
Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Results
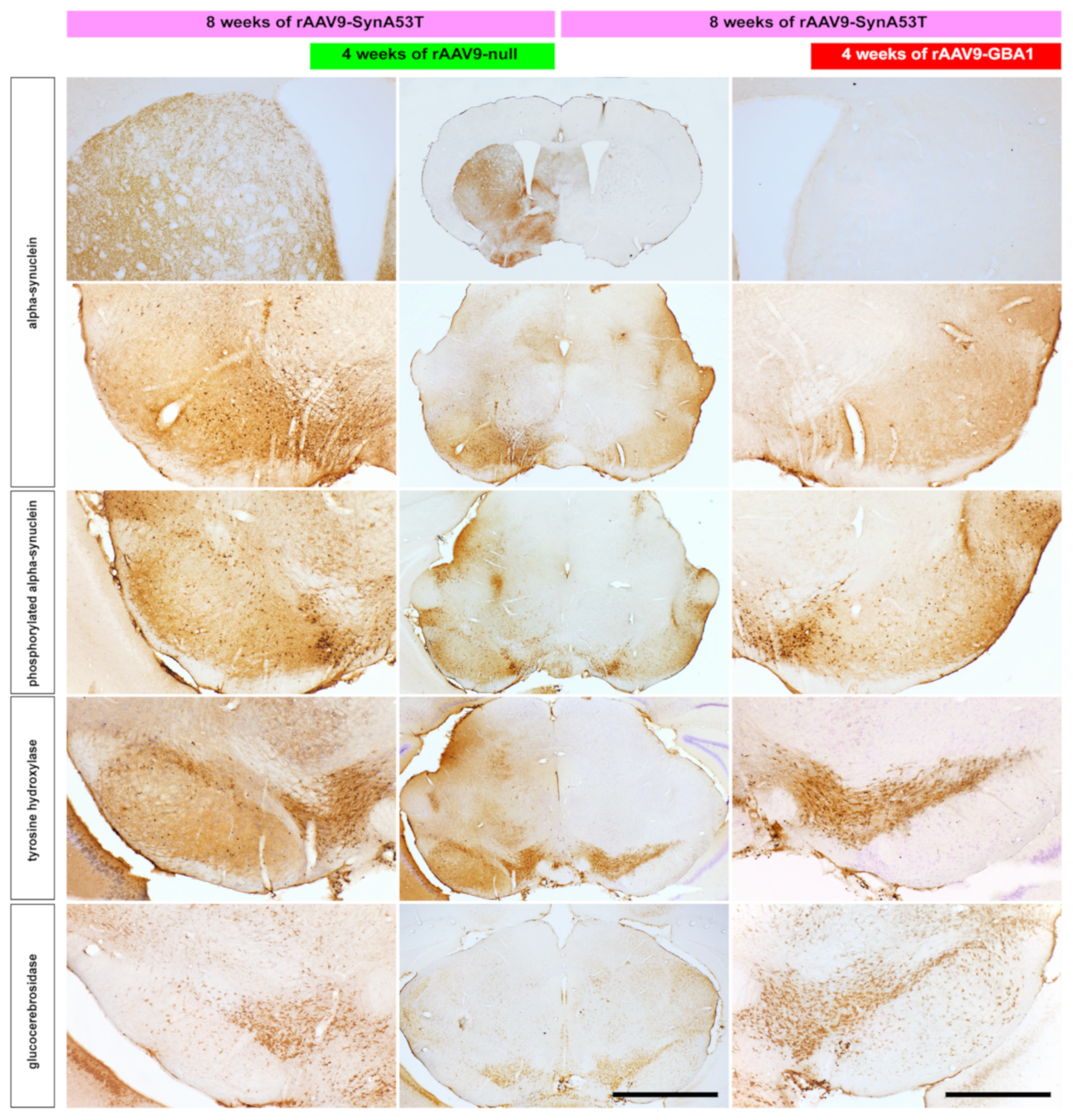
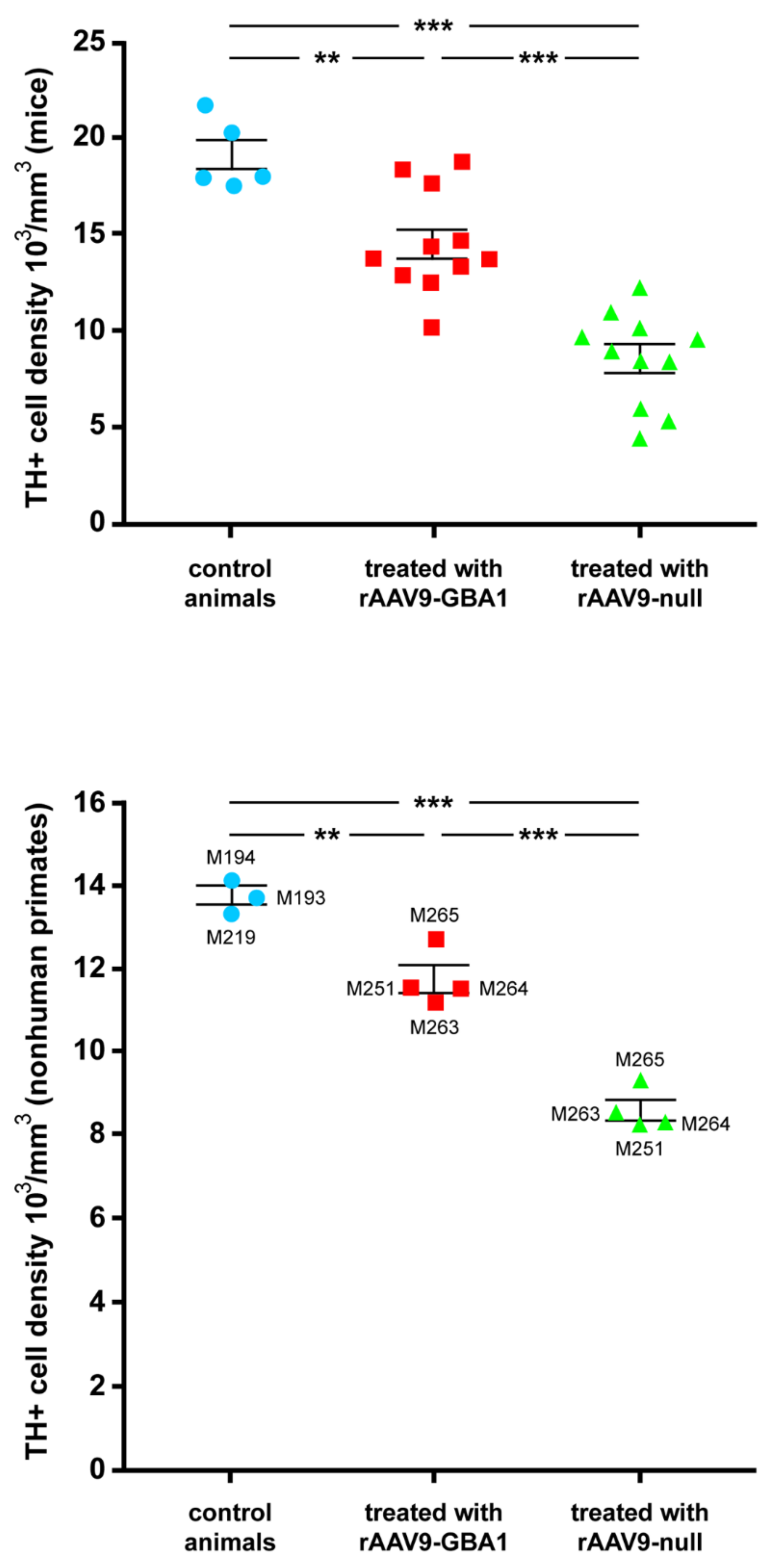
2.1. Experiments Conducted in Mice
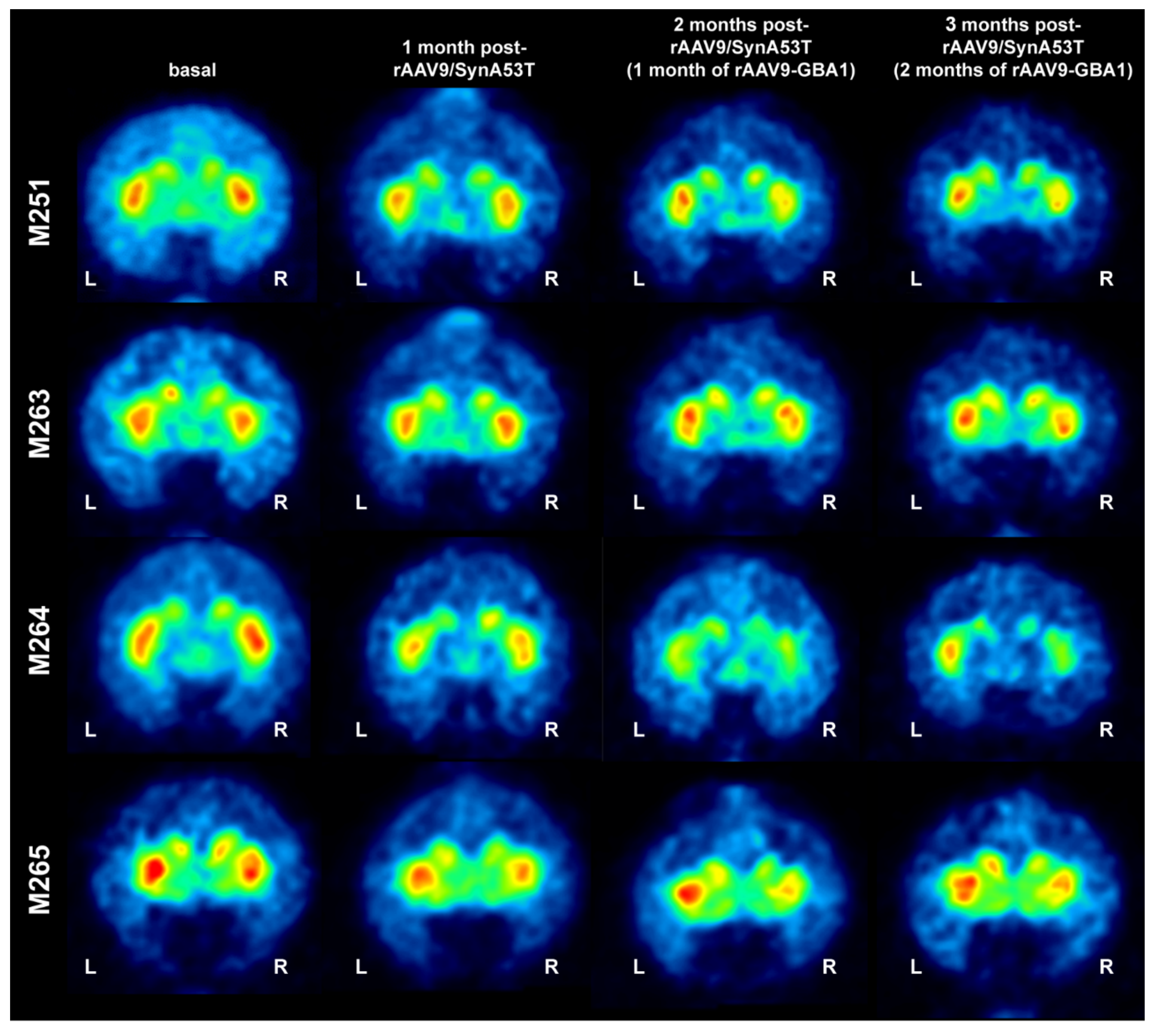
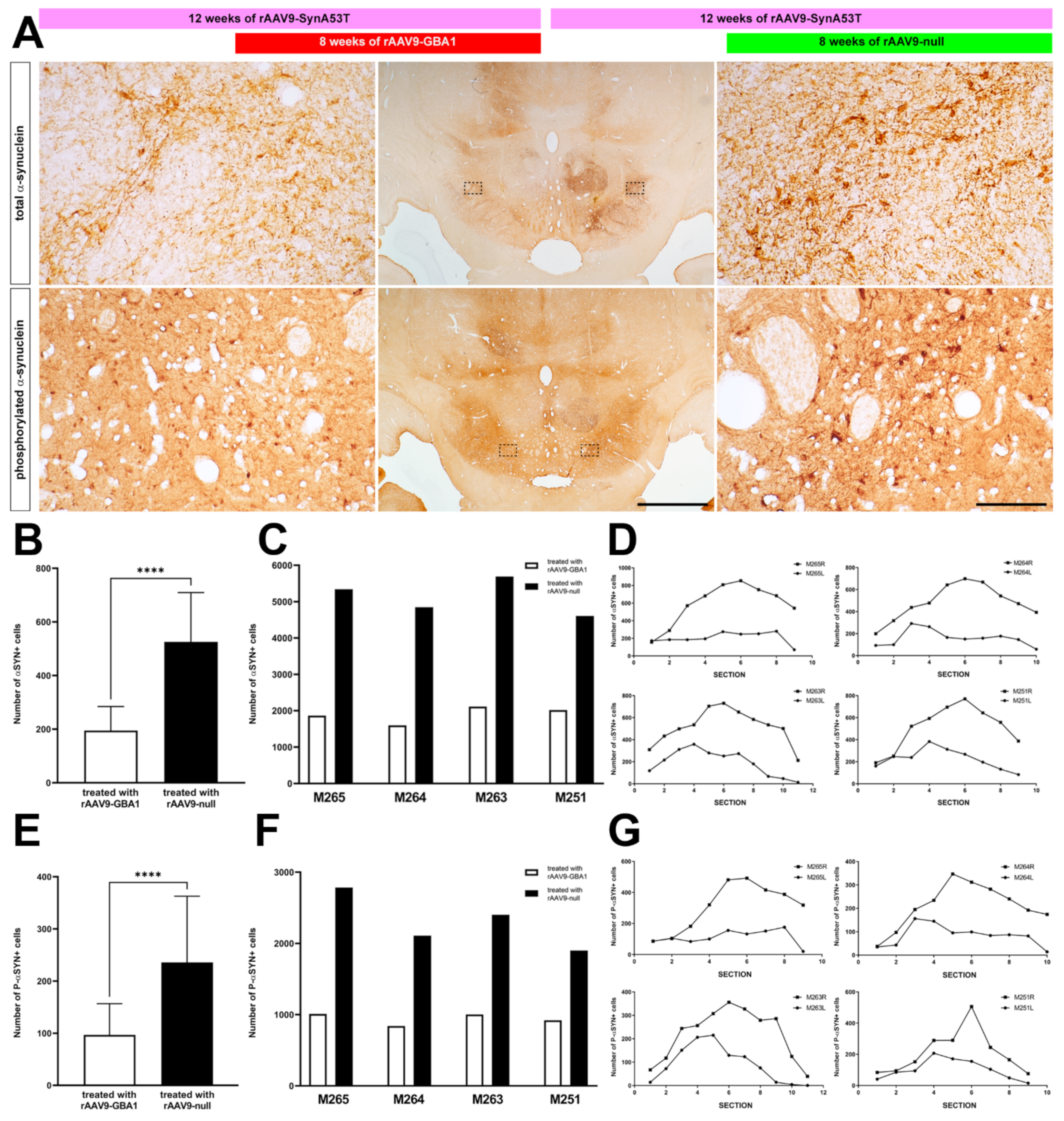
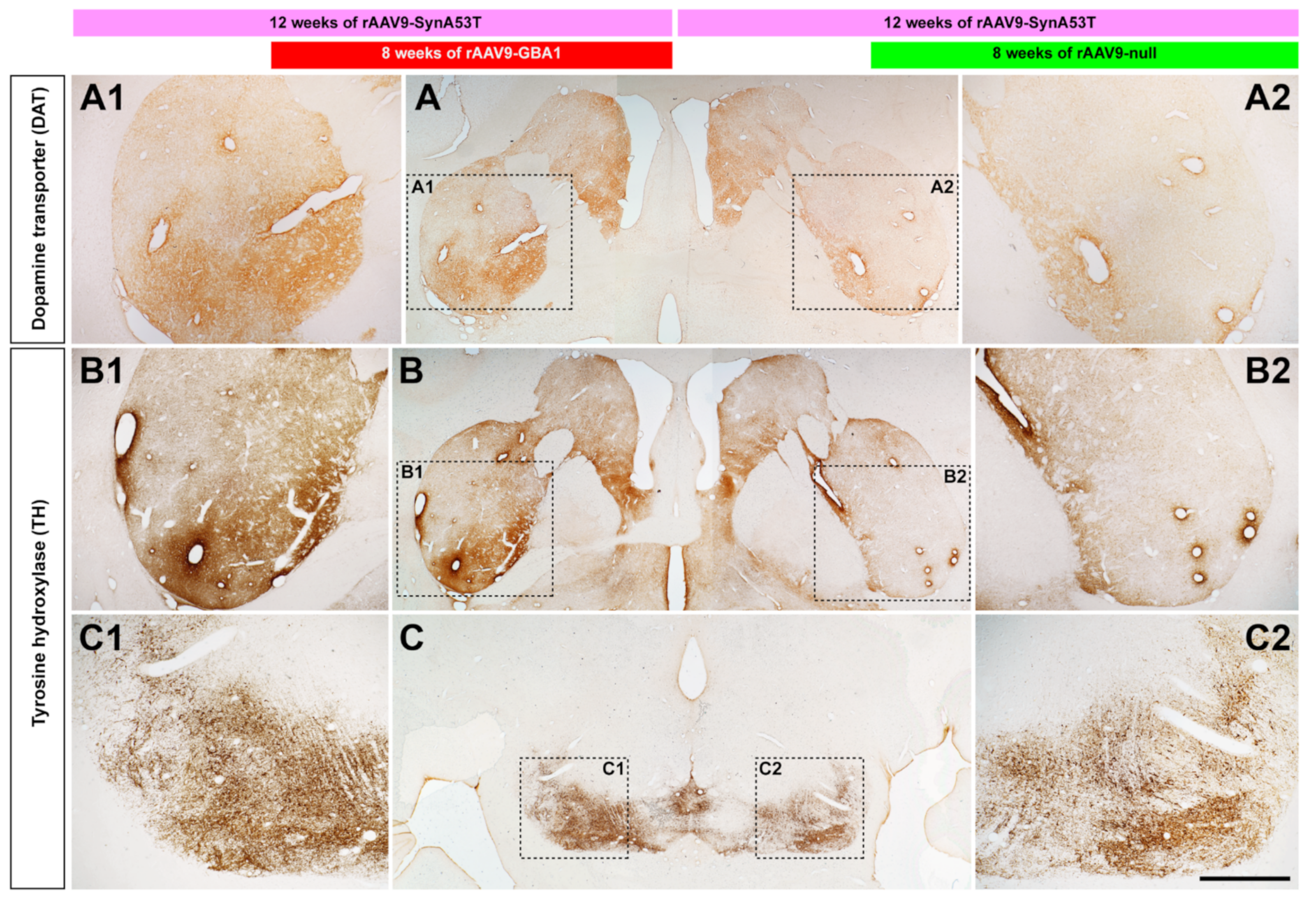
2.2. Experiments Conducted in NHPs
2.2.1. MicroPET Neuroimage Studies
2.2.2. GCase Expression Levels
2.2.3. rAAV-Mediated GCase Enhancement Is Linked to α-Syn Clearance
2.2.4. Neuroprotection of the Nigrostriatal System
3. Discussion
3.1. Considerations on rAAV-Mediated Animal Models of PD-Like Synucleinopathies
3.2. Targeting the GCase-Synuclein Pathway for the Treatment of Synucleinopathy
3.3. Available Choices for Enhancing GCase Enzymatic Activity
4. Materials and Methods
4.1. Viral Vector Production
4.2. Rodent Experiments
4.2.1. Intraparenchymal Deliveries of rAAVs
4.2.2. Necropsy and Tissue Processing
4.3. Nonhuman Primate Experiments
4.3.1. Intraparenchymal Deliveries of rAAVs
4.3.2. MicroPET Scans
4.3.3. Necropsy and Tissue Processing
4.3.4. Unbiased Stereological Estimation of TH+ Neurons in the SNpc
4.3.5. Unbiased Estimation of α-Syn+ Neurons in the SNpc
4.3.6. Estimation of GCase Expression Levels by Optical Density
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase Mutations in Clinical and Pathologically Proven Parkinson’s Disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase Deficiency in Substantia Nigra of Parkinson Disease Brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in IPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Li, Y.; Sekine, T.; Funayama, M.; Li, L.; Yoshino, H.; Nishioka, K.; Tomiyama, H.; Hattori, N. Clinicogenetic Study of GBA Mutations in Patients with Familial Parkinson’s Disease. Neurobiol. Aging 2014, 35, 935.e3–8. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Cilia, R.; Cerri, S.; Pezzoli, G.; Schapira, A.H.V.; Mullin, S.; Lanciego, J.L. Glucocerebrosidase Mutations and Synucleinopathies: Toward a Model of Precision Medicine. Mov. Disord. 2019, 34, 9–21. [Google Scholar] [CrossRef]
- Sidransky, E. Gaucher Disease and Parkinsonism. Mol. Genet. Metab. 2005, 84, 302–304. [Google Scholar] [CrossRef]
