
Mitochondrial Dysfunction in Podocytes Caused by CRIF1 Deficiency Leads to Progressive Albuminuria and Glomerular Sclerosis in Mice

 , ,
, , 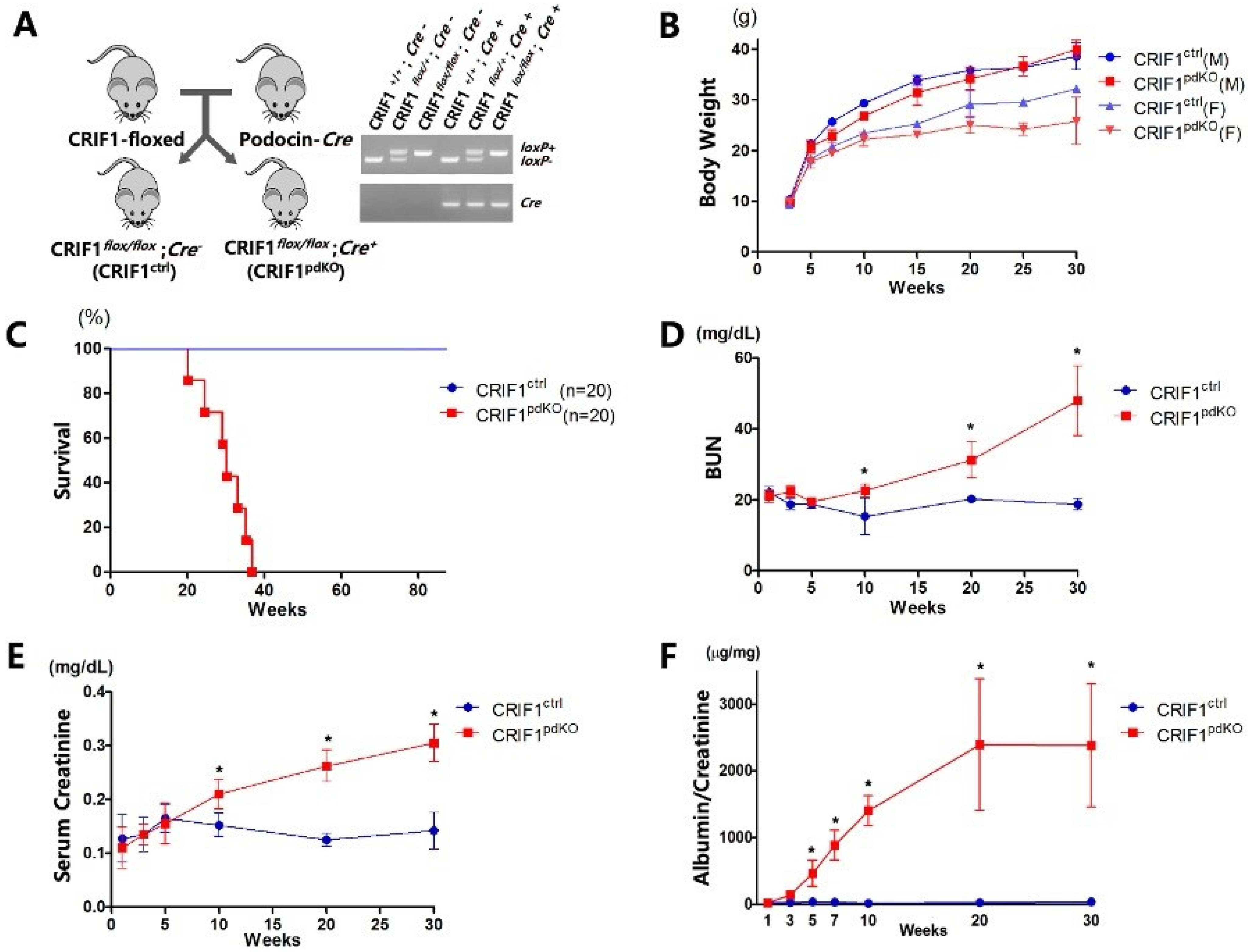{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. CRIF1pdKO Mice Showed Massive Albuminuria and Progressive Renal Failure
2.2. CRIF1pdKO Mice Showed Progressive Glomerular Sclerosis and Tubular Fibrosis
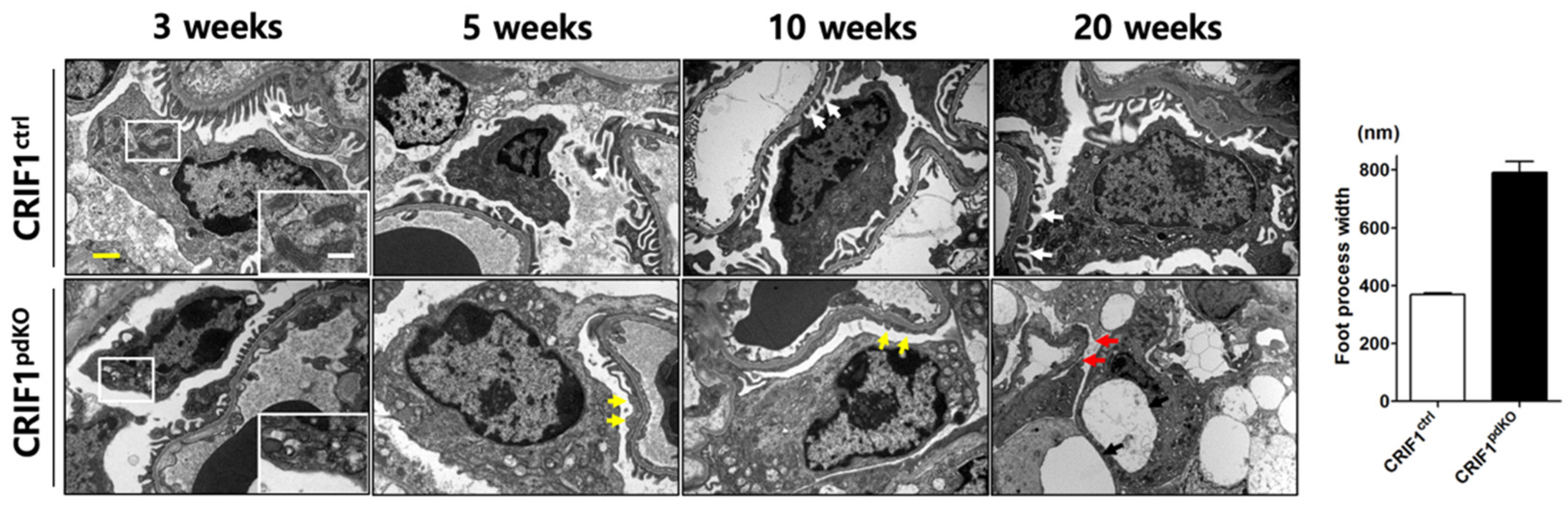
2.3. CRIF1pdKO Mice Showed Progressive Podocyte Injury
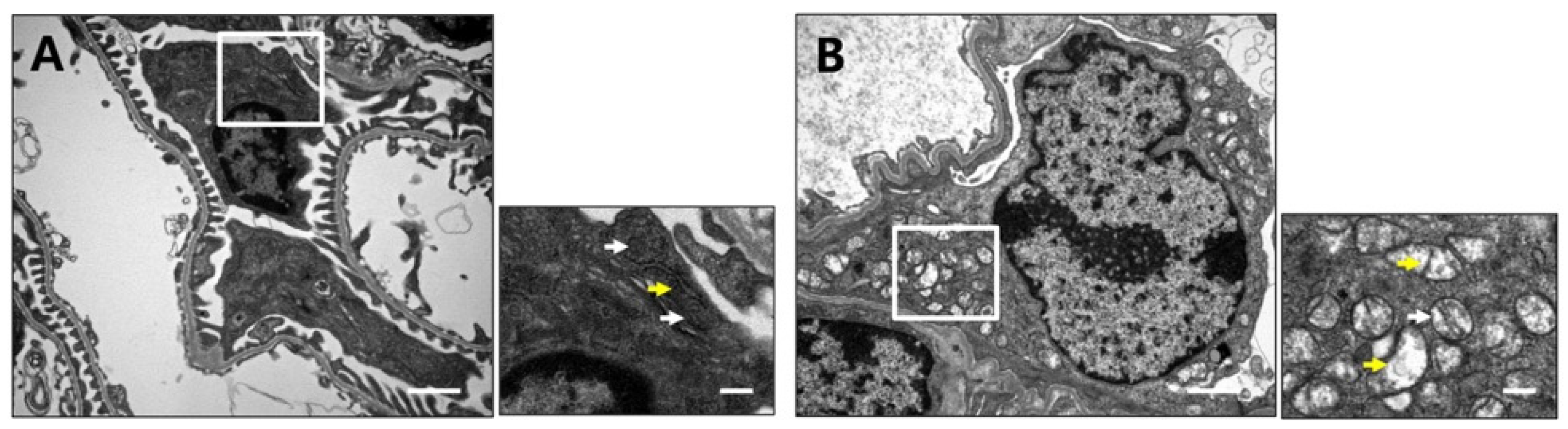
2.4. Podocyt-Specific CRIF1 Deletion Causes Structural Abnormality of Mitochondria
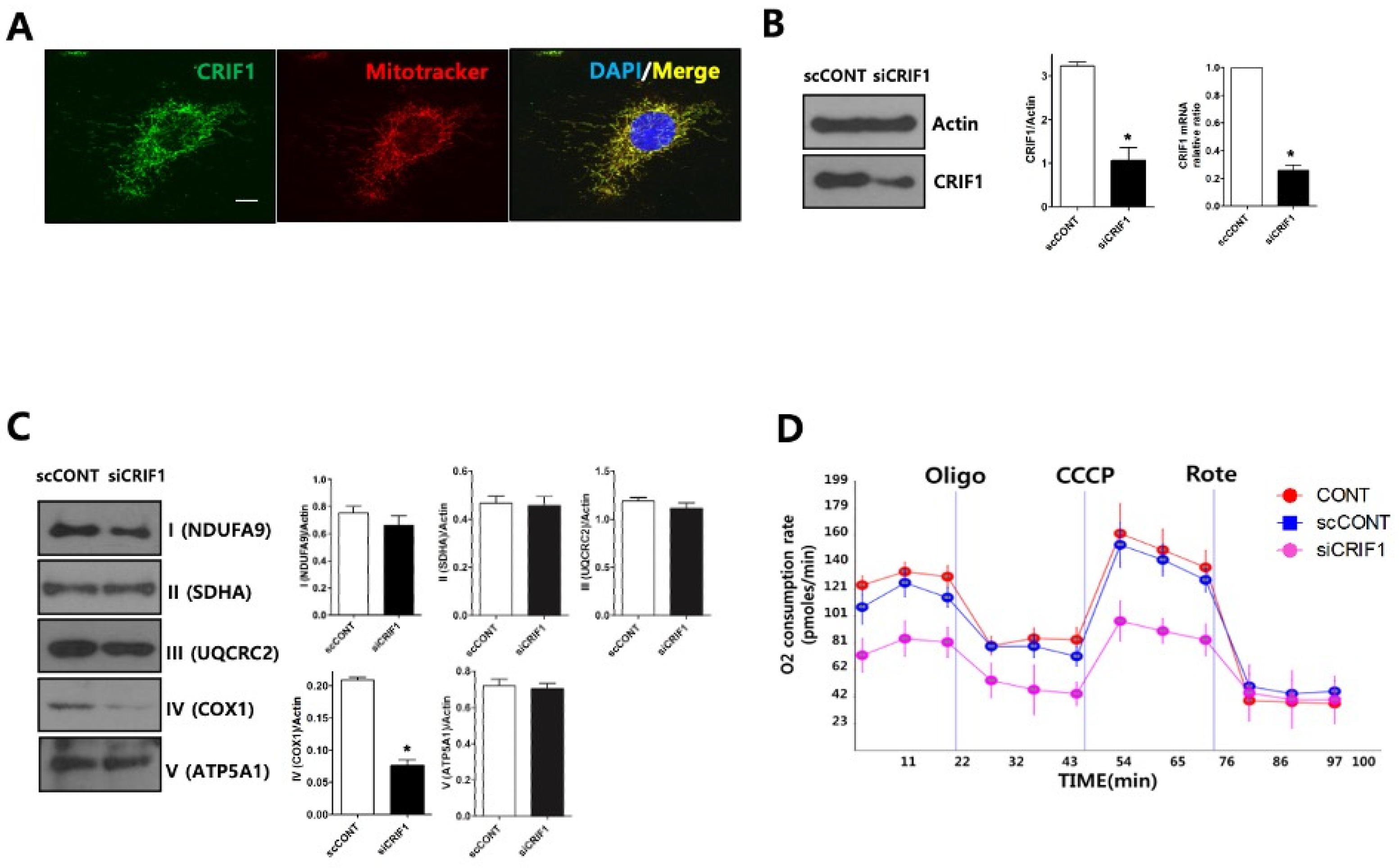
2.5. CRIF1 Deletion of Podocyte Leads to Mitochondrial Dysfunction
2.6. CRIF1 Deletion of Podocyte Leads to Loss and Aggregation of F-Actin
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Blood and Urinary Measurements
4.3. Histological Evaluation
4.4. Transmission Electron Microscopy
4.5. Immunostaining and Confocal Microscopy
4.6. Podocyte Culture and siRNA Transfection
4.7. OCR
4.8. Quantitative RT-PCR
4.9. Immunoblotting
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| mtDNA | mitochondrial DNA |
| ATP | Adenosine triphosphate |
| OxPhos | oxidative phosphorylation |
| CRIF1 | CR6-interacting factor-1 |
| FSGS | focal segmental glomerular sclerosis |
| OCRs | oxygen consumption rates |
| CCCP | chain decoupler carbonyl cyanide m-chlorophenylhydrazone |
| ZO-1 | Zonula occludens-1 |
| SDHA | Succinate Dehydrogenase Complex Flavoprotein Subunit A |
| NDUFA9 | NADH:Ubiquinone Oxidoreductase Subunit A9 |
