
Genes Implicated in Familial Parkinson’s Disease Provide a Dual Picture of Nigral Dopaminergic Neurodegeneration with Mitochondria Taking Center Stage

 ,
,  , , and
, , and
Abstract
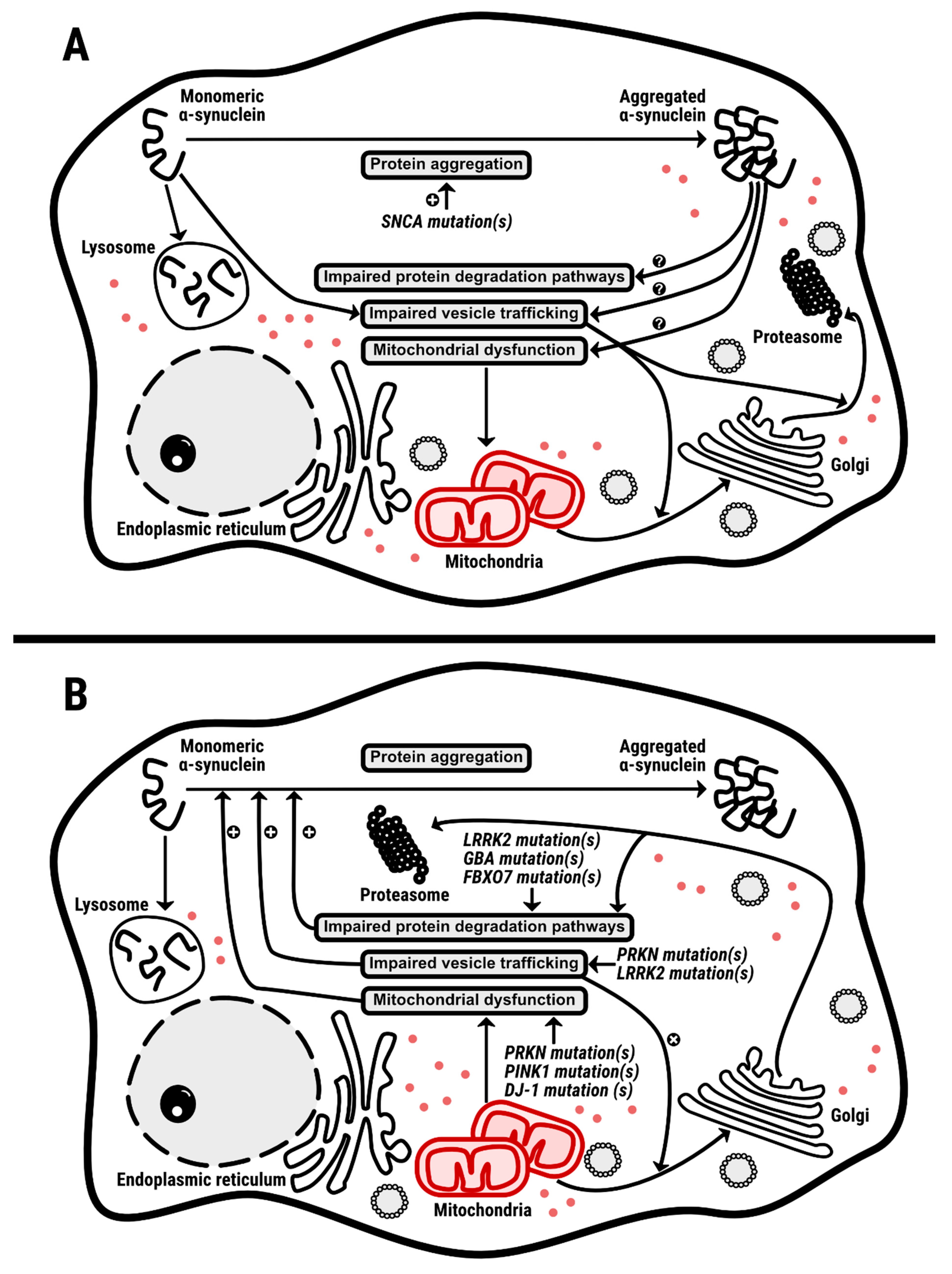
1. Introduction
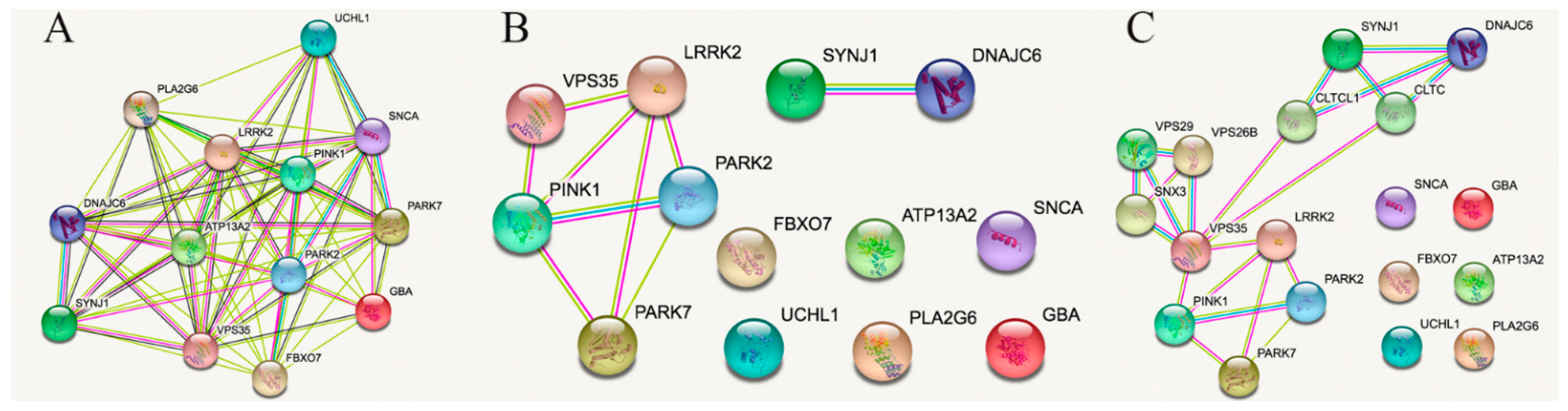
2. STRING Analysis Restricted to Connections Obtained from Experimental, Gene Fusion, Protein Analogy or Proximity Data
3. Lessons from Interactions among PARK2, PARK7, PINK1, LRRK2 and VPS35
4. Lessons from Interactions between and from SYNJ1 and DNAJC6
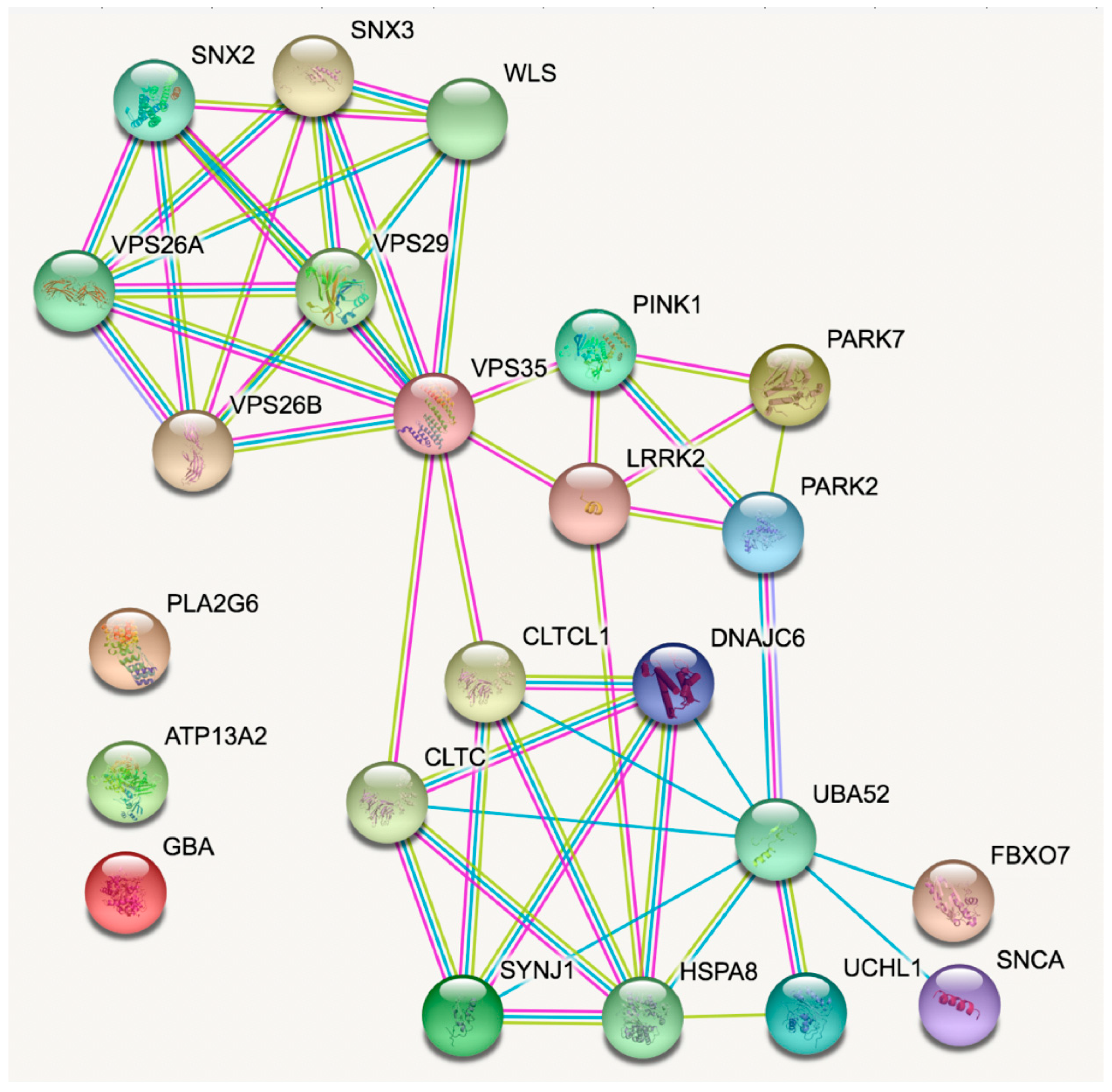
5. Enriching the Network
6. Lessons from Transgenic Animals

{kind=link}
{kind=link}
{kind=link}
| Animal Model (s) | In Vivo | In Vitro | Expression Level | Main Findings | Ref. |
|---|---|---|---|---|---|
| Mice harbouring Sncatm1Nbm mutation (n.s.n) | X | Snca KO mice | Modulates microglial activation phenotype Increased levels of reactive marker proteins | [49] | |
| Mice harbouring Sncatm1Nbm mutation (n.s.n) | X | Snca KO mice | Altered palmitate metabolism | [50] | |
| Mice harbouring Sncatm1Nbm mutation (n.s.n) | X | Snca KO mice | Mitochondrial lipid abnormality Electron transport chain impairment | [51] | |
| Mice harbouring Sncatm1Nbm mutation (n.s.n) | X | X | Snca KO mice | Synaptic vesicle depletion | [52] |
| C57BL/6N-Sncatm1Mjff/J | X | X | Snca KO mice | Non-altered mitochondrial bioenergetics | [53] |
| B6; 129 × 1-Sncatm1Rosl/J | X | Snca KO mice | α-syn restricts RNA viral infections in the brain | [54] | |
| B6; 129 × 1-Sncatm1Rosl/J | X | Snca KO mice | ROS and NOS-2 decreased in mature erythrocytes | [55] | |
| B6; 129 × 1-Sncatm1Rosl/J | X | Snca KO mice | No modification in pale body-like inclusion Altered proteasome function | [56] | |
| B6; 129 × 1-Sncatm1Rosl/J | X | Snca KO mice | Inhibition of intrasynaptic vesicle mobility Maintains recycling-pool homeostasis | [57] | |
| B6; 129 × 1-Sncatm1Rosl/J | X | Snca KO mice | Cognitive impairments | [58] | |
| B6; 129 × 1-Sncatm1Rosl/J | X | Snca KO mice | Vulnerability of peripheral catecholaminergic neurons to MPTP not regulated by α-synuclein | [59] | |
| B6; 129 × 1-Sncatm1Rosl/J | X | Snca KO mice | Resistant to mitochondrial toxins | [60] | |
| B6; 129 × 1-Sncatm1Rosl/J | X | X | Snca KO mice | Deficits in the nigrostriatal dopamine system | [61] |
| B6; 129 × 1-Sncatm1Rosl/J C57Bl/6JOlaHsd | X | X | Snca KO mice | Decreased impulsivity | [62] |
| B6; 129 × 1-Sncatm1Rosl/J C57Bl/6JOlaHsd | X | Snca KO mice | Neuromuscular pathology | [63] | |
| B6; 129 × 1-Sncatm1Rosl/J C57Bl/6JOlaHsd | X | X | Snca KO mice | Decreased reuptake of dopamine in the dorsal striatum | [64] |
| B6; 129 × 1-Sncatm1Rosl/J (Snca KO) B6-TgHSNCGtm1VLB (Sncg KO) | X | X | -α-syn (Snca) KO mice -ɣ-syn (Sncg) KO mice | Altered dopamine metabolism | [65] |
| B6; 129 × 1-Sncatm1Rosl/J (Snca KO) B6-TgHSNCGtm1VLB (Sncg KO) | X | X | -α-syn (Snca) KO mice -ɣ-syn (Sncg) KO mice -α-ɣ-syn double KO mice | Increased striatal dopamine release Hyperdopaminergic signs | [66] |
| Triple-synuclein-KO (TKO) | X | X | α-syn (Snca), ß-syn (Sncb) and ɣ-syn (Sncg) triple KO mice | Functional alterations to the nigrostriatal system | [67] |
| Triple-synuclein-KO (TKO) | X | X | α-syn (Snca), ß-syn (Sncb) and ɣ-syn (Sncg) triple KO mice | Altered synaptic vesicle endocytosis α -, ß -, or γ-synucleins are functionally redundant | [68] |
| Triple-synuclein-KO (TKO) | X | X | α-syn (Snca), ß-syn (Sncb) and ɣ-syn (Sncg) triple KO mice | Age-dependent neuronal dysfunction | [69] |
| Triple-synuclein-KO (TKO) See paper for further details on model(s) | X | X | α-syn (Snca), ß-syn (Sncb) and ɣ-syn (Sncg) triple KO mice. Mice overexpressing human SNCA mutants PARK1/hA30P or PARK4/hα-syn | Effects on presynaptic architecture | [70] |
| B6;129 × 1-Sncatm1Rosl/J B6.CgTg (SNCA) OVX37Rwm Sncatm1Rosl/J | X | X | -Snca KO mice -SNCA overexpressing mice | Blockade of TrkB neurotrophic effect | [71] |
| B6;DBA-Tg (Thy1-SNCA) 61Ema | X | SNCA overexpressing mice | Impairment of mitochondrial function Elevated ROS in brain mitochondria | [72] | |
| B6;DBA-Tg (Thy1-SNCA) 61Ema | X | SNCA overexpressing mice | Alterations in corticostriatal synaptic plasticity | [73] | |
| B6;DBA-Tg (Thy1-SNCA) 61Ema | X | X | SNCA overexpressing mice | Alterations in calcium buffering capacity | [74] |
| (Thy1)-(WT)a-syn | X | SNCA overexpressing mice | Enhanced axonal degeneration after peripheral nerve lesion | [75] | |
| B6.Cg-Tg (SNCA) OVX37Rwm Sncatm1Rosl/J | X | X | SNCA overexpressing mice | Deficits in dopaminergic transmission precede neuronal loss | [76] |
| B6.Cg-Tg (SNCA) OVX37Rwm Sncatm1Rosl/J | X | SNCA overexpressing mice | Impairment of macroautophagy in dopaminergic neurons | [77] | |
| B6; 129 × 1-Sncatm1Rosl/J neurons transfected with SNCA or TsixK-SNCA | X | Cultured neurons from Snca KO mice overexpressing SNCA or TsixK-SNCA | α-syn multimers attenuate synaptic vesicle recycling | [78] | |
