
Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
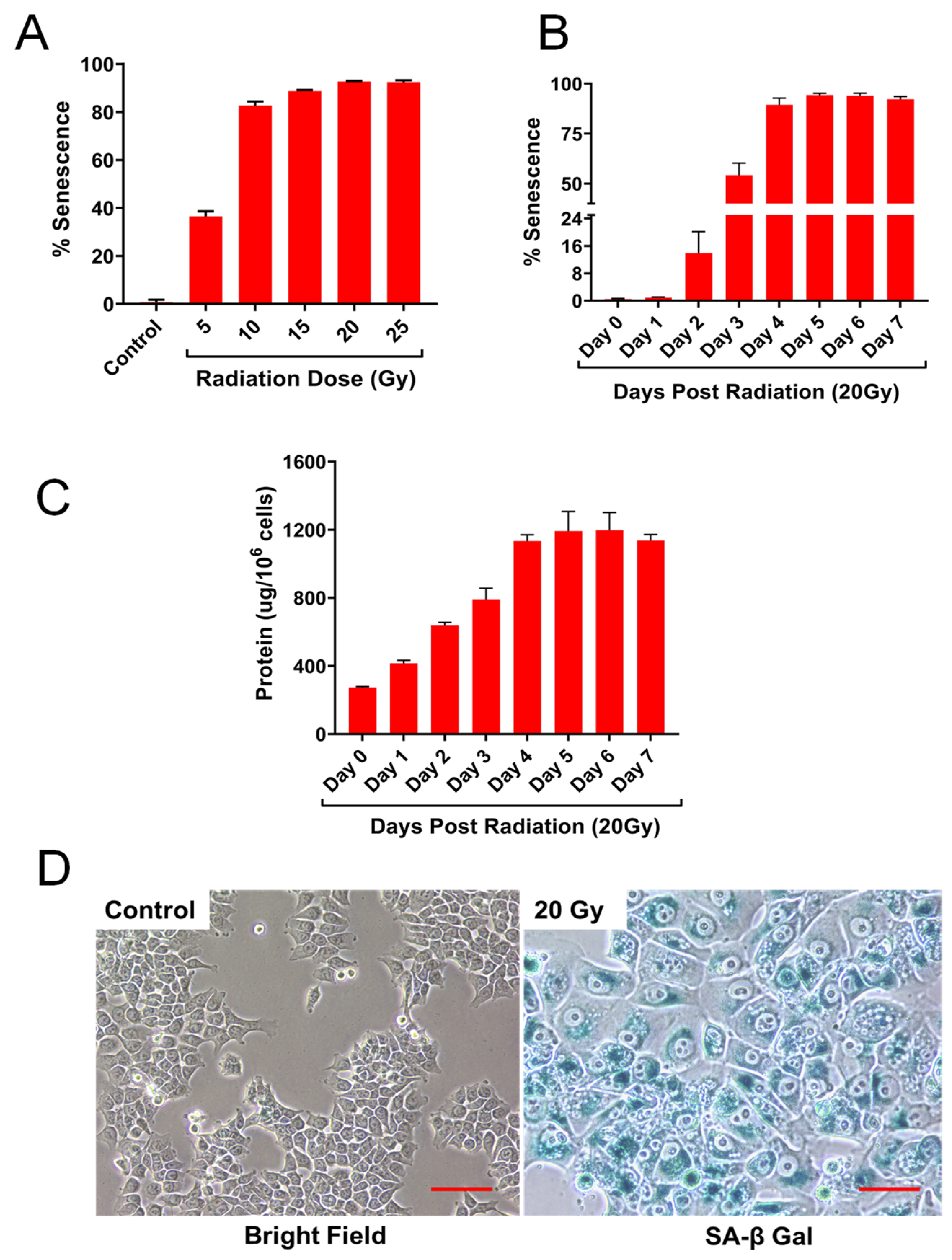
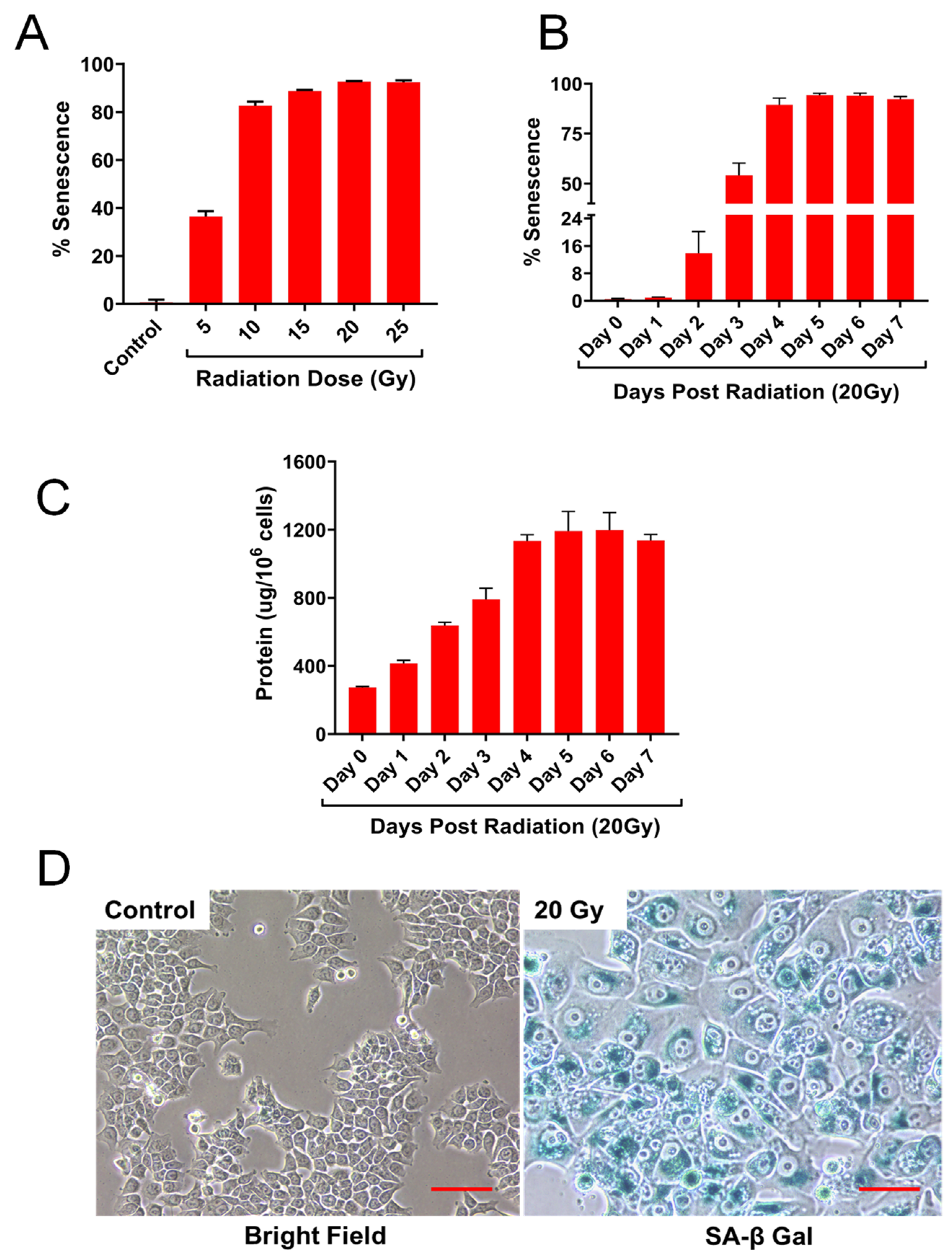
2.1. HCT-116 Cells Exhibit Cellular Senescence Phenotype Following Ionizing Radiation
2.2. IR Dose and Time Dependent Senescence Responses by HCT-116 Cells
2.3. Detection of Senescence-Associated Proteins, P53 and p21 by Immunoblot Analysis
2.4. Secretion of Cytokines and Chemokines by Senescent HCT-116 (SCC) Cell Cultures
2.5. Secretion of Extracellular Matrix Proteins, MMPs and TIMPs, and Angiogenic and Growth Factors by Senescent HCT-116 (SCC) Cell Cultures
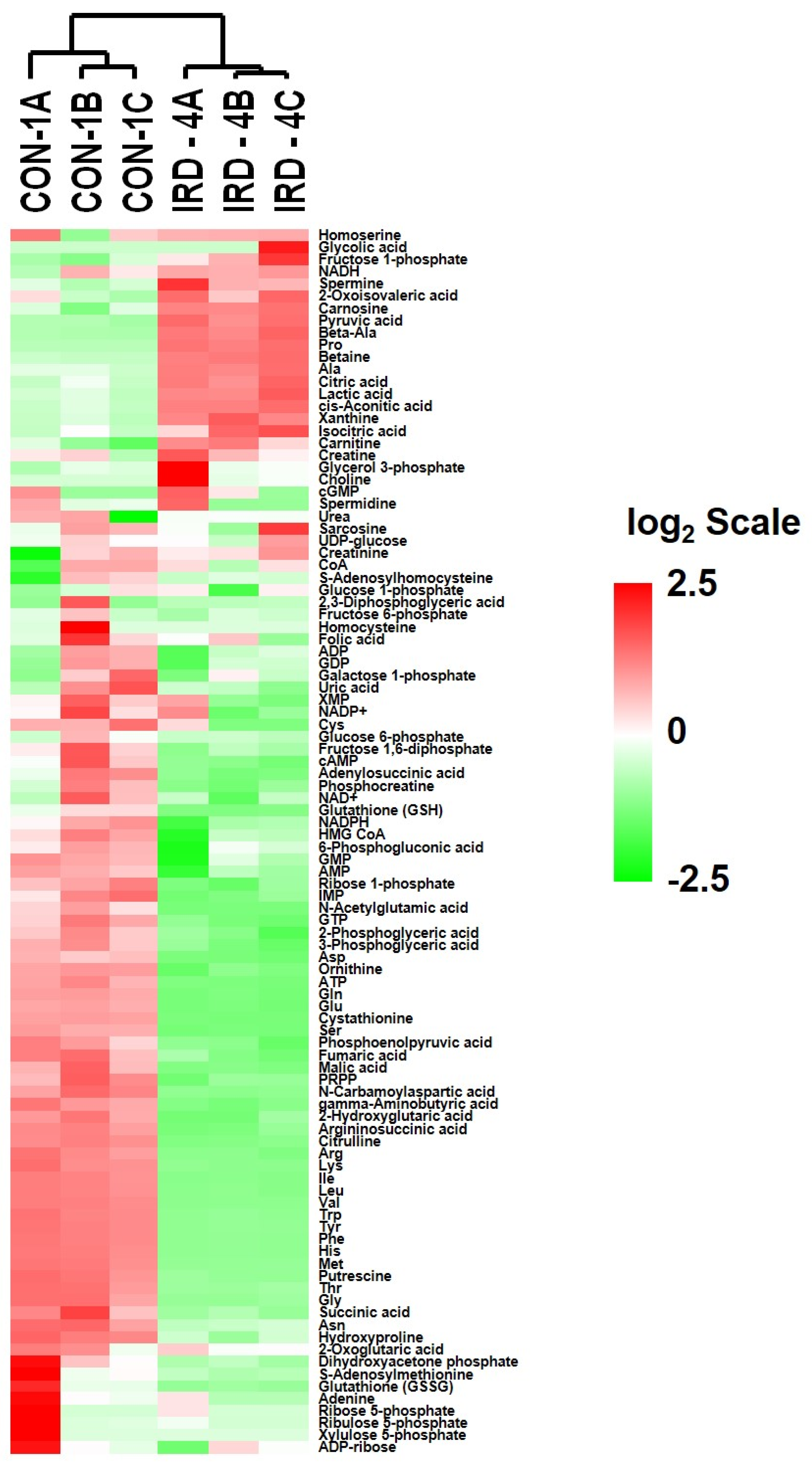
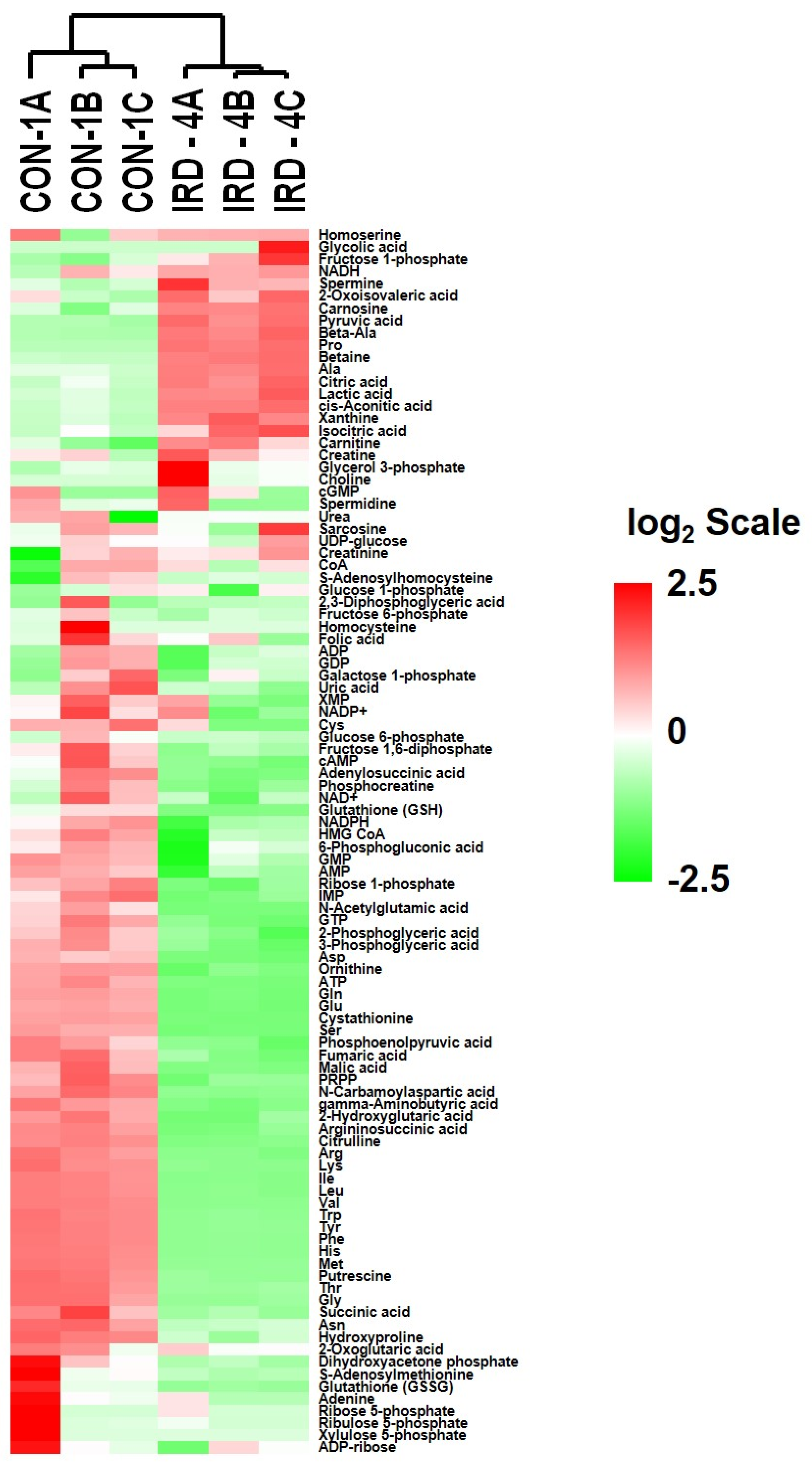
2.6. Analysis of Metabolites in Control and Senescent HCT-116 Cells (SCC)
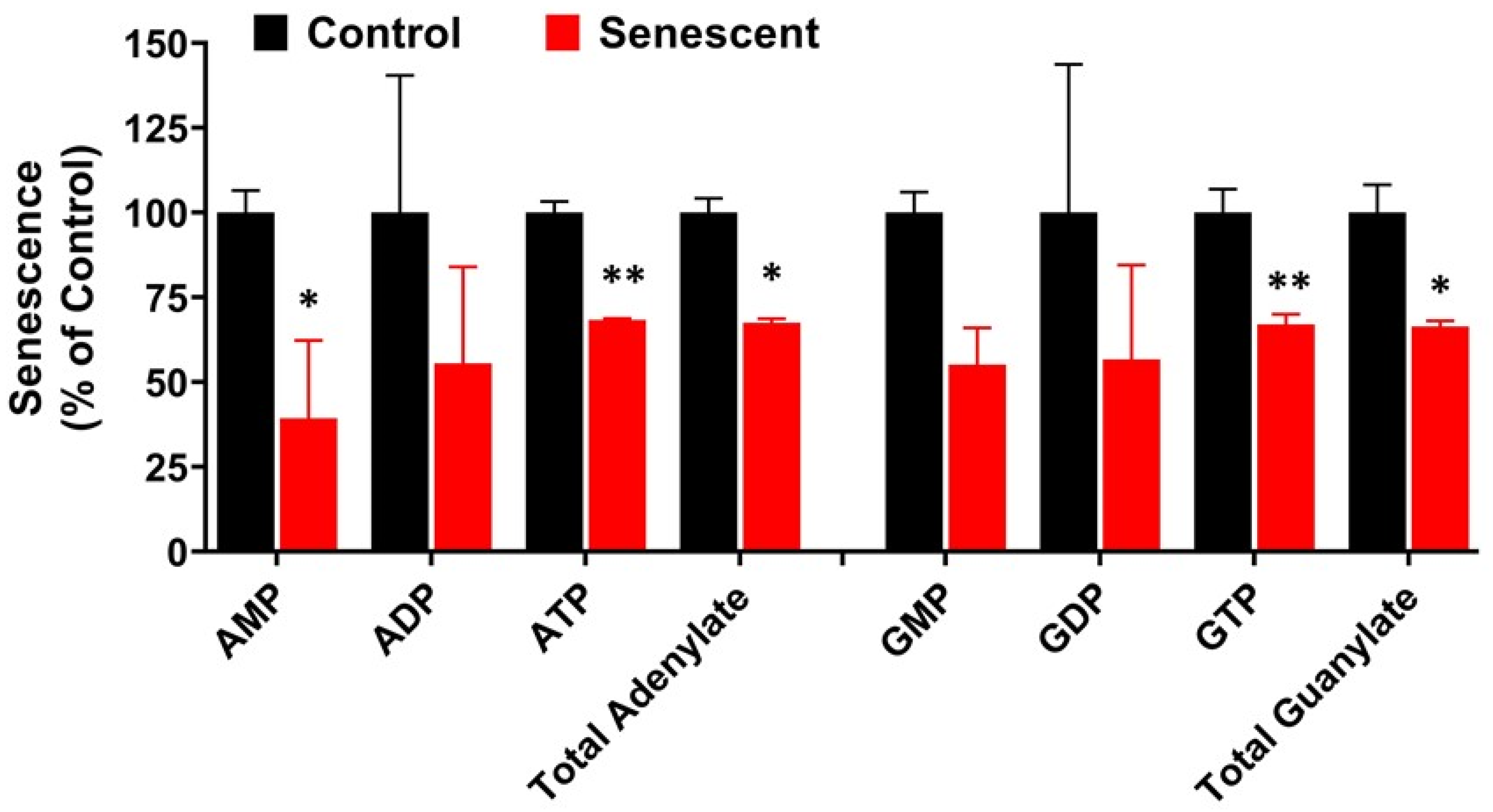
2.7. Adenylate, Guanylate Energy Charge and Creatine Levels in Senescent HCT-116 Cells (SCC)
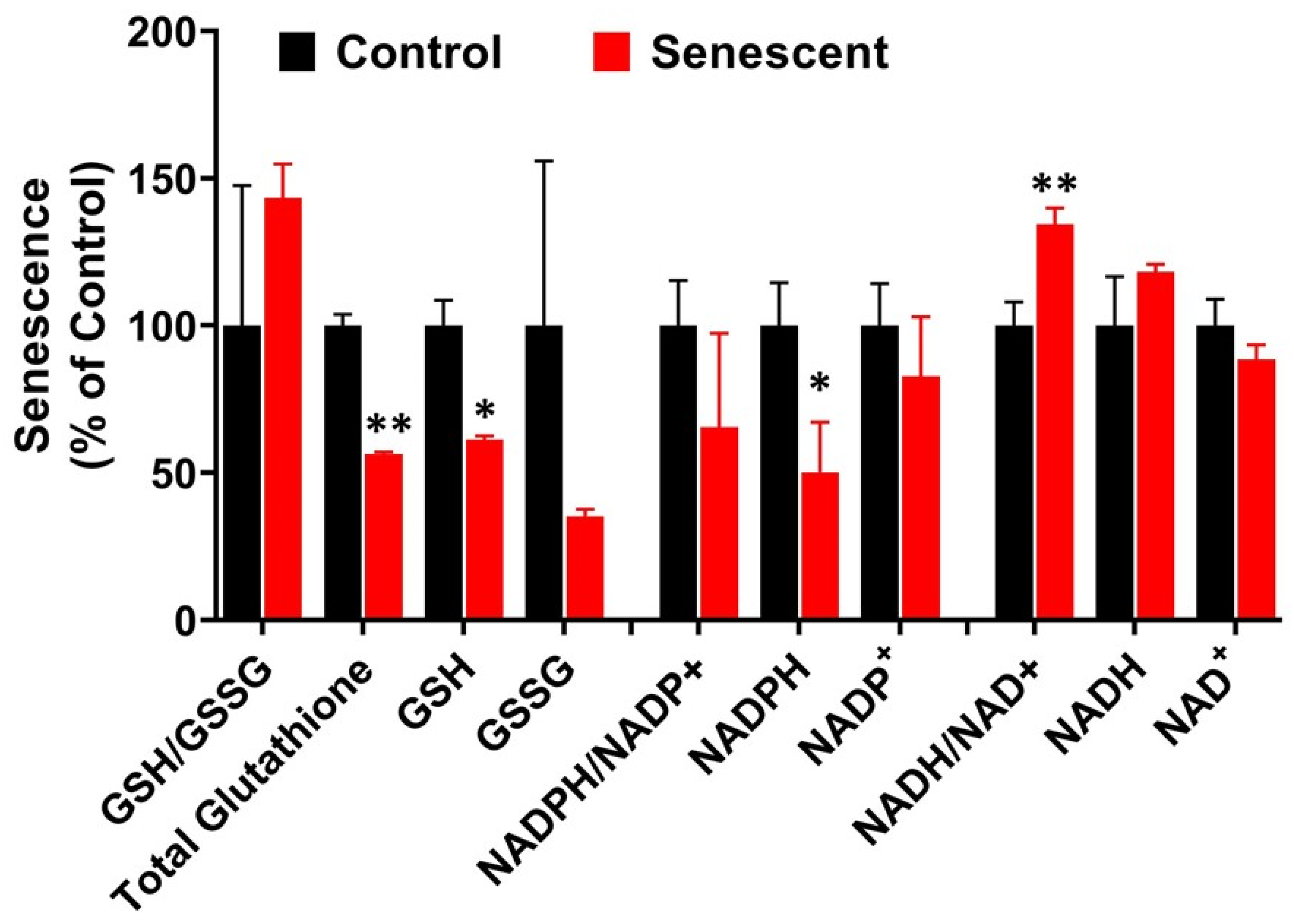
2.8. Redox System Metabolites, NAD, NADP and Glutathione in Senescent HCT-116 Cells (SCC)
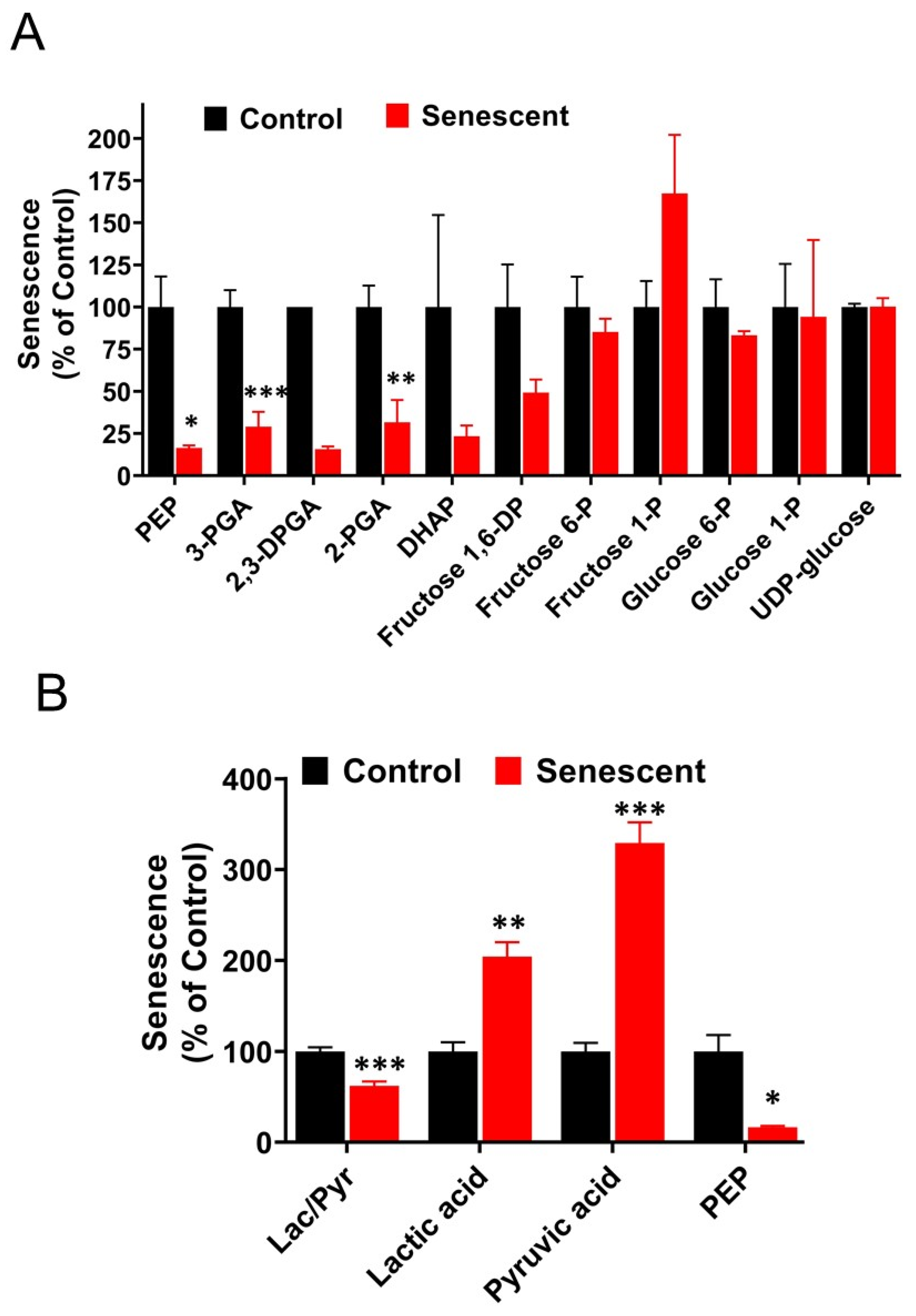
2.9. Aerobic Glycolysis with Accumulation of Pyruvate and Lactate in Senescent HCT-116 Cells (SCC)
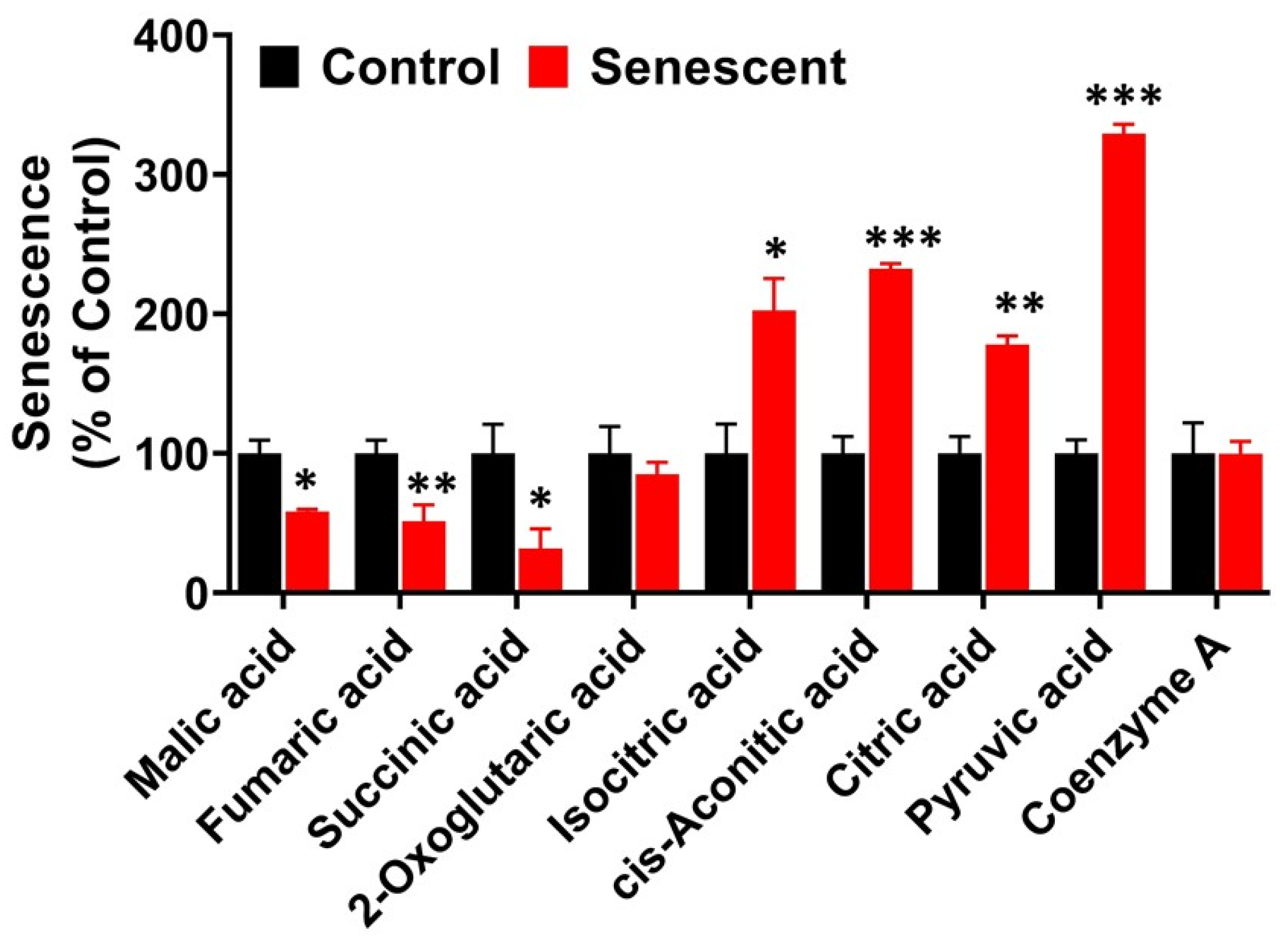
2.10. TCA (Citric Acid) Cycle Metabolites in Senescent HCT-116 Cells
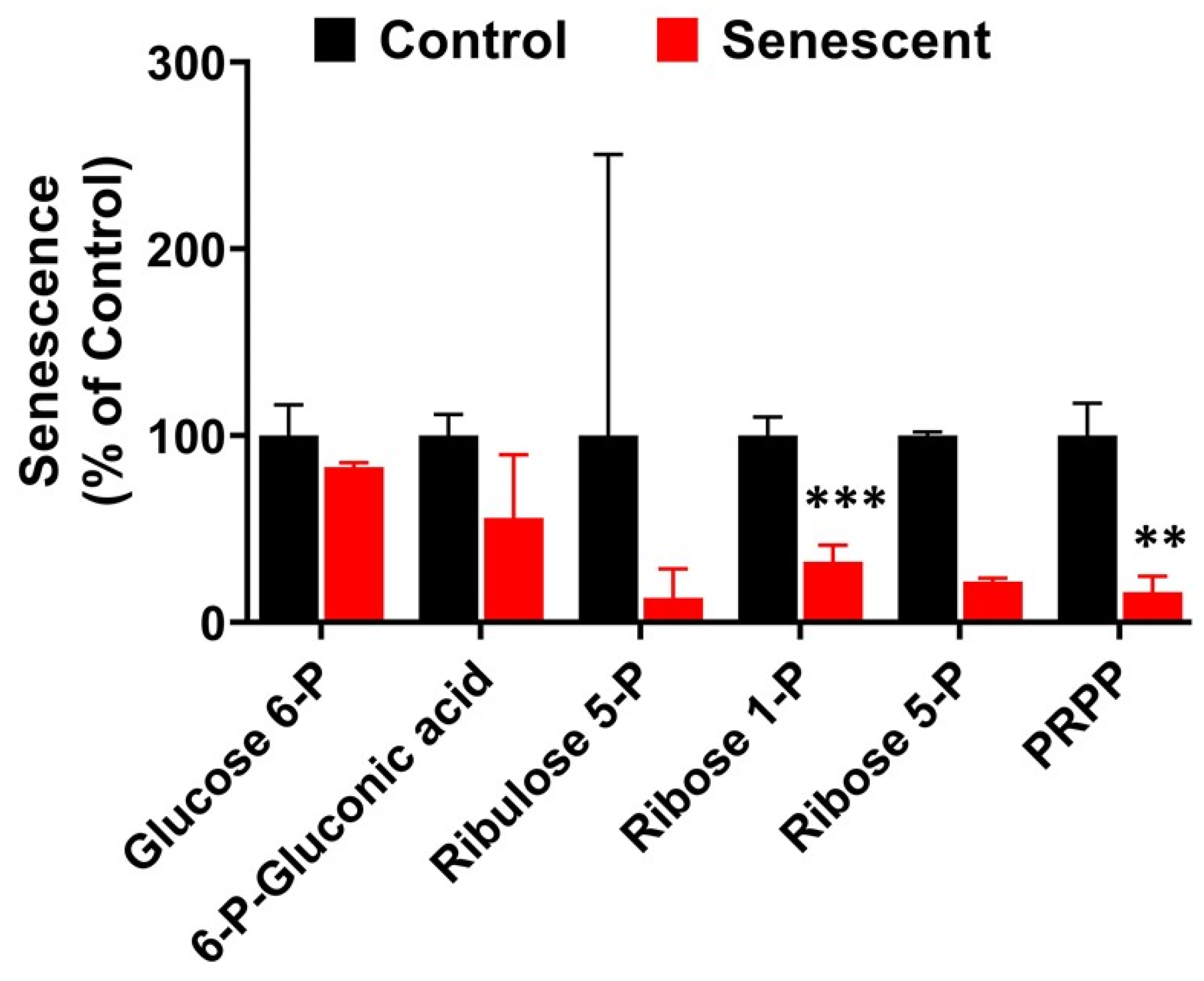
2.11. Pentose Phosphate Pathway Metabolites in Senescent HCT-116 Cells (SCC)
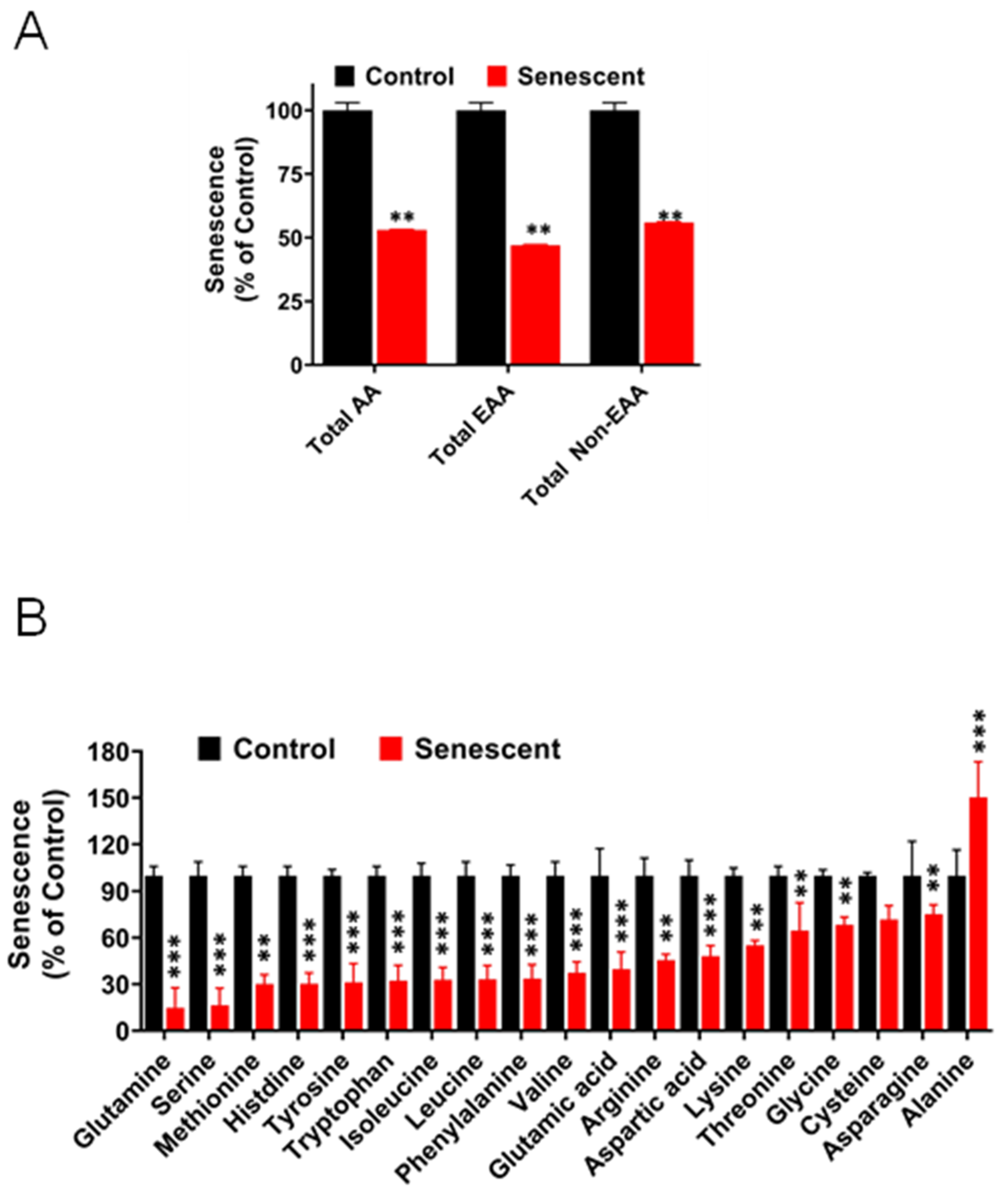
2.12. Levels of Amino Acids Decreased Significantly in Senescent HCT-116 Cells (SCC)
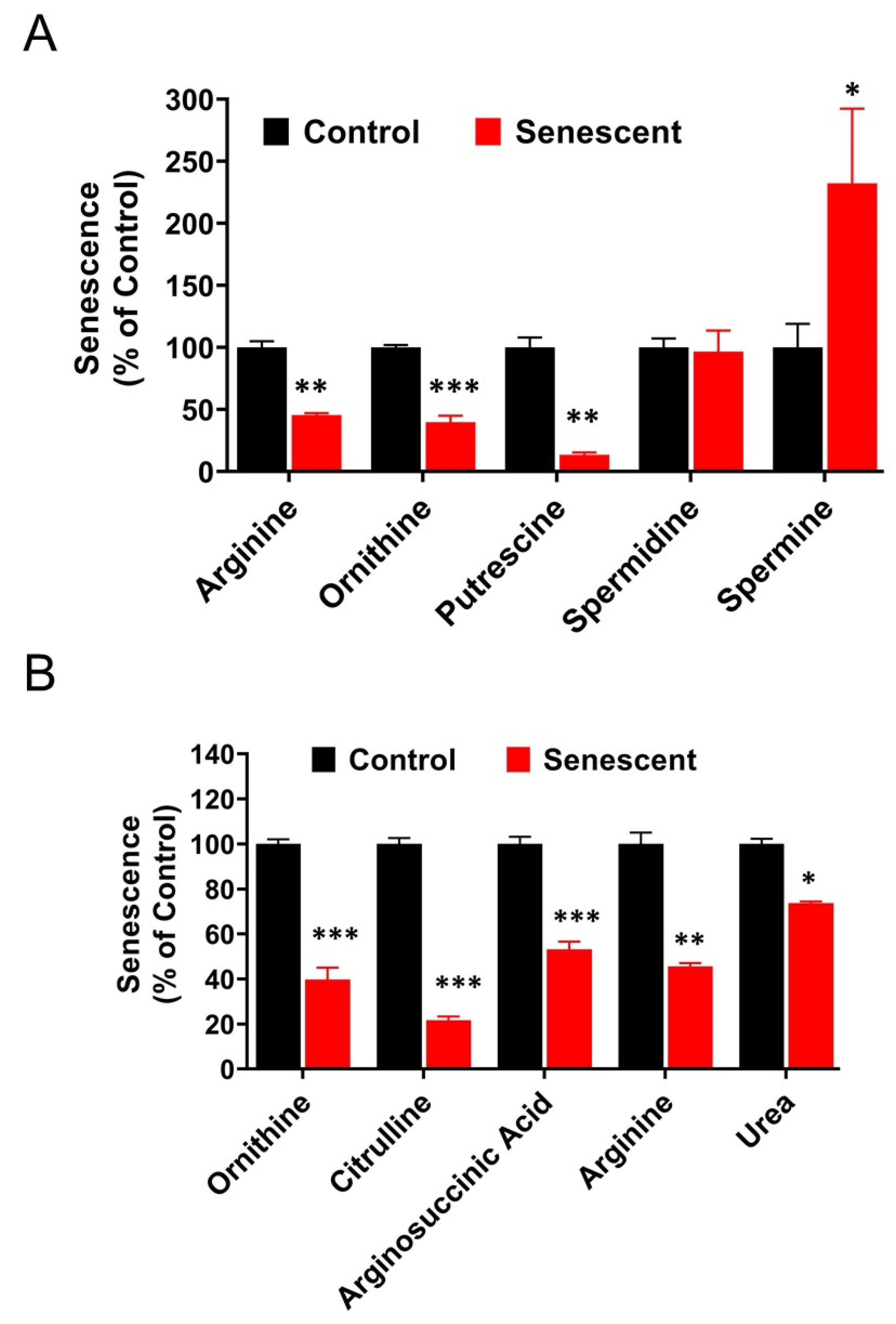
2.13. Polyamine Metabolites Are Differentially Regulated in Senescent HCT-116 Cells (SCC)
3. Discussion
4. Material & Methods
4.1. Cell Cultures
4.2. Radiation Treatments
4.3. Senescence-Associated Beta Galactosidase (SA-β Gal) Detection
4.4. Protein Extraction and Quantitation
4.5. Western Immunoblot Analysis of Proteins
4.6. Radiation Treatments and Collection of Culture Supernatant Samples
4.7. Multiplex Luminescence Analysis of Secreted Proteins in Culture Supernatants
4.8. Preparation of Proliferating (Control) and Senescent Cells for Metabolite Extraction
4.9. Analysis of Intracellular Metabolites by CE-TOFMS and CE-QqQMS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| ECM | Extracellular matrix CRC |
| HCT-116 | Human colon cancer cells |
| SCC | Senescent cancer cells |
| SCCVII | Mouse squamous cell carcinoma cells |
| IR | Ionizing radiation |
| cGy | cGray units, radiation intensity |
| SA- β-Gal | Senescence-associated β-galactosidase |
| NAD | Nicotinamide adenine dinucleotide |
| NADH | Nicotinamide adenine dinucleotide hydride |
| TCA | Tricarboxylic acid cycle (citric acid cycle) |
| PPP | Pentose phosphate pathway |
| Aldolase | Fructose, 1, 6 biphosphate aldolase |
| PRPP | 5-Phospho-D ribose 1-pyrophosphoric acid |
| VEGF | Vascular endothelial growth factor |
| MCT | Monocarboxylate transporter |
| SASP | Senescence-associated secretory phenotype |
| TGF | Transforming growth factor |
| PDGF | Platelet derived growth factor |
| IL | Interleukin |
| MMPs | Matrix metalloproteases |
| TIMPs | Tissue inhibitors of MMPs |
References
- Haugstetter, A.M.; Loddenkemper, C.; Lenze, D.; Gröne, J.; Standfuss, C.; Petersen, I.; Dörken, B.; Schmitt, C.A. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 2010, 103, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.C.; Wang, Q.; Grobman, L.; Chu, E.; Wu, D.Y. Accelerated cellular senescence in solid tumor therapy. Exp. Oncol. 2012, 34, 298–305. [Google Scholar]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; Di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; David, G. Pro- and anti-tumorigenic functions of the senescence-associated secretory phenotype. Expert Opin. Ther. Targets 2019, 23, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Liao, E.-C.; Hsu, Y.-T.; Chuah, Q.-Y.; Lee, Y.-J.; Hu, J.-Y.; Huang, T.-C.; Yang, P.-M.; Chiu, S.-J. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis. 2014, 5, e1255. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.C.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol. Cell. Proteom. 2016, 15, 2924–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Russo, A.; Kuppusamy, P.; Krishna, M.C. Radiation, Radicals, and Images. Ann. N. Y. Acad. Sci. 2006, 899, 28–43. [Google Scholar] [CrossRef]
- Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar]
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 2008, 76, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and Functional Characteristic of Senescent Cancer Cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef]
- Graf, W.; Dahlberg, M.; Osman, M.; Holmberg, L.; Påhlman, L.; Glimelius, B. Short-term preoperative radiotherapy results in down-staging of rectal cancer: A study of 1316 patients. Radiother. Oncol. 1997, 43, 133–137. [Google Scholar] [CrossRef]
- Bernier, J.; Hall, E.J.; Giaccia, A. Radiation oncology: A century of achievements. Nat. Rev. Cancer 2004, 4, 737–747. [Google Scholar] [CrossRef]
- Citrin, D.E.; Mitchell, J.B. Altering the Response to Radiation: Sensitizers and Protectors. Semin. Oncol. 2014, 41, 848–859. [Google Scholar] [CrossRef] [Green Version]
- Citrin, D.E. Recent Developments in Radiotherapy. N. Engl. J. Med. 2017, 377, 1065–1075. [Google Scholar] [CrossRef]
- Haddock, M.G. Intraoperative radiation therapy for colon and rectal cancers: A clinical review. Radiat. Oncol. 2017, 12, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Prim. 2015, 1, 15065. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantor, J.R.; Sabatini, D.M. Cancer Cell Metabolism: One Hallmark, Many Faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Scott, A.; Cameron, M.; Murray, D. Induction of accelerated senescence by gamma radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiat. Res. 2005, 163, 53–62. [Google Scholar] [CrossRef] [PubMed]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; De Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent Human Fibroblasts Show Increased Glycolysis and Redox Homeostasis with Extracellular Metabolomes That Overlap with Those of Irreparable DNA Damage, Aging, and Disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef] [PubMed]
- James, E.N.L.; Bennett, M.H.; Parkinson, E.K. The induction of the fibroblast extracellular senescence metabolome is a dynamic process. Sci. Rep. 2018, 8, 12148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Atkinson, D.E. Energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 1968, 7, 4030–4034. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Acosta, J.C.; Gil, J. Senescence: A new weapon for cancer therapy. Trends Cell Biol. 2012, 22, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Yoshie, O. The Chemokine Superfamily Revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-beta—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Zhang, R. Metabolic alterations accompanying oncogene-induced senescence. Mol. Cell. Oncol. 2014, 1, e963481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Olson, K.A.; Jiang, L.; Hawkins, A.J.; Van Vranken, J.G.; Xie, J.; Egnatchik, R.A.; Earl, E.G.; DeBerardinis, R.J.; Rutter, J. A Role for the Mitochondrial Pyruvate Carrier as a Repressor of the Warburg Effect and Colon Cancer Cell Growth. Mol. Cell 2014, 56, 400–413. [Google Scholar] [CrossRef] [Green Version]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. eLife 2014, 3, e03342. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A Metabolic Key Player in Cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef] [Green Version]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef]
- Meng, Y.; Efimova, E.V.; Hamzeh, K.W.; Darga, T.E.; Mauceri, H.J.; Fu, Y.-X.; Kron, S.J.; Weichselbaum, R.R. Radiation-inducible Immunotherapy for Cancer: Senescent Tumor Cells as a Cancer Vaccine. Mol. Ther. 2012, 20, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Mikuła-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Książek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell. Mol. Life Sci. 2020, 77, 213–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2020, 9, 1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef] [PubMed]
- AbdelGawad, I.Y.; Sadak, K.T.; Lone, D.W.; Dabour, M.S.; Niedernhofer, L.J.; Zordoky, B.N. Molecular mechanisms and cardiovascular implications of cancer therapy-induced senescence. Pharmacol. Ther. 2021, 221, 107751. [Google Scholar] [CrossRef] [PubMed]
- Brattain, M.G.; Fine, W.D.; Khaled, F.M.; Thompson, J.; Brattain, D.E. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res. 1981, 41, 1751–1756. [Google Scholar]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagineni, C.N.; Naz, S.; Choudhuri, R.; Chandramouli, G.V.R.; Krishna, M.C.; Brender, J.R.; Cook, J.A.; Mitchell, J.B. Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells. Int. J. Mol. Sci. 2021, 22, 4835. https://doi.org/10.3390/ijms22094835
Nagineni CN, Naz S, Choudhuri R, Chandramouli GVR, Krishna MC, Brender JR, Cook JA, Mitchell JB. Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells. International Journal of Molecular Sciences. 2021; 22(9):4835. https://doi.org/10.3390/ijms22094835
Chicago/Turabian StyleNagineni, Chandrasekharam N., Sarwat Naz, Rajani Choudhuri, Gadisetti V. R. Chandramouli, Murali C. Krishna, Jeffrey R. Brender, John A. Cook, and James B. Mitchell. 2021. "Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells" International Journal of Molecular Sciences 22, no. 9: 4835. https://doi.org/10.3390/ijms22094835
APA StyleNagineni, C. N., Naz, S., Choudhuri, R., Chandramouli, G. V. R., Krishna, M. C., Brender, J. R., Cook, J. A., & Mitchell, J. B. (2021). Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells. International Journal of Molecular Sciences, 22(9), 4835. https://doi.org/10.3390/ijms22094835