| UQCRC2 | Cytochrome b-c1 complex subunit 2 |
| COX1 | Cytochrome c oxidase I |
| ATP5A1 | ATP synthase F1 subunit alpha |
References
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef]
- Hagiwara, M.; Yamagata, K.; Capaldi, R.A.; Koyama, A. Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis. Kidney Int. 2006, 69, 1146–1152. [Google Scholar] [CrossRef]
- Doleris, L.M.; Hill, G.S.; Chedin, P.; Nochy, D.; Bellanne-Chantelot, C.; Hanslik, T.; Bedrossian, J.; Caillat-Zucman, S.; Cahen-Varsaux, J.; Bariety, J. Focal segmental glomerulosclerosis associated with mitochondrial cytopathy. Kidney Int. 2000, 58, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef]
- Yu, S.M.; Nissaisorakarn, P.; Husain, I.; Jim, B. Proteinuric Kidney Diseases: A Podocyte’s Slit Diaphragm and Cytoskeleton Approach. Front. Med. 2018, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Ishibe, S. Podocyte endocytosis in the regulation of the glomerular filtration barrier. Am. J. Physiol. Renal. Physiol. 2015, 309, F398–F405. [Google Scholar] [CrossRef]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Mibus, A.L.; Forbes, J.M. Oxidative stress and advanced glycation in diabetic nephropathy. Ann. N. Y. Acad. Sci. 2008, 1126, 190–193. [Google Scholar] [CrossRef]
- Hotta, O.; Inoue, C.N.; Miyabayashi, S.; Furuta, T.; Takeuchi, A.; Taguma, Y. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 2001, 59, 1236–1243. [Google Scholar] [CrossRef]
- Schell, C.; Huber, T.B. The Evolving Complexity of the Podocyte Cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 3166–3174. [Google Scholar] [CrossRef]