| S129 mutations performed on mice harbouring Sncatm1Nbm mutation (n.s.n.) | X | X | S129D-SNCA (phosphomimetic) or S129A- SNCA (non-phosphorylatable) overexpressing Snca KO mice | No abnormalities detected (in vivo) | [79] |
| FVB;129S6-Sncatm1Nbm Tg (SNCA*A53T) 1Nbm Tg (SNCA*A53T) 2Nbm/J FVB;129S6-Sncatm1Nbm Tg (SNCA*A30P) 1Nbm Tg (SNCA*A30P) 2Nbm/J | X | X | A53T-SNCA or A30P-SNCA overexpressing Snca KO mice | Enteric nervous system abnormalities | [80] |
| M83KO mice resulting from crossing M83 line with B6; 129X1-Sncatm1Rosl/J line | X | A30P-SNCA overexpressing Snca KO mice | Dopaminergic neurodegeneration | [81] | |
| B6.Cg-Sncatm1Rosl Tg (SNCA*A30P) 192Rwm/J | X | X | A30P-SNCA overexpressing Snca KO mice | Region-specific deficits in dopamine signaling | [82] |
| FVB;129-Tg (Prnp-SNCA*A53T) AAub/J | X | X | A53T-SNCA overexpressing mice | Dysfunctional neurotransmission Impaired synaptic plasticity | [83] |
| FVB;129-Tg (Prnp-SNCA*A53T) AAub/J | X | X | A53T-SNCA overexpressing mice | Neuronal dysfunction in the absence of aggregate formation Behavioral alterations | [84] |
| NTac:SD-Tg (SNCA*A53T) 268Cjli | X | X | A53T-SNCA overexpressing mice | Dynamic changes in striatal mGluR1 but not mGluR5 | [85] |
| B6;C3-Tg (Prnp-SNCA*A53T) 83Vle/J (also known as:A53T α-synuclein transgenic line M83) | X | X | Brain inoculation with brain homogenates from older Tg mice or with human α-syn fibrils in Tg A53T-SNCA overexpressing mice | Inoculation initiates a rapidly progressive neurodegenerative α-synucleinopathy | [86] |
| NTac:SD-Tg (SNCA*E46K) 70CJLi | X | X | E46K-SNCA overexpressing rats | Enhanced vulnerability to mitochondrial impairment | [87] |
| Double transgenic Uchl1tm1Dgen:Thy1-maSN | X | X | Uch-L1 KO + Snca overexpressing mice | Excess α-syn worsens disease in mice lacking Uch-L1 | [88] |
| B6; 129×1-Sncatm1Rosl/J C57B/6jxSJL F3, Tg5093 | X | -Snca KO mice -A53T-SNCA overexpressing mice | α-syn expression levels do not significantly affect proteasome function | [89] | |
| See paper for details of the animal model(s) | X | -A53T-SNCA transfected in WT neurons -Snca KO mice | Altered fatty acid composition of dopaminergic neuron PUFAs enhance α-syn oligomerization | [90] | |
| B6;129-Sncatm1Sud Sncbtm1.1Sud/J See also paper for further details | X | -Cultured neurons from Snca KO mice overexpressing SNCA -See paper for other models used | Inhibition of synaptic vesicle reclustering after endocytosis | [91] | |
| B6;129×1-Sncatm1Rosl/J | X | X | α-syn fibrils gut-injected in Snca KO mice | α-syn transneuronal propagation from the gut to the brain | [92] |
| WT vs. KOM2 | X | Glial cytoplasmic inclusions-α-syn or Lewy Bodies-α-syn injected in WT mice vs. mice that express human α-syn only in oligodendrocites (KOM2) | Cellular milieu affects pathology of α-syn | [93] |
| Gene | Animal Model (s) | In Vivo | In Vitro | Expression Level | Main Findings | Ref. |
|---|---|---|---|---|---|---|
| PRKN | B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Independent regulation of parkin ubiquitination and alpha-synuclein clearance | [96] |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Accelerated microtubule aging in dopaminergic neurons | [97] | |
| B6.129S4-Prkntm1Shn/J | X | Prkn KO mice | Myotubular atrophy Impaired mitochondrial function and smaller myofiber area | [98] | ||
| B6.129S4-Prkntm1Shn/J | X | Prkn KO mice | Parkin mediates the ubiquitination of VPS35 Reduced WASH complexes | [99] | ||
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | ER stress and induced inflammation levels | [100] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Behavioral impairments Amplified EtOH-induced dopaminergic neurodegeneration, oxidative stress and apoptosis Dysfunction of mitochondrial autophagy | [101] | |
| B6.129S4-Prkntm1Shn/J | X | Prkn KO mice | Parkin promotes proteasomal degradation of Synaptotagmin IV | [102] | ||
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | SNPH Cargo vesicle generation not affected | [103] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Exacerbated mitochondrial dysfunction in neurons | [104] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Increased sensitivity to myocardial infarction Reduced survival after the infarction Reduced mitophagy | [105] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Parkin antagonizes the death potential of FAF1 | [106] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Acutely sensitivity to oxidative stress Inability to maintain Mcl-1 levels Death of dopaminergic neurons | [107,108] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Aberrant behavioral response to dopamine replacement therapy in PD | [109] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Resisted weight gain, steatohepatitis, and insulin resistance Abolished hepatic fat uptake | [110] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Reductions in the total catecholamine release Impaired LTP and LTD Normal levels of dopamine receptors and dopamine transporters | [111] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Requiring inflammatory stimulus for nigral DA neuron loss | [112] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Reduced respiratory capacity mitochondria (in striatal cells) Delayed weight gain Lower protection against ROS | [113] | |