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s Syndrome in Type I Gaucher Disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and Dementia in GBA-Associated Parkinson’s Disease: The Mutation Matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Scriver, C.; Beaudet, A.; Sly, W.; Valle, D.; Childs, B.; Kinzler, K.; Vogelstein, B. The Metabolic and Molecular Bases of Inherited Disease, 4 Volume Set; Mcgraw-Hill: New York, NY, USA, 2001; ISBN 978-0-07-913035-8. [Google Scholar]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal Fluid Lysosomal Enzymes and Alpha-Synuclein in Parkinson’s Disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase Activity in Parkinson’s Disease with and without GBA Mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive Decline of Glucocerebrosidase in Aging and Parkinson’s Disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase Gene Therapy Prevents α-Synucleinopathy of Midbrain Dopamine Neurons. Neurobiol. Dis. 2015, 82, 495–503. [Google Scholar] [CrossRef]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS Expression of Glucocerebrosidase Corrects Alpha-Synuclein Pathology and Memory in a Mouse Model of Gaucher-Related Synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS Glucocerebrosidase Activity as a Therapeutic Strategy for Parkinsonism and Other Gaucher-Related Synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef]
- Dopeso-Reyes, I.G.; Sucunza, D.; Rico, A.J.; Pignataro, D.; Marín-Ramos, D.; Roda, E.; Rodríguez-Pérez, A.I.; Labandeira-García, J.L.; Lanciego, J.L. Glucocerebrosidase Expression Patterns in the Non-Human Primate Brain. Brain Struct. Funct. 2018, 223, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Lanciego, J.L.; Luquin, N.; Obeso, J.A. Functional Neuroanatomy of the Basal Ganglia. Cold Spring Harb. Perspect. Med. 2012, 2, a009621. [Google Scholar] [CrossRef]
