-intrinsic epigenetic modulation of antibody responses by dietary fiber-derived short-chain fatty acids. Nat. Commun. 2020, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.C.; Brown, J.; Gong, M.; Ge, Y.; Zadeh, M.; Li, W.; Croker, B.P.; Michailidis, G.; Garrett, T.J.; Mohamadzadeh, M.; et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Pachucki, R.J.; Corradetti, C.; Kohler, L.; Ghadiali, J.; Gallo, P.M.; Nicastro, L.; Tursi, S.A.; Gallucci, S.; Tukel, C.; Caricchio, R. Persistent Bacteriuria and Antibodies Recognizing Curli/eDNA Complexes From Escherichia coli Are Linked to Flares in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2020, 72, 1872–1881. [Google Scholar] [CrossRef]
- Azzouz, D.; Omarbekova, A.; Heguy, A.; Schwudke, D.; Gisch, N.; Rovin, B.H.; Caricchio, R.; Buyon, J.P.; Alekseyenko, A.V.; Silverman, G.J. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann. Rheum Dis. 2019, 78, 947–956. [Google Scholar] [CrossRef]
- Manfredo Vieira, S.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359, 1156–1161. [Google Scholar] [CrossRef]
- Melbye, P.; Olsson, A.; Hansen, T.H.; Sondergaard, H.B.; Bang Oturai, A. Short-chain fatty acids and gut microbiota in multiple sclerosis. Acta Neurol. Scand. 2019, 139, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef]
- Mizuno, M.; Noto, D.; Kaga, N.; Chiba, A.; Miyake, S. The dual role of short fatty acid chains in the pathogenesis of autoimmune disease models. PLoS ONE 2017, 12, e0173032. [Google Scholar] [CrossRef]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Hart, J.; Roalstad, S.; Graves, J.; Lynch, S.; Waubant, E.; Centers, U.S.N.o.P.M. Gut microbiota composition and relapse risk in pediatric MS: A pilot study. J. Neurol. Sci. 2016, 363, 153–157. [Google Scholar] [CrossRef]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Hart, J.; Roalstad, S.; Graves, J.; Spencer, C.M.; Lynch, S.V.; Zamvil, S.S.; Waubant, E.; et al. Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol. 2016, 16, 182. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Mangalam, A.; Shahi, S.K.; Luckey, D.; Karau, M.; Marietta, E.; Luo, N.; Choung, R.S.; Ju, J.; Sompallae, R.; Gibson-Corley, K.; et al. Human Gut-Derived Commensal Bacteria Suppress CNS Inflammatory and Demyelinating Disease. Cell Rep. 2017, 20, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Cosorich, I.; Dalla-Costa, G.; Sorini, C.; Ferrarese, R.; Messina, M.J.; Dolpady, J.; Radice, E.; Mariani, A.; Testoni, P.A.; Canducci, F.; et al. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci. Adv. 2017, 3, e1700492. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Mestre, L.; Carrillo-Salinas, F.J.; Mecha, M.; Feliu, A.; Espejo, C.; Alvarez-Cermeno, J.C.; Villar, L.M.; Guaza, C. Manipulation of Gut Microbiota Influences Immune Responses, Axon Preservation, and Motor Disability in a Model of Progressive Multiple Sclerosis. Front. Immunol. 2019, 10, 1374. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K.H. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef]
- Ochoa-Reparaz, J.; Kasper, L.H. The influence of gut-derived CD39 regulatory T cells in CNS demyelinating disease. Transl. Res. 2017, 179, 126–138. [Google Scholar] [CrossRef][Green Version]
- Pant, A.B.; Wang, Y.; Mielcarz, D.W.; Kasper, E.J.; Telesford, K.M.; Mishra, M.; Haque, A.; Channon, J.Y.; Kasper, L.H.; Begum-Haque, S. Alteration of CD39+Foxp3+ CD4 T cell and cytokine levels in EAE/MS following anti-CD52 treatment. J. Neuroimmunol. 2017, 303, 22–30. [Google Scholar] [CrossRef]