| B6.129S4-Prkntm1Shn/J | X | X | Prkn KO mice | Increased extracellular dopamine concentration in the striatum Deficits in behavioral tests | [114] | |
| Double-mutant “TwinkPark” mice, resulting from crossing B6.129S4-Prkntm1Shn/J line with Twinkledup/+ line | X | X | Prkn KO (enhanced in the substantia nigra) mice | Decrease of mitochondrial DNA Low mitochondrial function and membrane potential Neurobehavioral deficits | [115] | |
| Crossing B6.129S4-Prkntm1Shn/J line with a Mcl-1 +/− line (n.s.n.) | X | X | Prkn KO + Mcl-1 +/− mice | Dopaminergic neuron loss Motor impairments | [107] | |
| B6.129S4-Prkntm1Shn/J B6.129S4-Pink1tm1Shn/J | X | X | -Prkn KO mice -Pink1 KO mice | Inflammation rescued by STING-mediated action | [116] | |
| B6.129S4-Prkntm1Shn/J B6.129S4-Pink1tm1Shn/J | X | X | -Prkn KO mice -Pink1 KO mice | No repression of mitochondrial antigen presentation | [117] | |
| B6.129S4-Prkntm1Shn/J LEH-Pink1tm1sage | X | -Prkn KO mice -Pink1 KO rats | Mitophagy of damaged mitochondria in axons requires PINK1 and Parkin | [118] | ||
| Crossing B6.129S4-Prkntm1Shn/J line and B6.129S4-Pink1tm1Shn/J line | X | Prkn/Pink1 double KO mice | Higher levels of ATP synthase Denervated neuromuscular junctions | [119] | ||
| B6.129S4-Prkntm1Shn/J B6.129S4-Pink1tm1Shn/J B6.Cg-Park7tm1Shn/J | X | X | -Prkn KO mice -Pink1 KO mice -Dj-1 KO mice | Aberrant striatal synaptic plasticity in rodent models of autosomal recessive PD | [120] | |
| Crossing DASYN53 double-transgenic (tetO-SNCA*A53T) E2Cai/J line + DAT-PF-tTA) mice with B6.129S4-Prkntm1Shn/J line or with Pink1tm1Zhzh mutation line (n.s.n.) | X | X | Overexpressing human A53T-SNCA in DA neurons and KO for either Prkn or Pink1 | Pervasive mitochondrial macroautophagy defects Dopamine neuron degeneration | [121] | |
| B6;129-Pink1tm1Aub/J | X | Pink1 KO mice | Altered spontaneous EPSCs | [122] | ||
| B6;129-Pink1tm1Aub/J | X | Pink1 KO mice | Mitochondrial recruitment of parkin not affected | [123] | ||
| B6;129-Pink1tm1Aub/J | X | X | Pink1 KO mice | Progressive mitochondrial dysfunction in absence of neurodegeneration | [124] | |
| LRRK2 | B6.129X1(FVB)-Lrrk2tm1.1Cai/J | X | Lrrk2 KO mice | Alterations in protein synthesis Alterations in degradation pathways | [125] | |
| B6.129X1(FVB)-Lrrk2tm1.1Cai/J | X | X | Lrrk2 KO mice | LRRK2 regulates synaptogenesis and dopamine receptor activation | [126] | |
| B6.129X1(FVB)-Lrrk2tm1.1Cai/J | X | Lrrk2 KO mice | LRRK2 regulates ER-Golgi export | [127] | ||
| B6.129X1(FVB)-Lrrk2tm1.1Cai/J | X | Lrrk2 KO mice | Neurons have more motile axonal and dendritic growth | [128] | ||
| C57BL/6-Lrrk2tm1Mjfa/J | X | X | Lrrk2 KO mice | LRRK2 modulates microglial phenotype and dopaminergic neurodegeneration | [129] | |
| C57BL/6-Lrrk2tm1Mjfa/J | X | Lrrk2 KO mice | Stress-Related Gastrointestinal Dysmotility | [130] | ||
| C57BL/6-Lrrk2tm1Mjfa/J | X | Lrrk2 KO mice | LRRK2 is required for Rip2 localization to DCVs | [131] | ||
| C57BL/6-Lrrk2tm1Mjfa/J | X | Lrrk2 KO mice | Significant increase of ceramide levels Direct effects on GBA1 | [132] | ||
| B6;129-Lrrk2tm2.1Shn/J | X | X | Lrrk2 KO mice | Impairment of Autophagy | [133] | |
| B6;129-Lrrk2tm2.1Shn/J B6;129-Lrrk2tm3.1Shn/J | X | Lrrk2 KO mice | Impairment of protein degradation pathways Apoptotic cell death | [134] | ||
| C57BL/6-Lrrk2tm1.1Mjff/J | X | X | Lrrk2 KO mice | No obvious bone alteration phenotypes | [135] | |
| B6.Cg-Tg(Lrrk2)6Yue/J | X | X | Lrrk2 overexpressing mice | Autophagy suppression | [136] | |
| B6.FVB-Tg (LRRK2) WT1Mjfa/J | X | X | LRRK2 overexpressing mice | Behavioral hypoactivity Altered dopamine-dependent short-term plasticity | [137] | |
| STOCK Tg (tetO-LRRK2*G2019S) E3Cai/J | X | G2019S-LRRK2 overexpressing mice | Perturbed homeostasis Altered neuronal morphogenesis | [138] | ||
| B6.Cg-Tg (Lrrk2*G2019S) 2Yue/J | X | G2019S-LRRK2 overexpressing mice | Reduction in lysosomal pH Increased expression of lysosomal ATPases | [139] | ||
| B6.FVB-Tg (LRRK2*G2019S) 1Mjfa/J | X | X | G2019S-LRRK2 overexpressing mice | Synapsis gain-of-function effect of the G2019S mutation | [140] | |
| NTac:SD-Tg (LRRK2*G2019S) 571CJLi | X | X | G2019S-Lrrk2 overexpressing rats | Altered bone marrow myelopoiesis Peripheral myeloid cell differentiation | [141] | |
| NTac:SD-Tg (LRRK2*G2019S) 571CJLi | X | X | G2019S-Lrrk2 overexpressing rats | Enhanced α-syn gene-induced neurodegeneration | [142] | |
| K-14Cre-positive Gbalnl/lnl | X | Gba KO mice (except in skin) | Reduced cerebral vascularization | [143] | ||
| PINK1 | B6.129S4-Pink1tm1Shn/J | X | Pink1 KO mice | Pink1 is not required for ubiquitination of mitochondrial proteins | [144] | |
| B6.129S4-Pink1tm1Shn/J | X | X | Pink1 KO mice | Reduced motor activity Slower locomotor activity time Absence of nigrostriatal dopamine loss | [145] | |
| B6.129S4-Pink1tm1Shn/J | X | Pink1 KO mice | Impaired mitochondrial trafficking Fragmented mitochondria | [146] | ||