- Pignataro, D.; Sucunza, D.; Rico, A.J.; Dopeso-Reyes, I.G.; Roda, E.; Rodríguez-Perez, A.I.; Labandeira-Garcia, J.L.; Broccoli, V.; Kato, S.; Kobayashi, K.; et al. Gene Therapy Approaches in the Non-Human Primate Model of Parkinson’s Disease. J. Neural. Transm. 2018, 125, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Marmion, D.J.; Kordower, J.H. α-Synuclein Nonhuman Primate Models of Parkinson’s Disease. J. Neural. Transm. 2018, 125, 385–400. [Google Scholar] [CrossRef]
- Kirik, D.; Rosenblad, C.; Burger, C.; Lundberg, C.; Johansen, T.E.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Parkinson-like Neurodegeneration Induced by Targeted Overexpression of Alpha-Synuclein in the Nigrostriatal System. J. Neurosci. 2002, 22, 2780–2791. [Google Scholar] [CrossRef] [PubMed]
- Koprich, J.B.; Johnston, T.H.; Reyes, M.G.; Sun, X.; Brotchie, J.M. Expression of Human A53T Alpha-Synuclein in the Rat Substantia Nigra Using a Novel AAV1/2 Vector Produces a Rapidly Evolving Pathology with Protein Aggregation, Dystrophic Neurite Architecture and Nigrostriatal Degeneration with Potential to Model the Pathology of Parkinson’s Disease. Mol. Neurodegener. 2010, 5, 43. [Google Scholar] [CrossRef]
- Koprich, J.B.; Johnston, T.H.; Huot, P.; Reyes, M.G.; Espinosa, M.; Brotchie, J.M. Progressive Neurodegeneration or Endogenous Compensation in an Animal Model of Parkinson’s Disease Produced by Decreasing Doses of Alpha-Synuclein. PLoS ONE 2011, 6, e17698. [Google Scholar] [CrossRef]
- Kirik, D.; Annett, L.E.; Burger, C.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Nigrostriatal Alpha-Synucleinopathy Induced by Viral Vector-Mediated Overexpression of Human Alpha-Synuclein: A New Primate Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2884–2889. [Google Scholar] [CrossRef]
- Eslamboli, A.; Romero-Ramos, M.; Burger, C.; Bjorklund, T.; Muzyczka, N.; Mandel, R.J.; Baker, H.; Ridley, R.M.; Kirik, D. Long-Term Consequences of Human Alpha-Synuclein Overexpression in the Primate Ventral Midbrain. Brain 2007, 130, 799–815. [Google Scholar] [CrossRef]