- Wang, Y.; Begum-Haque, S.; Telesford, K.M.; Ochoa-Reparaz, J.; Christy, M.; Kasper, E.J.; Kasper, D.L.; Robson, S.C.; Kasper, L.H. A commensal bacterial product elicits and modulates migratory capacity of CD39(+) CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes 2014, 5, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Yanaba, K.; Bouaziz, J.D.; Fujimoto, M.; Tedder, T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Investig. 2008, 118, 3420–3430. [Google Scholar] [CrossRef]
- Ochoa-Reparaz, J.; Mielcarz, D.W.; Haque-Begum, S.; Kasper, L.H. Induction of a regulatory B cell population in experimental allergic encephalomyelitis by alteration of the gut commensal microflora. Gut Microbes 2010, 1, 103–108. [Google Scholar] [CrossRef]
- Probstel, A.K.; Zhou, X.; Baumann, R.; Wischnewski, S.; Kutza, M.; Rojas, O.L.; Sellrie, K.; Bischof, A.; Kim, K.; Ramesh, A.; et al. Gut microbiota-specific IgA(+) B cells traffic to the CNS in active multiple sclerosis. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Reina-Bueno, M.; Gonzalez-Lopez, J.R.; Lopez-Lopez, D.; Calvo-Lobo, C.; Ballesteros-Mora, M.; Rodriguez-Moreno, I.; Munuera-Martinez, P.V. Development and Validation of the Overall Foot Health Questionnaire for Patients with Rheumatoid Arthritis: A Cross-Sectional Descriptive Analysis. Medicina (Kaunas) 2019, 55, 290. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, H.; Fan, D.; Liu, M.; Cao, J.; Xia, Y.; Ju, D.; Xiao, C.; Guan, Q. Interactions between Gut Microbiota and Immunomodulatory Cells in Rheumatoid Arthritis. Mediat. Inflamm. 2020, 2020, 1430605. [Google Scholar] [CrossRef] [PubMed]
- Maerz, J.K.; Trostel, C.; Lange, A.; Parusel, R.; Michaelis, L.; Schafer, A.; Yao, H.; Low, H.C.; Frick, J.S. Bacterial Immunogenicity Is Critical for the Induction of Regulatory B Cells in Suppressing Inflammatory Immune Responses. Front. Immunol. 2019, 10, 3093. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Mannaa, M.; Kim, Y.; Kim, J.; Kim, G.T.; Seo, Y.S. Comparative Analysis of Fecal Microbiota Composition Between Rheumatoid Arthritis and Osteoarthritis Patients. Genes 2019, 10, 748. [Google Scholar] [CrossRef]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Alpizar-Rodriguez, D.; Lesker, T.R.; Gronow, A.; Gilbert, B.; Raemy, E.; Lamacchia, C.; Gabay, C.; Finckh, A.; Strowig, T. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann. Rheum Dis. 2019, 78, 590–593. [Google Scholar] [CrossRef]
- Maeda, Y.; Takeda, K. Role of Gut Microbiota in Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 60. [Google Scholar] [CrossRef]
- Pianta, A.; Arvikar, S.; Strle, K.; Drouin, E.E.; Wang, Q.; Costello, C.E.; Steere, A.C. Evidence of the Immune Relevance of Prevotella copri, a Gut Microbe, in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 964–975. [Google Scholar] [CrossRef]
- Liu, X.; Zou, Q.; Zeng, B.; Fang, Y.; Wei, H. Analysis of fecal Lactobacillus community structure in patients with early rheumatoid arthritis. Curr. Microbiol. 2013, 67, 170–176. [Google Scholar] [CrossRef]