- Kim, S.J.; Kwon, M.C.; Ryu, M.J.; Chung, H.K.; Tadi, S.; Kim, Y.K.; Kim, J.M.; Lee, S.H.; Park, J.H.; Kweon, G.R.; et al. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab. 2012, 16, 274–283. [Google Scholar] [CrossRef]
- Ryu, M.J.; Kim, S.J.; Kim, Y.K.; Choi, M.J.; Tadi, S.; Lee, M.H.; Lee, S.E.; Chung, H.K.; Jung, S.B.; Kim, H.J.; et al. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet. 2013, 9, e1003356. [Google Scholar] [CrossRef]
- Kwon, M.C.; Koo, B.K.; Moon, J.S.; Kim, Y.Y.; Park, K.C.; Kim, N.S.; Kwon, M.Y.; Kong, M.P.; Yoon, K.J.; Im, S.K.; et al. Crif1 is a novel transcriptional coactivator of STAT3. EMBO J. 2008, 27, 642–653. [Google Scholar] [CrossRef]
- Moeller, M.J.; Sanden, S.K.; Soofi, A.; Wiggins, R.C.; Holzman, L.B. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 2003, 35, 39–42. [Google Scholar] [CrossRef]
- Zhou, Y.; Bian, X.; Fang, L.; He, W.; Dai, C.; Yang, J. Aristolochic acid causes albuminuria by promoting mitochondrial DNA damage and dysfunction in podocyte. PLoS ONE 2013, 8, e83408. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.B.; Pippin, J.W.; Krofft, R.D.; Shankland, S.J. Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int. 2006, 70, 1962–1973. [Google Scholar] [CrossRef]
- Yamagata, K.; Muro, K.; Usui, J.; Hagiwara, M.; Kai, H.; Arakawa, Y.; Shimizu, Y.; Tomida, C.; Hirayama, K.; Kobayashi, M.; et al. Mitochondrial DNA mutations in focal segmental glomerulosclerosis lesions. J. Am. Soc. Nephrol. 2002, 13, 1816–1823. [Google Scholar] [CrossRef]