- Koprich, J.B.; Johnston, T.H.; Reyes, G.; Omana, V.; Brotchie, J.M. Towards a Non-Human Primate Model of Alpha-Synucleinopathy for Development of Therapeutics for Parkinson’s Disease: Optimization of AAV1/2 Delivery Parameters to Drive Sustained Expression of Alpha Synuclein and Dopaminergic Degeneration in Macaque. PLoS ONE 2016, 11, e0167235. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, D.; Sucunza, D.; Vanrell, L.; Lopez-Franco, E.; Dopeso-Reyes, I.G.; Vales, A.; Hommel, M.; Rico, A.J.; Lanciego, J.L.; Gonzalez-Aseguinolaza, G. Adeno-Associated Viral Vectors Serotype 8 for Cell-Specific Delivery of Therapeutic Genes in the Central Nervous System. Front. Neuroanat. 2017, 11, 2. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Schapira, A.H.V. The Relationship between Glucocerebrosidase Mutations and Parkinson Disease. J. Neurochem. 2016, 139 (Suppl. 1), 77–90. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.H.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase Mutations Influence the Natural History of Parkinson’s Disease in a Community-Based Incident Cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.-K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-Associated Parkinson’s Disease: Reduced Survival and More Rapid Progression in a Prospective Longitudinal Study. Mov. Disord. 2015, 30, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Creese, B.; Bell, E.; Johar, I.; Francis, P.; Ballard, C.; Aarsland, D. Glucocerebrosidase Mutations and Neuropsychiatric Phenotypes in Parkinson’s Disease and Lewy Body Dementias: Review and Meta-Analyses. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Goker-Alpan, O.; Masdeu, J.C.; Kohn, P.D.; Ianni, A.; Lopez, G.; Groden, C.; Chapman, M.C.; Cropp, B.; Eisenberg, D.P.; Maniwang, E.D.; et al. The Neurobiology of Glucocerebrosidase-Associated Parkinsonism: A Positron Emission Tomography Study of Dopamine Synthesis and Regional Cerebral Blood Flow. Brain 2012, 135, 2440–2448. [Google Scholar] [CrossRef]
- Tsuang, D.; Leverenz, J.B.; Lopez, O.L.; Hamilton, R.L.; Bennett, D.A.; Schneider, J.A.; Buchman, A.S.; Larson, E.B.; Crane, P.K.; Kaye, J.A.; et al. GBA Mutations Increase Risk for Lewy Body Disease with and without Alzheimer Disease Pathology. Neurology 2012, 79, 1944–1950. [Google Scholar] [CrossRef]