- Chiang, H.I.; Li, J.R.; Liu, C.C.; Liu, P.Y.; Chen, H.H.; Chen, Y.M.; Lan, J.L.; Chen, D.Y. An Association of Gut Microbiota with Different Phenotypes in Chinese Patients with Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 1770. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.M.; Sefik, E.; Upadhyay, R.; Weissleder, R.; Benoist, C.; Mathis, D. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 6696–6701. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Klinger, C.N.; Felix, K.M.; Bradley, C.P.; Wu, E.; Tran, N.L.; Umesaki, Y.; Wu, H.J. Gut Microbiota Drive Autoimmune Arthritis by Promoting Differentiation and Migration of Peyer’s Patch T Follicular Helper Cells. Immunity 2016, 44, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Zacca, E.R.; Onofrio, L.I.; Acosta, C.D.V.; Ferrero, P.V.; Alonso, S.M.; Ramello, M.C.; Mussano, E.; Onetti, L.; Cadile, I.I.; Stancich, M.I.; et al. PD-L1(+) Regulatory B Cells Are Significantly Decreased in Rheumatoid Arthritis Patients and Increase After Successful Treatment. Front. Immunol. 2018, 9, 2241. [Google Scholar] [CrossRef]
- Piper, C.J.M.; Rosser, E.C.; Oleinika, K.; Nistala, K.; Krausgruber, T.; Rendeiro, A.F.; Banos, A.; Drozdov, I.; Villa, M.; Thomson, S.; et al. Aryl Hydrocarbon Receptor Contributes to the Transcriptional Program of IL-10-Producing Regulatory B Cells. Cell Rep. 2019, 29, 1878–1892.E7. [Google Scholar] [CrossRef]
- Sherr, D.H.; Monti, S. The role of the aryl hydrocarbon receptor in normal and malignant B cell development. Semin. Immunopathol. 2013, 35, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Lester, D.; Abe, K. The effect of restricting access to lethal methods for suicide: A study of suicide by domestic gas in Japan. Acta Psychiatr. Scand. 1989, 80, 180–182. [Google Scholar] [CrossRef]
- Rosser, E.C.; Piper, C.J.M.; Matei, D.E.; Blair, P.A.; Rendeiro, A.F.; Orford, M.; Alber, D.G.; Krausgruber, T.; Catalan, D.; Klein, N.; et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 2020, 31, 837–851.e10. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, P.; Bonifacio, E.; Ziegler, A.G. Predicting type 1 diabetes. Curr. Diab. Rep. 2005, 5, 98–103. [Google Scholar] [CrossRef]
- Yang, M.; Charlton, B.; Gautam, A.M. Development of insulitis and diabetes in B cell-deficient NOD mice. J. Autoimmun. 1997, 10, 257–260. [Google Scholar] [CrossRef]
- Hu, C.Y.; Rodriguez-Pinto, D.; Du, W.; Ahuja, A.; Henegariu, O.; Wong, F.S.; Shlomchik, M.J.; Wen, L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J. Clin. Investig. 2007, 117, 3857–3867. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sun, L.; Zhang, S.; Zhao, X.; Gang, X.; Wang, G. Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front. Endocrinol. 2020, 11, 125. [Google Scholar] [CrossRef]
- Paun, A.; Yau, C.; Meshkibaf, S.; Daigneault, M.C.; Marandi, L.; Mortin-Toth, S.; Bar-Or, A.; Allen-Vercoe, E.; Poussier, P.; Danska, J.S. Association of HLA-dependent islet autoimmunity with systemic antibody responses to intestinal commensal bacteria in children. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botía-Sánchez, M.; Alarcón-Riquelme, M.E.; Galicia, G. B Cells and Microbiota in Autoimmunity. Int. J. Mol. Sci. 2021, 22, 4846. https://doi.org/10.3390/ijms22094846
Botía-Sánchez M, Alarcón-Riquelme ME, Galicia G. B Cells and Microbiota in Autoimmunity. International Journal of Molecular Sciences. 2021; 22(9):4846. https://doi.org/10.3390/ijms22094846
Chicago/Turabian StyleBotía-Sánchez, María, Marta E. Alarcón-Riquelme, and Georgina Galicia. 2021. "B Cells and Microbiota in Autoimmunity" International Journal of Molecular Sciences 22, no. 9: 4846. https://doi.org/10.3390/ijms22094846
APA StyleBotía-Sánchez, M., Alarcón-Riquelme, M. E., & Galicia, G. (2021). B Cells and Microbiota in Autoimmunity. International Journal of Molecular Sciences, 22(9), 4846. https://doi.org/10.3390/ijms22094846