| B6.129S4-Pink1tm1Shn/J | X | X | Pink1 KO mice | Hypersensitivity to MPTP-induced dopaminergic neuronal loss | [147] | |
| B6.129S4-Pink1tm1Shn/J | X | Pink1 KO mice | No significant change in Ca2+ currents | [148] | ||
| B6.129S4-Pink1tm1Shn/J | X | X | Pink1 KO mice | Pathological cardiac hypertrophy Greater levels of oxidative stress Impaired mitochondrial function | [149] | |
| B6.129S4-Pink1tm1Shn/J | X | X | Pink1 KO mice | Impairments of corticostriatal LTP and LTD Impaired dopamine release | [150] | |
| n.s.n. | X | Pink1 KO mice | Intestinal infection triggers Parkinson’s disease-like symptoms | [151] | ||
| Crossing B6;129-Pink1tm1Aub/J line with dOTC line | X | X | Pink1 KO mice overexpressing OTC in DA neurons | Enhanced neurodegeneration in a model of mitochondrial stress | [152] | |
| B6.129S4-Pink1tm1Shn/J B6.129S4-Prkntm1Shn/J | X | -Pink1 KO mice -Prkn KO mice | Enhanced sensitivity to group II mGlu receptor activation | [153] | ||
| B6.129S4-Pink1tm1Shn/J B6.129S4-Prkntm1Shn/J | X | -Pink1 KO mice -Prkn KO mice | Reduced mitochondria functions Altered mitophagy in macrophages | [154] | ||
| FVB;129-Pink1tm1Aub Tg(Prnp-SNCA*A53T)AAub/J | X | A53T-SNCA overexpressing Pink1 KO mice | Altered mitochondrial biogenesis | [155] | ||
| FVB;129-Pink1tm1Aub Tg(Prnp-SNCA*A53T)AAub/J | X | X | A53T-SNCA overexpressing Pink1 KO mice | Exacerbated synucleinopathy | [156] | |
| FVB;129-Pink1tm1Aub Tg(Prnp-SNCA*A53T)AAub/J | X | X | A53T-SNCA overexpressing Pink1 KO mice | Potentiation of neurotoxicity | [157] | |
| Atad3afl/fl Mx1CrePink1 −/− mice, resulting from crossing B6.129S4-Pink1tm1Shn/J line with Atad3afl/fl Mx1Cre line | X | X | Pink1 KO + conditional Atad3a KO mice | Aberrant stem-cell and progenitor homeostasis Pink1-dependent mitophagy | [158] | |
| B6N.129S6(Cg)-Atp13a2tm1Pjsch/J | X | X | Atp13a2 KO mice | Autophagy impairment Reduced HDAC6 activity | [159] | |
| B6N.129S6(Cg)-Atp13a2tm1Pjsch/J | X | Atp13a2 KO mice | Harmful gliosis | [160] | ||
| B6N.129S6(Cg)-Atp13a2tm1Pjsch/J | X | X | Atp13a2 KO mice | Neuronal ceroid lipofuscinosis Limited α-syn accumulation Sensorimotor deficits | [161] |
7. α-Synuclein: Main Character or Supporting Actor?
8. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lesage, S.; Lunati, A.; Houot, M.; Ben Romdhan, S.; Clot, F.; Tesson, C.; Mangone, G.; Toullec, B.L.; Courtin, T.; Larcher, K.; et al. Characterization of Recessive Parkinson Disease in a Large Multicenter Study. Ann. Neurol. 2020, 88, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Oñatibia-Astibia, A.; Franco, R.; Martínez-Pinilla, E. Health benefits of methylxanthines in neurodegenerative diseases. Mol. Nutr. Food Res. 2017, 61, 1600670. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Wu, C.; Tilley, B.C.; Lohmann, K.; Klein, C.; Payami, H.; Wills, A.M.; Aminoff, M.J.; Bainbridge, J.; Dewey, R.; et al. Caffeine, creatine, GRIN2A and Parkinson’s disease progression. J. Neurol. Sci. 2017, 375, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Sipetic, S.B.; Vlajinac, H.D.; Maksimovic, J.M.; Marinkovic, J.M.; Dzoljic, E.D.; Ratkov, I.S.; Kostic, V.S. Cigarette smoking, coffee intake and alcohol consumption preceding Parkinson’s disease: A case-control study. Acta Neuropsychiatr. 2012, 24, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Petrovitch, H. Current Evidence for Neuroprotective Effects of Nicotine and Caffeine Against Parkinsons Disease. Drugs Aging 2001, 18, 797–806. [Google Scholar] [CrossRef]
- Ross, G.W.; RD, A.; Petrovitch, H.; Al, E.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; et al. Association of coffee and caffeine intake with the risk of parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Mateo, D.; Giméanez-Roldan, S. Premorbid smoking, alcohol consumption, and coffee drinking habits in Parkinson’s disease: A case-control study. Mov. Disord. 1992, 7, 339–344. [Google Scholar] [CrossRef]
- Bronstein, J.; Carvey, P.; Chen, H.; Cory-Slechta, D.; DiMonte, D.; Duda, J.; English, P.; Goldman, S.; Grate, S.; Hanssen, J.; et al. Meeting report: Consensus Statement—Parkinson’s disease and the environment: Collaborative on health and the environment and Parkinson’s action network (CHE PAN) conference 26–28 June 2007. Environ. Health Perspect. 2009, 117, 117–121. [Google Scholar] [CrossRef]
- Chen, J.-F. Adenosine receptor control of cognition in normal and disease. Int. Rev. Neurobiol. 2014, 119, 257–307. [Google Scholar] [PubMed]
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Birkmayer, W.; Hornykiewicz, O. The L-dihydroxyphenylalanine (L-DOPA) effect in Parkinson’s syndrome in man: On the pathogenesis and treatment of Parkinson akinesis. Arch. Psychiatr. Nervenkr. Z. Gesamte Neurol. Psychiatr. 1962, 203, 560–574. [Google Scholar] [CrossRef]
- Hornykiewicz, O. Die topische Lokalization und des Verhalten von Noradrenalin und Dopamin (3-Hydroxytyramin) in der Substantia nigra der normalen und Parkinson Kranken Menschen. Wien. Klin. Wochschr. 1963, 75, 309–312. [Google Scholar]