- Beavan, M.; McNeill, A.; Proukakis, C.; Hughes, D.A.; Mehta, A.; Schapira, A.H.V. Evolution of Prodromal Clinical Markers of Parkinson Disease in a GBA Mutation-Positive Cohort. JAMA Neurol. 2015, 72, 201–208. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Mirelman, A.; Postuma, R.B.; Arnulf, I.; Bar-Shira, A.; Dauvilliers, Y.; Desautels, A.; Gagnon, J.-F.; Leblond, C.S.; Frauscher, B.; et al. GBA Mutations Are Associated with Rapid Eye Movement Sleep Behavior Disorder. Ann. Clin. Transl. Neurol. 2015, 2, 941–945. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D. Advances in Markers of Prodromal Parkinson Disease. Nat. Rev. Neurol. 2016, 12, 622–634. [Google Scholar] [CrossRef]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Factor, S.A.; Wood-Siverio, C.; et al. GBA Variants Are Associated with a Distinct Pattern of Cognitive Deficits in Parkinson’s Disease. Mov. Disord. 2016, 31, 95–102. [Google Scholar] [CrossRef]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016, 73, 1217–1224. [Google Scholar] [CrossRef]
- Alfonso, P.; Rodríguez-Rey, J.C.; Gañán, A.; Pérez-Calvo, J.I.; Giralt, M.; Giraldo, P.; Pocoví, M. Expression and Functional Characterization of Mutated Glucocerebrosidase Alleles Causing Gaucher Disease in Spanish Patients. Blood Cells Mol. Dis. 2004, 32, 218–225. [Google Scholar] [CrossRef]
- Beutler, E.; Gelbart, T.; Scott, C.R. Hematologically Important Mutations: Gaucher Disease. Blood Cells Mol. Dis. 2005, 35, 355–364. [Google Scholar] [CrossRef]
- Malini, E.; Grossi, S.; Deganuto, M.; Rosano, C.; Parini, R.; Dominisini, S.; Cariati, R.; Zampieri, S.; Bembi, B.; Filocamo, M.; et al. Functional Analysis of 11 Novel GBA Alleles. Eur. J. Hum. Genet. 2014, 22, 511–516. [Google Scholar] [CrossRef]
- Grace, M.E.; Ashton-Prolla, P.; Pastores, G.M.; Soni, A.; Desnick, R.J. Non-Pseudogene-Derived Complex Acid Beta-Glucosidase Mutations Causing Mild Type 1 and Severe Type 2 Gaucher Disease. J. Clin. Investig. 1999, 103, 817–823. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Salvioli, R.; Tatti, M.; Scarpa, S.; Moavero, S.M.; Ciaffoni, F.; Felicetti, F.; Kaneski, C.R.; Brady, R.O.; Vaccaro, A.M. The N370S (Asn370-->Ser) Mutation Affects the Capacity of Glucosylceramidase to Interact with Anionic Phospholipid-Containing Membranes and Saposin C. Biochem. J. 2005, 390, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Liou, B.; Kazimierczuk, A.; Zhang, M.; Scott, C.R.; Hegde, R.S.; Grabowski, G.A. Analyses of Variant Acid Beta-Glucosidases: Effects of Gaucher Disease Mutations. J. Biol. Chem. 2006, 281, 4242–4253. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; et al. Effectiveness of Enzyme Replacement Therapy in 1028 Patients with Type 1 Gaucher Disease after 2 to 5 Years of Treatment: A Report from the Gaucher Registry. Am. J. Med. 2002, 113, 112–119. [Google Scholar] [CrossRef]
- Connock, M.; Frew, E.; Evans, B.-W.; Bryan, S.; Cummins, C.; Fry-Smith, A.; Li Wan Po, A.; Sandercock, J. The Clinical Effectiveness and Cost-Effectiveness of Newer Drugs for Children with Epilepsy. A Systematic Review. Health Technol. Assess. 2006, 10, iii. [Google Scholar] [CrossRef]
- Brady, R.O.; Yang, C.; Zhuang, Z. An Innovative Approach to the Treatment of Gaucher Disease and Possibly Other Metabolic Disorders of the Brain. J. Inherit. Metab. Dis. 2013, 36, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a Pharmacological Chaperone for Mutant Glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef]
- Luan, Z.; Li, L.; Higaki, K.; Nanba, E.; Suzuki, Y.; Ohno, K. The Chaperone Activity and Toxicity of Ambroxol on Gaucher Cells and Normal Mice. Brain Dev. 2013, 35, 317–322. [Google Scholar] [CrossRef]
- Jung, O.; Patnaik, S.; Marugan, J.; Sidransky, E.; Westbroek, W. Progress and Potential of Non-Inhibitory Small Molecule Chaperones for the Treatment of Gaucher Disease and Its Implications for Parkinson Disease. Expert Rev. Proteomics 2016, 13, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Ko, W.K.D.; Li, Q.; Bezard, E.; Schapira, A.H.V. Oral Ambroxol Increases Brain Glucocerebrosidase Activity in a Nonhuman Primate. Synapse 2017, 71. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The Pharmacological Chaperone Isofagomine Increases the Activity of the Gaucher Disease L444P Mutant Form of Beta-Glucosidase. FEBS J. 2010, 277, 1618–1638. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide Synthase Inhibition Alleviates Aberrations in Synucleinopathy Models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Franklin, K.B.J.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Lanciego, J.L.; Vázquez, A. The Basal Ganglia and Thalamus of the Long-Tailed Macaque in Stereotaxic Coordinates. A Template Atlas Based on Coronal, Sagittal and Horizontal Brain Sections. Brain Struct. Funct. 2012, 217, 613–666. [Google Scholar] [CrossRef] [PubMed]
- Quincoces, G.; Collantes, M.; Catalán, R.; Ecay, M.; Prieto, E.; Martino, E.; Blesa, F.J.; Alvarez-Erviti, L.; Areses, P.; Arbizu, J.; et al. Quick and simple synthesis of (11)C-(+)-alpha-dihydrotetrabenazine to be used as a PET radioligand of vesicular monoamine transporters. Rev. Esp. Med. Nucl. 2008, 27, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Juri, C.; Collantes, M.; Peñuelas, I.; Prieto, E.; Iglesias, E.; Martí-Climent, J.; Arbizu, J.; Zubieta, J.L.; Rodríguez-Oroz, M.C.; et al. Progression of Dopaminergic Depletion in a Model of MPTP-Induced Parkinsonism in Non-Human Primates. An (18)F-DOPA and (11)C-DTBZ PET Study. Neurobiol. Dis. 2010, 38, 456–463. [Google Scholar] [CrossRef]
- Collantes, M.; Prieto, E.; Peñuelas, I.; Blesa, J.; Juri, C.; Martí-Climent, J.M.; Quincoces, G.; Arbizu, J.; Riverol, M.; Zubieta, J.L.; et al. New MRI, 18F-DOPA and 11C-(+)-Alpha-Dihydrotetrabenazine Templates for Macaca Fascicularis Neuroimaging: Advantages to Improve PET Quantification. Neuroimage 2009, 47, 533–539. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward-ITR | 5′-GGAACCCCTAGTGATGGAGTT-3′ |
|---|---|
| Reverse-ITR | 5′-CGGCCTCAGTGAGCGA-3′ |
| Forward-ASYN | 5′-AGAAGACAGTGGAGGGAGCA-3′ |
| Reverse-ASYN | 5′TGTCAGGATCCACAGGCATA-3′ |
| Forward-GBA | 5′-CCTGGATGCTTATGCTGAGC-3′ |
| Reverse-GBA | 5′-CCAGATACCAGTGCACAGCAATC-3′ |
| Antibody | Dilution | Incubation Time | Supplier | Reference |
|---|---|---|---|---|
| Goat anti-tyrosine hydroxylase | 1:500 | overnight | MyBioSource | MBS421729 |
| Rabbit anti-tyrosine hydroxylase | 1:100 | overnight | Millipore | AB152 |
| Mouse anti-glucocerebrosidase | 1:500 | overnight | Abcam | Ab55080 |
| Rabbit anti-glucocerebrosidase | 1:500 | overnight | Thermo Fisher | PAS-21347 |
| Goat anti-α-synuclein | 1:300 | overnight | Abcam | Ab2080 |
| Mouse anti-α-synuclein | 1:500 | overnight | Abcam | Ab27766 |
| Mouse anti-phospho-α-synuclein | 1:1000 | overnight | Wako | 015-25291 |
| Rat anti-dopamine transporter | 1:500 | overnight | Millipore | MAB369 |
| Donkey anti-goat IgG biotinylated | 1:600 | 120 min | Jackson | 705-065-147 |
| Donkey anti-rabbit IgG biotinylated | 1:600 | 120 min | Jackson | 711-065-152 |
| Donkey anti-mouse IgG biotinylated | 1:600 | 120 min | Jackson | 715-066-150 |
| Goat anti-rat IgG biotinylated | 1:600 | 120 min | Jackson | 112-066-003 |
| Vectastain ABC HRP standard kit | 1:1000 | 60 min | Vector Labs | PK-4000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sucunza, D.; Rico, A.J.; Roda, E.; Collantes, M.; González-Aseguinolaza, G.; Rodríguez-Pérez, A.I.; Peñuelas, I.; Vázquez, A.; Labandeira-García, J.L.; Broccoli, V.; et al. Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques. Int. J. Mol. Sci. 2021, 22, 4825. https://doi.org/10.3390/ijms22094825
Sucunza D, Rico AJ, Roda E, Collantes M, González-Aseguinolaza G, Rodríguez-Pérez AI, Peñuelas I, Vázquez A, Labandeira-García JL, Broccoli V, et al. Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques. International Journal of Molecular Sciences. 2021; 22(9):4825. https://doi.org/10.3390/ijms22094825
Chicago/Turabian StyleSucunza, Diego, Alberto J. Rico, Elvira Roda, María Collantes, Gloria González-Aseguinolaza, Ana I. Rodríguez-Pérez, Iván Peñuelas, Alfonso Vázquez, José L. Labandeira-García, Vania Broccoli, and et al. 2021. "Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques" International Journal of Molecular Sciences 22, no. 9: 4825. https://doi.org/10.3390/ijms22094825
APA StyleSucunza, D., Rico, A. J., Roda, E., Collantes, M., González-Aseguinolaza, G., Rodríguez-Pérez, A. I., Peñuelas, I., Vázquez, A., Labandeira-García, J. L., Broccoli, V., & Lanciego, J. L. (2021). Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques. International Journal of Molecular Sciences, 22(9), 4825. https://doi.org/10.3390/ijms22094825