- Zhu, C.; Huang, S.; Yuan, Y.; Ding, G.; Chen, R.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: A therapeutic target of PPARgamma. Am. J. Pathol. 2011, 178, 2020–2031. [Google Scholar] [CrossRef]
- Inoue, K.; Nakada, K.; Ogura, A.; Isobe, K.; Goto, Y.; Nonaka, I.; Hayashi, J.I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000, 26, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Gucer, S.; Talim, B.; Asan, E.; Korkusuz, P.; Ozen, S.; Unal, S.; Kalkanoglu, S.H.; Kale, G.; Caglar, M. Focal segmental glomerulosclerosis associated with mitochondrial cytopathy: Report of two cases with special emphasis on podocytes. Pediatr. Dev. Pathol. 2005, 8, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Dhoopun, A.R.; Yuan, Y.; Huang, S.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am. J. Physiol. Renal. Physiol. 2013, 305, F520–F531. [Google Scholar] [CrossRef]
- Xie, L.; Zhu, X.; Hu, Y.; Li, T.; Gao, Y.; Shi, Y.; Tang, S. Mitochondrial DNA oxidative damage triggering mitochondrial dysfunction and apoptosis in high glucose-induced HRECs. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4203–4209. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Izquierdo, A.; Lopez-Luna, P.; Ortega, A.; Romero, M.; Guitierrez-Tarres, M.A.; Arribas, I.; Alvarez, M.J.; Esbrit, P.; Bosch, R.J. The parathyroid hormone-related protein system and diabetic nephropathy outcome in streptozotocin-induced diabetes. Kidney Int. 2006, 69, 2171–2177. [Google Scholar] [CrossRef]
- Mundel, P.; Reiser, J.; Zuniga Mejia Borja, A.; Pavenstadt, H.; Davidson, G.R.; Kriz, W.; Zeller, R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 1997, 236, 248–258. [Google Scholar] [CrossRef]
- Greiber, S.; Muller, B.; Daemisch, P.; Pavenstadt, H. Reactive oxygen species alter gene expression in podocytes: Induction of granulocyte macrophage-colony-stimulating factor. J. Am. Soc. Nephrol. 2002, 13, 86–95. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, K.R.; Jeong, J.Y.; Shin, J.A.; Chang, Y.-K.; Suh, K.-S.; Lee, K.W.; Choi, D.E. Mitochondrial Dysfunction in Podocytes Caused by CRIF1 Deficiency Leads to Progressive Albuminuria and Glomerular Sclerosis in Mice. Int. J. Mol. Sci. 2021, 22, 4827. https://doi.org/10.3390/ijms22094827
Na KR, Jeong JY, Shin JA, Chang Y-K, Suh K-S, Lee KW, Choi DE. Mitochondrial Dysfunction in Podocytes Caused by CRIF1 Deficiency Leads to Progressive Albuminuria and Glomerular Sclerosis in Mice. International Journal of Molecular Sciences. 2021; 22(9):4827. https://doi.org/10.3390/ijms22094827
Chicago/Turabian StyleNa, Ki Ryang, Jin Young Jeong, Jin Ah Shin, Yoon-Kyung Chang, Kwang-Sun Suh, Kang Wook Lee, and Dae Eun Choi. 2021. "Mitochondrial Dysfunction in Podocytes Caused by CRIF1 Deficiency Leads to Progressive Albuminuria and Glomerular Sclerosis in Mice" International Journal of Molecular Sciences 22, no. 9: 4827. https://doi.org/10.3390/ijms22094827
APA StyleNa, K. R., Jeong, J. Y., Shin, J. A., Chang, Y.-K., Suh, K.-S., Lee, K. W., & Choi, D. E. (2021). Mitochondrial Dysfunction in Podocytes Caused by CRIF1 Deficiency Leads to Progressive Albuminuria and Glomerular Sclerosis in Mice. International Journal of Molecular Sciences, 22(9), 4827. https://doi.org/10.3390/ijms22094827