- Birkmayer, W.; Hornykiewicz, O. Additional experimental studies on L-DOPA in Parkinson’s syndrome and reserpine parkinsonism. Arch. Psychiatr. Nervenkr. 1964, 206, 367–381. [Google Scholar] [CrossRef]
- Holzer, G.; Hornykiewicz, O. Über den Dopamin-(Hydroxytyramin-)Stoffwechsel im Gehirn der Ratte. Naunyn-Schmiedeberg’s Arch. Exp. Pathol. Pharmakol. 1959, 237, 27–33. [Google Scholar]
- Hornykiewicz, O. The discovery of dopamine deficiency in the parkinsonian brain. J. Neural Transm. Suppl. 2006, 70, 9–15. [Google Scholar]
- Olanow, C.W.; Agid, Y.; Mizuno, Y.; Albanese, A.; Bonuccelli, U.; Bonucelli, U.; Damier, P.; De Yebenes, J.; Gershanik, O.; Guttman, M.; et al. Levodopa in the treatment of Parkinson’s disease: Current controversies. Mov. Disord. 2004, 19, 997–1005. [Google Scholar] [CrossRef]
- Walter, B.L.; Vitek, J.L. Surgical treatment for Parkinson’s disease. Lancet Neurol. 2004, 3, 719–728. [Google Scholar] [CrossRef]
- Conley, S.C.; Kirchner, J.T. Medical and surgical treatment of Parkinson’s disease: Strategies to slow symptom progression and improve quality of life. Postgrad. Med. 1999, 106, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Durif, F. Treating and preventing levodopa-induced dyskinesias: Current and future strategies. Drugs Aging 1999, 14, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Lozano, A.M.; Montgomery, E.; Lang, A.E. Pallidotomy and deep brain stimulation of the pallidum and subthalamic nucleus in advanced Parkinson’s disease. Mov. Disord. 1998, 13 (Suppl. 1), 73–82. [Google Scholar]
- Arle, J.E.; Alterman, R.L. Surgical options in Parkinson’s disease. Med. Clin. N. Am. 1999, 83, 483–498. [Google Scholar] [CrossRef]
- Limousin-Dowsey, P.; Pollak, P.; Blercom, N.; Krack, P.; Benazzouz, A.; Benabid, A.-L. Thalamic, subthalamic nucleus and internal pallidum stimulation in Parkinson’s disease. J. Neurol. 1999, 246, II42–II45. [Google Scholar] [CrossRef]
- Gross, C.E.; Boraud, T.; Guehl, D.; Bioulac, B.; Bezard, E. From experimentation to the surgical treatment of Parkinson’s disease: Prelude or suite in basal ganglia research? Prog. Neurobiol. 1999, 59, 509–532. [Google Scholar] [CrossRef]
- Gross, R.E.; Lozano, A.M. Advances in neurostimulation for movement disorders. Neurol. Res. 2000, 22, 247–258. [Google Scholar] [CrossRef]
- Caparros-Lefebvre, D.; Blond, S.; Vermersch, P.; Pecheux, N.; Guieu, J.D.; Petit, H. Chronic thalamic stimulation improves tremor and levodopa induced dyskinesias in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1993, 56, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2012, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Burn, D. Spectrum of Parkinson’s disease, Parkinson’s dementia, and Lewy body dementia. Neurol. Clin. 2000, 18, 865–883. [Google Scholar] [CrossRef]
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Noyce, A.J.; Byrne, M.; Savica, R.; Marras, C. Infection and risk of Parkinson’s disease. J. Parkinsons. Dis. 2021, 11, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Lunati, A.; Lesage, S.; Brice, A. The genetic landscape of Parkinson’s disease. Rev. Neurol. 2018, 174, 628–643. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A. New Genes Causing Hereditary Parkinson’s Disease or Parkinsonism. Curr. Neurol. Neurosci. Rep. 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Haas, A.L.; Warms, J.V.; Hershko, A.; Rose, I.A. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J. Biol. Chem. 1982, 257, 2543–2548. [Google Scholar] [CrossRef]
- Ciechanover, A.; Finley, D.; Varshavsky, A. The ubiquitin-mediated proteolytic pathway and mechanisms of energy-dependent intracellular protein degradation. J. Cell. Biochem. 1984, 24, 27–53. [Google Scholar] [CrossRef]
- Hershko, A.; Leshinsky, E.; Ganoth, D.; Heller, H. ATP-dependent degradation of ubiquitin-protein conjugates. Proc. Natl. Acad. Sci. USA 1984, 81, 1619–1623. [Google Scholar] [CrossRef]
- Bonifati, V.; Dekker, M.C.J.; Vanacore, N.; Fabbrini, G.; Squitieri, F.; Marconi, R.; Antonini, A.; Brustenghi, P.; Dalla Libera, A.; De Mari, M.; et al. Autosomal recessive early onset parkinsonism is linked to three loci: PARK2, PARK6, and PARK. Neurol. Sci. 2002, 23, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; Zhang, L.; Troncoso, J.; Lee, M.K.; Hattori, N.; Mizuno, Y.; Dawson, T.M.; Dawson, V.L. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Genet. 2005, 14, 71–84. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef]
- Exner, N.; Treske, B.; Paquet, D.; Holmström, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.H.; Gasser, T.; et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 2007, 27, 12413–12418. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-Selected Transport from the Mitochondria to Peroxisomes Is Mediated by Vesicular Carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef]
- Lee, D.W.; Zhao, X.; Yim, Y.I.; Eisenberg, E.; Greene, L.E. Essential role of cyclin-G-associated kinase (auxilin-2) in developing and mature mice. Mol. Biol. Cell 2008, 19, 2766–2776. [Google Scholar] [CrossRef]
- Pishvaee, B.; Costaguta, G.; Yeung, B.G.; Ryazantsev, S.; Greener, T.; Greene, L.E.; Eisenberg, E.; McCaffery, J.M.; Payne, G.S. A yeast DNA J protein required for uncoating of clathrin-coated vesicles in vivo. Nat. Cell Biol. 2000, 2, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Toret, C.P.; Lee, L.; Sekiya-Kawasaki, M.; Drubin, D.G. Multiple pathways regulate endocytic coat disassembly in Saccharomyces cerevisiae for optimal downstream trafficking. Traffic 2008, 9, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Surguchev, A.A.; Surguchov, A. Synucleins and gene expression: Ramblers in a crowd or cops regulating traffic? Front. Mol. Neurosci. 2017, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Floden, A.M.; Murphy, E.J.; Combs, C.K. α-Synuclein Expression Modulates Microglial Activation Phenotype. J. Neurosci. 2006, 26, 10558–10563. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Faergeman, N.J.; Cole, N.B.; Castagnet, P.I.; Nussbaum, R.L.; Murphy, E.J. α-Synuclein Gene Deletion Decreases Brain Palmitate Uptake and Alters the Palmitate Metabolism in the Absence of R -Synuclein Palmitate Binding. Biochemistry 2005, 44, 8251–8259. [Google Scholar] [CrossRef]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barcelo, G.C.; Nussbaum, R.L. Mitochondrial Lipid Abnormality and Electron Transport Chain Impairment in Mice Lacking α-Synuclein. Mol. Cell. Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef] [PubMed]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; Mcilwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic Vesicle Depletion Correlates with Attenuated Synaptic α-Synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef]
- Pathak, D.; Berthet, A.; Bendor, J.T.; Yu, K.; Sellnow, R.C.; Orr, L.; Nguyen, M.K.; Edwards, R.H.; Manfredsson, F.P.; Nakamura, K. Loss of α-Synuclein Does Not Affect Mitochondrial Bioenergetics in Rodent Neurons. Eneuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E. α-Synuclein Expression Restricts RNA Viral Infections in the Brain. J. Virol. 2016, 90, 2767–2782. [Google Scholar] [CrossRef] [PubMed]
- Renella, R.; Schlehe, J.S.; Selkoe, D.J.; Williams, D.A.; LaVoie, M.J. Genetic deletion of the GATA1-regulated protein α-synuclein reduces oxidative stress and nitric oxide synthase levels in mature erythrocytes. Am. J. Hematol. 2014, 89, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Paine, S.M.L.; Anderson, G.; Bedford, K.; Lawler, K.; Mayer, R.J.; Lowe, J.; Bedford, L. Pale Body-Like Inclusion Formation and Neurodegeneration following Depletion of 26S Proteasomes in Mouse Brain Neurones are Independent of α-Synuclein. PLoS ONE 2013, 8, e54711. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Roy, S. α-Synuclein Inhibits Intersynaptic Vesicle Mobility and Maintains Recycling-Pool Homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef] [PubMed]
- Kokhan, V.S.; Afanasyeva, M.A.; Van, G.I. α-Synuclein knockout mice have cognitive impairments. Behav. Brain Res. 2012, 231, 226–230. [Google Scholar] [CrossRef]
- Cano-jaimez, M.; Pérez-sánchez, F.; Milán, M.; Buendía, P.; Ambrosio, S.; Fariñas, I. Neurobiology of Disease Vulnerability of peripheral catecholaminergic neurons to MPTP is not regulated by α-synuclein. Neurobiol. Dis. 2010, 38, 92–103. [Google Scholar] [CrossRef]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking α-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef]
- Abeliovich, A.; Schmitz, Y.; Farin, I.; Choi-lundberg, D.; Ho, W.; Castillo, P.E.; Shinsky, N.; Manuel, J.; Verdugo, G.; Armanini, M.; et al. Mice Lacking α-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Peña-Oliver, Y.; Buchman, V.L.; Dalley, J.W.; Robbins, T.W.; Schumann, G.; Ripley, T.L.; King, S.L.; Stephens, D.N. Deletion of α-synuclein decreases impulsivity in mice. Genes Brain Behav. 2012, 11, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, A.; Yavich, L. Neuroscience Letters Neuromuscular pathology in mice lacking α-synuclein. Neurosci. Lett. 2011, 487, 350–353. [Google Scholar] [CrossRef]
- Chadchankar, H.; Ihalainen, J.; Tanila, H.; Yavich, L. Decreased reuptake of dopamine in the dorsal striatum in the absence of α-synuclein. Brain Res. 2011, 1382, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Al-Wandi, A.; Ninkina, N.; Millership, S.; Williamson, S.J.M.; Jones, P.A.; Buchman, V.L. Absence of α-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 2010, 31, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Senior, S.L.; Ninkina, N.; Deacon, R.; Bannerman, D.; Vladimir, L.; Cragg, S.J.; Wade-martins, R. Europe PMC Funders Group Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both α-synuclein and gamma- synuclein. J. Neurosci. 2011, 27, 947–957. [Google Scholar]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional Alterations to the Nigrostriatal System in Mice Lacking All Three Members of the Synuclein Family. J. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef] [PubMed]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins Regulate the Kinetics of Synaptic Vesicle Endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M. αβγ -Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [PubMed]
- Vargas, K.J.; Schrod, N.; Davis, T.; Fernandez-Busnadiego, R.; Taguchi, Y.V.; Laugks, U.; Lucic, V.; Chandra, S.S. Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep. 2017, 18, 161–173. [Google Scholar] [CrossRef]
- Su, S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J. TrkB neurotrophic activities are blocked by α-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar]
- Sarafian, T.A.; Ryan, C.M.; Souda, P.; Masliah, E.; Kar, U.K.; Vinters, H.V.; Mathern, G.W.; Faull, K.F.; Whitelegge, J.P.; Watson, J.B. Impairment of Mitochondria in Adult Mouse Brain Overexpressing Predominantly Full-Length, N-Terminally Acetylated Human α-Synuclein. PLoS ONE 2013, 8, e63557. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.B.; Hatami, A.; David, H.; Masliah, E.; Roberts, K.; Evans, C.E.; Levine, M.S. Aleterations in corticostriatal synaptic plasticity in mice overexpressing human α-synuclein. NSC 2009, 159, 501–513. [Google Scholar]
- Reznichenko, L.; Cheng, Q.; Nizar, K.; Gratiy, S.L.; Saisan, P.A.; Rockenstein, E.M.; Patrick, C.; Spencer, B.; Desplats, P.; Dale, A.M.; et al. In Vivo Alterations in Calcium Buffering Capacity in Transgenic Mouse Model of Synucleinopathy. J. Neurosci. 2012, 32, 9992–9998. [Google Scholar] [CrossRef] [PubMed]
- Siebert, H.; Kahle, P.J.; Kramer, M.L.; Isik, T.; Schlüter, O.M.; Schulz-Schaeffer, W.J.; Brück, W. Over-expression of α-synuclein in the nervous system enhances axonal degeneration after peripheral nerve lesion in a transgenic mouse strain. J. Neurochem. 2010, 114, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Janezic, S.; Threlfell, S.; Dodson, P.D.; Dowie, M.J.; Taylor, T.N.; Potgieter, D.; Parkkinen, L.; Senior, S.L.; Anwar, S.; Ryan, B.; et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA 2013, 110, E4016–E4025. [Google Scholar] [CrossRef]
- Hunn, B.H.M.; Vingill, S.; Threlfell, S.; Bannerman, D.M.; Cragg, S.J.; Wade-martins, R.; Hunn, B.H.M.; Vingill, S.; Threlfell, S.; Alegre-abarrategui, J.; et al. Impairment of Macroautophagy in Dopamine Neurons Has Opposing Effects on Parkinsonian Pathology and Behavior Article Impairment of Macroautophagy in Dopamine Neurons Has Opposing Effects on Parkinsonian Pathology and Behavior. Cell Rep. 2019, 29, 920–931.e7. [Google Scholar] [CrossRef]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.J.; Roy, S. α-Synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol. 2014, 24, 2319–2326. [Google Scholar] [CrossRef]
- Escobar, V.D.; Kuo, Y.-M.; Orrison, B.M.; Giasson, B.I.; Nussbaum, R.L. Transgenic mice expressing S129 phosphorylation mutations in α-synuclein. Neurosci. Lett. 2014, 563, 96–100. [Google Scholar] [CrossRef][Green Version]
- Kuo, Y.; Li, Z.; Jiao, Y.; Gaborit, N.; Pani, A.K.; Orrison, B.M.; Bruneau, B.G.; Giasson, B.I.; Smeyne, R.J.; Gershon, M.D.; et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated α-synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet. 2010, 19, 1633–1650. [Google Scholar] [CrossRef]
- Gao, H.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M. Neuroinflammation and Oxidation / Nitration of α-Synuclein Linked to Dopaminergic Neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef]
- Taylor, T.N.; Potgieter, D.; Anwar, S.; Senior, S.L.; Janezic, S.; Threlfell, S.; Ryan, B.; Parkkinen, L.; Deltheil, T.; Cioroch, M.; et al. Region-specific deficits in dopamine, but not norepinephrine, signaling in a novel A30P α-synuclein BAC transgenic mouse. Neurobiol. Dis. 2014, 62, 193–207. [Google Scholar] [CrossRef]
- Kurz, A.; Double, K.L.; Lastres-becker, I.; Tozzi, A.; Tantucci, M.; Nuber, S.; Schlaudraff, F.; Bockhart, V.; Bonin, M.; Ferna, J. A53T-α-Synuclein Overexpression Impairs Dopamine Signaling and Striatal Synaptic Plasticity in Old Mice. PLoS ONE 2010, 5, e11464. [Google Scholar] [CrossRef]
- Gispert, S.; Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-clement, J.; Korf, H.; Deller, T.; Braak, H.; Auburger, G.; et al. Transgenic mice expressing mutant A53T human α-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell. Neurosci. 2003, 24, 419–429. [Google Scholar] [CrossRef]
- Yamasaki, T.; Fujinaga, M.; Kawamura, K.; Furutsuka, K.; Nengaki, N.; Shimoda, Y.; Shiomi, S.; Takei, M.; Hashimoto, H.; Yui, J.; et al. Dynamic Changes in Striatal mGluR1 But Not mGluR5 during Pathological Progression of Parkinson’s Disease in Human Alpha-Synuclein A53T Transgenic Rats: A Multi-PET Imaging Study. J. Neurosci. 2016, 36, 375–384. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; Brien, P.O.; Trojanowski, J.Q.; Lee, V.M.Y. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef]
- Cannon, J.R.; Geghman, K.D.; Tapias, V.; Sew, T.; Dail, M.K.; Li, C.; Greenamyre, J.T. Expression of human E46K-mutated α-synuclein in BAC-transgenic rats replicates early-stage Parkinson’s disease features and enhances vulnerability to mitochondrial impairment. Exp. Neurol. 2013, 240, 44–56. [Google Scholar] [CrossRef]
- Shimshek, D.R.; Schweizer, T.; Schmid, P.; Van Der Putten, P.H. Excess α-synuclein worsens disease in mice lacking ubiquitin carboxy-terminal hydrolase L1. Sci. Rep. 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Martín-Clemente, B.; Alvarez-Castelao, B.; Mayo, I.; Sierra, A.B.; Díaz, V.; Milán, M.; Fariñas, I.; Gómez-Isla, T.; Ferrer, I.; Castaño, J.G. α-Synuclein expression levels do not significantly affect proteasome function and expression in mice and stably transfected PC12 cell lines. J. Biol. Chem. 2004, 279, 52984–52990. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-joseph, I.; Mirick, G.E.; Serhan, C.N.; Selkoe, D.J. Altered Fatty Acid Composition of Dopaminergic Neurons Expressing α-Synuclein and Human Brains with α-Synucleinopathies. J. Biol. Chem. 2003, 278, 49874–49881. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.; Kam, T.; Dawson, V.L.; Dawson, T.M.; Ko, H.S.; Kim, S.; Kwon, S.; Kam, T.; Panicker, N.; et al. Transneuronal Propagation of Pathologic a -Synuclein from the Gut to the Brain Models Parkinson’ s Disease Article Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’ s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. α -synuclein strains in α -synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Lin, J.; Zheng, X.; Zhang, Z.; Zhuge, J.; Shao, Z.; Huang, C.; Jin, J.; Chen, X.; Chen, Y.; Wu, Y.; et al. Inhibition of LRRK2 restores parkin-mediated mitophagy and attenuates intervertebral disc degeneration. Osteoarthr. Cartil. 2021, 29, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Feng, S.T.; Wang, Y.T.; Yuan, Y.H.; Li, Z.P.; Chen, N.H.; Wang, Z.Z.; Zhang, Y. Mitophagy, a Form of Selective Autophagy, Plays an Essential Role in Mitochondrial Dynamics of Parkinson’s Disease. Cell. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Liu, X.; Hebron, M.; Shi, W.; Lonskaya, I.; Moussa, C.E.H. Ubiquitin specific protease-13 independently regulates parkin ubiquitination and α-synuclein clearance in alpha-synucleinopathies. Hum. Mol. Genet. 2019, 28, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Amadeo, A.; Calogero, A.M.; Casagrande, F.V.M.; De Gregorio, C.; Gioria, M.; Kuzumaki, N.; Costa, I.; Sassone, J.; Ciammola, A.; et al. Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol. Aging 2018, 61, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Peker, N.; Donipadi, V.; Sharma, M.; McFarlane, C.; Kambadur, R. Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am. J. Physiol. Cell Physiol. 2018, 315, C164–C185. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.T.; Glauser, L.; Tsika, E.; Jiang, H.; Islam, S.; Moore, D.J. Parkin mediates the ubiquitination of VPS35 and modulates retromer-dependent endosomal sorting. Hum. Mol. Genet. 2018, 27, 3189–3205. [Google Scholar] [CrossRef]
- Singh, K.; Han, K.; Tilve, S.; Wu, K.; Geller, H.M.; Sack, M.N. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia 2018, 66, 2427–2437. [Google Scholar] [CrossRef]
- Hwang, C.J.; Kim, Y.E.; Son, D.J.; Park, M.H.; Choi, D.Y.; Park, P.H.; Hellström, M.; Han, S.B.; Oh, K.W.; Park, E.K.; et al. Parkin deficiency exacerbate ethanol-induced dopaminergic neurodegeneration by P38 pathway dependent inhibition of autophagy and mitochondrial function. Redox Biol. 2017, 11, 456–468. [Google Scholar] [CrossRef]
- Kabayama, H.; Tokushige, N.; Takeuchi, M.; Kabayama, M.; Fukuda, M.; Mikoshiba, K. Parkin promotes proteasomal degradation of synaptotagmin IV by accelerating polyubiquitination. Mol. Cell. Neurosci. 2017, 80, 89–99. [Google Scholar] [CrossRef]
- Lin, M.Y.; Cheng, X.T.; Tammineni, P.; Xie, Y.; Zhou, B.; Cai, Q.; Sheng, Z.H. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 2017, 94, 595–610.e6. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyans, S.; Murphy, A.N.; Gustafsson, Å.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.W.; Park, M.Y.; Shin, J.; Kim, Y.R.; Yoo, S.E.; Kong, Y.Y.; Kwon, K.S.; Lee, Y.H.; Kim, E. Accumulation of the parkin substrate, FAF1, plays a key role in the dopaminergic neurodegeneration. Hum. Mol. Genet. 2013, 22, 1558–1573. [Google Scholar] [CrossRef] [PubMed]
- Ekholm-Reed, S.; Baker, R.; Campos, A.R.; Stouffer, D.; Henze, M.; Wolf, D.A.; Loring, J.F.; Thomas, E.A.; Reed, S.I. Reducing Mcl-1 gene dosage induces dopaminergic neuronal loss and motor impairments in Park2 knockout mice. Commun. Biol. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Ekholm-Reed, S.; Goldberg, M.S.; Schlossmacher, M.G.; Reed, S.I. Parkin-Dependent Degradation of the F-Box Protein Fbw7 Promotes Neuronal Survival in Response to Oxidative Stress by Stabilizing Mcl. Mol. Cell. Biol. 2013, 33, 3627–3643. [Google Scholar] [CrossRef] [PubMed]
- Berthet, A.; Bezard, E.; Porras, G.; Fasano, S.; Barroso-Chinea, P.; Dehay, B.; Martinez, A.; Thiolat, M.L.; Nosten-Bertrand, M.; Giros, B.; et al. L-DOPA impairs proteasome activity in parkinsonism through D 1 dopamine receptor. J. Neurosci. 2012, 32, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 3701–3712. [Google Scholar] [CrossRef]
- Kitada, T.; Pisani, A.; Karouani, M.; Haburcak, M.; