
Proapoptotic Bad Involved in Brain Development, When Severely Defected, Induces Dramatic Malformation in Zebrafish


Abstract
1. Introduction
2. Results
2.1. The Bad Gene Is a Material Factor Involved in PCD at an Early Developmental Stage
2.2. The Bad Gene Involved in Regulating Environmental Stress at an Early Developmental Stage
2.3. Loss-of-Bad-Mediated PCD Can Interrupt Cell Migration and the Formation of the Three Germ Layers, Especially the Ectoderm
2.4. Bad-Mediated PCD Can Regulate Brain Morphogenesis
2.5. Dramatic Malformation Was Observed in the Hindbrain during Knockdown of BAD
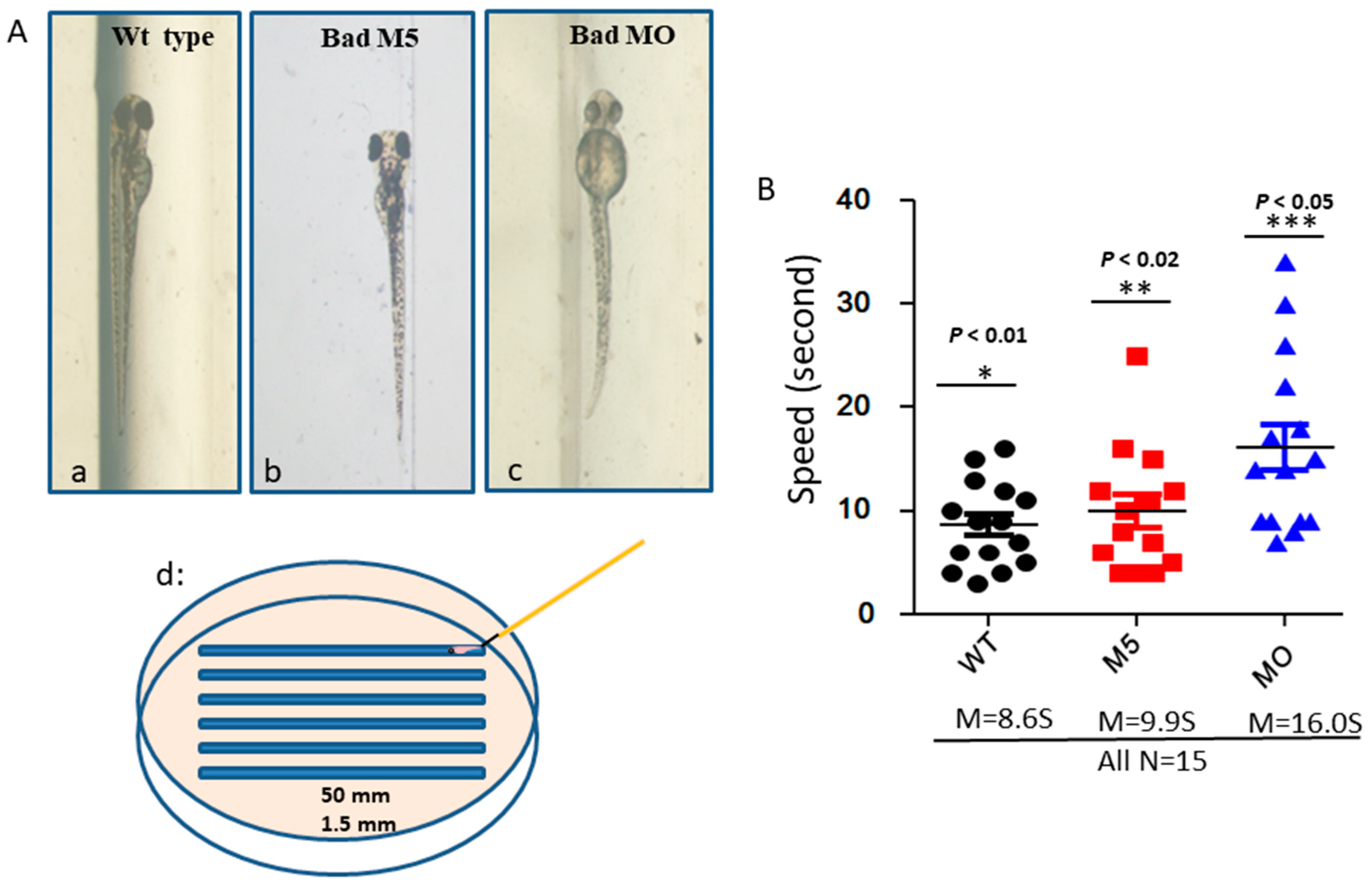
2.6. Investigation of the Correlation of Bad-Mediated Cell Death with Brain Function through the Swimming Ability Test
3. Discussion
3.1. Bad Loss-of-Function Can Enhance Cell Death Signaling
3.2. Reactive Oxygen Species (ROS) Act as Environmental Stress Factors That Regulate Cell Death during Early Embryonic Development
3.3. Loss of Bad-Mediated PCD Can Interfere with Ectoderm Development, Especially Brain Development, via Delayed Early Cell Migration
3.4. Bad Knockdown-Induced Brain Defect during Development
4. Materials and Methods
4.1. Experimental Fish
4.2. Apoptotic Cell Staining
4.3. ROS Detection
4.4. RNA Extraction
4.5. Reverse-Transcription Polymerase Chain Reaction (RT-PCR) Analysis
4.6. Quantitative (q)RT-PCR
4.7. Morpholino Oligonucleotides (MOs)
4.8. Western Blotting
4.9. Whole-Mount In Situ Hybridization
4.10. Swimming Activity Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nature Mol. Cell Biol. 2015, 16, 329–342. [Google Scholar] [CrossRef]
- White, K.; Grether, M.E.; Abrams, J.M.; Young, L.; Farrell, K.; Steller, H. Genetic control of programmed cell death in Drosophila. Science 1994, 264, 677–683. [Google Scholar] [CrossRef]
- Grether, M.E.; Abrams, J.M.; Agapite, J.; White, K.; Steller, H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995, 9, 1694–1708. [Google Scholar] [CrossRef]
- Chen, P.; Nordstrom, W.; Gish, B.; Abrams, J.M. Grim, a novel cell death gene in Drosophila. Genes Dev. 1996, 10, 1773–1782. [Google Scholar] [CrossRef]
- Ellis, R.E.; Yuan, J.Y.; Horvitz, H.R. Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 1991, 7, 663–698. [Google Scholar] [CrossRef] [PubMed]
- Suzanne, M.; Steller, H. Shaping organisms with apoptosis. Cell Death Differ. 2013, 20, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.S.; Barkett, M.; McCall, K. Stage-specific regulation of caspase activity in Drosophila oogenesis. Dev. Biol. 2003, 260, 113–123. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2010, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Monier, B.; Gettings, M.; Gay, G.; Mangeat, T.; Schott, S.; Guarner, A.; Suzanne, M. Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature 2015, 518, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Prdro, J.M.; Kroemer, G. Organelle-specific of cell death. Nat. Cell Biol. 2014, 16, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Zhan, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996, 87, 619–628. [Google Scholar]
- Datta, S.R.; Katsov, A.; Hu, L.; Petros, A.; Fesik, S.W.; Yaffe, M.B.; Greenberg, M.E. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 2000, 6, 41–51. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar] [CrossRef]
- Harada, H.; Becknell, B.; Wilm, M.; Mann, M.; Huang, L.J.; Taylor, S.S.; Scott, J.D.; Korsmeyer, S.J. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 1999, 3, 413–422. [Google Scholar] [CrossRef]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, A.; Ballif, B.A.; Richards, S.A.; Blenis, J. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr. Biol. 2000, 10, 127–135. [Google Scholar] [CrossRef]
- Ranger, A.M.; Zha, J.; Harada, H.; Datta, S.D.; Danial, N.N.; Gilmore, A.P.; Kutok, J.L.; Le Beau, M.M.; Greenberg, M.F.; Korsmeyer, S.J. Bad-deficient mice develop diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9324–9329. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Chang, M.S.; Chen, J.Y.; Yen, J.J.Y.; Lu, I.C.; Chou, C.M.; Huang, C.J. Cloning of zebrafish BAD, a BH3-only proapoptotic protein, whoseoverexpression leads to apoptosis in COS-1 cells and zebrafish embryos. Biochem. Biophys. Res. Comm. 2003, 304, 667. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book; A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio), 2nd ed.; University of Oregon Press: Eugene, OR, USA, 1993; p. 300. [Google Scholar]
- Wang, K.; Yin, X.M.; Chao, D.T.; Milliman, C.L.; Korsmeyer, S.J. BID: A novel BH3 domain-only death agonist. Genes Dev. 1996, 10, 2859–2869. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef]
- Yin, X.M.; Wang, K.; Gross, A.; Zhao, Y.; Zinkel, S.; Klocke, B.; Roth, K.A.; Korsmeyer, S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999, 400, 886–891. [Google Scholar] [CrossRef]
- Bouillet, P.; Purton, J.F.; Godfrey, D.I.; Zhang, L.C.; Coultas, L.; Puthalakath, H.; Pellegrini, M.; Cory, S.; Adams, J.M.; Strasser, A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 2002, 415, 922–926. [Google Scholar] [CrossRef]
- Doughty, M.L.; De Jager, P.L.; Korsmeyer, S.J.; Heintz, N. Neuro-degeneration in Lurcher mice occurs via multiple cell death path-ways. J. Neurosci. 2000, 20, 3687–3694. [Google Scholar] [CrossRef] [PubMed]
- Salas-Vidal, E.; Lomelí, H.; Castro-Obregón, S.; Cuervo, R.; Escalante-Alcalde, D.; Covarrubias, L. Reactive oxygen species participate in the control of mouse embryonic cell death. Exp. Cell Res. 1998, 238, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Effects of Oxidative Stress on Embryonic Development. Birth Defects Res. 2007, 81, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, L.; Hernández-García, D.; Schnabel, D.; Salas-Vidal, E.; Susana Castro-Obregón, S. Function of reactive oxygen species during animal development: Passive or active? Dev. Biol. 2008, 320, 1–11. [Google Scholar] [CrossRef]
- Hong, J.R.; Lin, G.H.; Lin, C.J.F.; Wang, W.P.; Lee, C.C.; Wu, J.L. The phosphatidylserine receptor is required for the engulfment of dead apoptotic cell and for normal development in zebrafish. Development 2004, 131, 5417–5427. [Google Scholar] [CrossRef]
- Li, M.O.; Sarkisian, M.R.; Mehal, W.Z.; Rakic, P.; Flavell, R.A. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 2003, 302, 1560–3156. [Google Scholar] [CrossRef]
- Shiu, W.L.; Huang, K.L.; Hung, J.C.; Li, H.C.; Wu, J.L.; Hong, J.R. Knockdown of zebrafish YY1a can downregulate the phosphatidylserine (PS) receptor expression, leading to abnormal brain and heart development. J. Biomed. 2016, 23, 31. [Google Scholar] [CrossRef]
- Cole, L.K.; Ross, L.S. Apoptosis in the developing zebrafish embryo. Dev. Biol. 2001, 240, 123–142. [Google Scholar] [CrossRef]
- Blader, P.; Strahle, U. Zebrafish developmental genetics and central nervous system development. Human Mol. Genet. 2000, 9, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Manova, K.; Tomihara-Newberger, C.; Wang, S.; Godelman, A.; Kalantry, S.; Witty-Blease, K.; De Leon, V.; Chen, W.S.; Lacy, E.; Bachvarova, R.F. Apoptosis in mouse embryos: Elevated levels in pregastrulae and in the distal anterior region of gastrulae of normal and mutant mice. Dev. Dyn. 1998, 213, 293–308. [Google Scholar] [CrossRef]
- Kuan, C.Y.; Roth, K.A.; Flavell, R.A.; Rakic, P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000, 23, 291–297. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Staley, K.; Chun, J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996, 122, 1165–1174. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Weiner, J.A.; Chun, J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J. Comp. Neurol. 1998, 396, 39–50. [Google Scholar] [CrossRef]
- Thomaidou, D.; Mione, M.C.; Cavanagh, J.F.; Parnavelas, J.G. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J. Neurosci. 1997, 17, 1075–1085. [Google Scholar] [CrossRef]
- Aloyz, R.S.; Bamji, S.X.; Pozniak, C.D.; Toma, J.G.; Atwal, J.; Kaplan, D.R.; Miller, F.D. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 1998, 143, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.N.; Tolkovsky, A.M. A role for MAPK/ERK insympathetic neuron survival: Protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 1999, 19, 664–673. [Google Scholar] [CrossRef]
- Slack, R.S.; Belliveau, D.J.; Rosenberg, M.; Atwal, J.; Lochmu, L.H.; Aloyz, R.; Haghighi, A.; Lach, B.; Seth, P.; Cooper, E.; et al. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J. Cell Biol. 1996, 135, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.D.; Pozniak, C.D.; Walsh, G.S. Neuronal life and death: An essential role for the p53 family. Cell Death Differ. 2000, 7, 880–888. [Google Scholar] [CrossRef]
- Levine, A.L.; Hu, W.; Feng, Z. The P53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef]
- Tedeschi, A.; Giovanni, S.D. The non-apoptotic role of p53 in neuronal biology: Enlightening the dark side of the moon. EMBO Rep. 2009, 10, 576–583. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death preceptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Chatoo, W.; Abdouh, M.; Bernier, G. p53 Pro-Oxidant Activity in the Central Nervous System: Implication in Aging and Neurodegenerative Diseases. Antioxi. Redox Signal. 2011, 15, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Hong, J.R. Betanodavirus B2 causes ATP depletion-induced cell death via mitochondrial targeting and Complex II inhibition in vitro and in vivo. J. Biol. Chem. 2010, 285, 39801–39810. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Wu, J.L. Induction of apoptotic death in cells via Bad gene expression by infectious pancreatic necrosis virus infection. Cell Death Differ. 2002, 9, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Thisse, C.; Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008, 3, 59–69. [Google Scholar] [CrossRef]
- Thisse, B.; Pflumio, S.; Fthauer, M.; Loppin, B.; Heyer, V.; Degrave, A.; Woehl, R.; Lux, A.; Steffan, T.; Charbonnier, X.Q.; et al. Expression of the Zebrafish Genome during Embryogenesis (NIH R01 RR15402). ZFIN Direct Data Submission. 2001. Available online: https://zfin.org/ZDB-PUB-010810-1 (accessed on 1 May 2021).
- Thisse, B.; Thisse, C. Fast Release Clones: A High Throughput Expression Analysis. ZFIN Direct Data Submission. 2004. Available online: https://zfin.org/ZDB-PUB-040907-1 (accessed on 1 May 2021).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward 5′→3′ | Reverse 5′→3′ |
|---|---|---|
| p53 | CTACTAAACTACATGTGCAATAGCAG | CTGAGGCAGGCACCACATCACT |
| bad | ATGGCACATATGTTTAATATCTCTGA | CTACTCTGCGGGGCGCGA |
| fabp7a | CTCTCAACATGGTCGATGCATT | CTGGACATTATGCCTTCTCGTA |
| pax2a | CTTCTAACAGGCACATCCCAT | CATTAACCCTCACTAAAGGGAACTATCCGTTCAAAGCCCG |
| Gene | Forward 5′→3′ | Reverse 5′→3′ |
|---|---|---|
| β-actin | ACTGTATTGTCTGGTGGTAC | TACTCCTGCTTGCTAATCC |
| p53 | ACCACTGGGACCAAACGTAG | CAG AGTCGCTTCTTCCTTCG |
| Caspase-8 | CCAGACAATCTGGATGAACTTTAC | TGCAAACTGCTTTATCTCATCT |
| pva1b5 | ATGGCACTTGCAGGAATCCTGA | TGTTGGTCTCGGCCTCTGTGAG |
| crybb1 | ATGTCTCAGACCGCCAAATCCG | GCCCTGGAAGTTCTCCTGGTCA |
| pax7a | CCAGGAACAGTTCCTCGAATGATG | CCGTGATGGGCCATTTCCAC |
| irx4a | GCGGACAAGGCTACGGGAATT | AGCGTTTTCCTGCGGGTCC |
| fabp7a | TGTGCCACTTGGAAACTGGTTGAC | CCCAGTTTGAAGGAGATCTCGGTG |
| Catalase | TAAAGGAGCAGGAGCGTTTGGCTA | TTCACTGCGAAACCACGAGGATCT |
| Mn–sod | CCGGACTATGTTAAGGCCATCT | ACACTCGGTTGCTCTCTTTTCTCT |
| Cu/Zn–sod | GTCGTCTGGCTTGTGGAGTG | TGTCAGCGGGCTAGTGCTT |
| nrf2a | GAGCGGGAGAAATCACACAGAATG | CAGGAGCTGCATGCACTCATCG |
| nrf2b | GGCAGAGGGAGGAGGAGACCAT | AAACAGCAGGGCAGACAACAAGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, J.-C.; Wu, J.-L.; Hong, J.-R. Proapoptotic Bad Involved in Brain Development, When Severely Defected, Induces Dramatic Malformation in Zebrafish. Int. J. Mol. Sci. 2021, 22, 4832. https://doi.org/10.3390/ijms22094832
Hung J-C, Wu J-L, Hong J-R. Proapoptotic Bad Involved in Brain Development, When Severely Defected, Induces Dramatic Malformation in Zebrafish. International Journal of Molecular Sciences. 2021; 22(9):4832. https://doi.org/10.3390/ijms22094832
Chicago/Turabian StyleHung, Jo-Chi, Jen-Leih Wu, and Jiann-Ruey Hong. 2021. "Proapoptotic Bad Involved in Brain Development, When Severely Defected, Induces Dramatic Malformation in Zebrafish" International Journal of Molecular Sciences 22, no. 9: 4832. https://doi.org/10.3390/ijms22094832
APA StyleHung, J.-C., Wu, J.-L., & Hong, J.-R. (2021). Proapoptotic Bad Involved in Brain Development, When Severely Defected, Induces Dramatic Malformation in Zebrafish. International Journal of Molecular Sciences, 22(9), 4832. https://doi.org/10.3390/ijms22094832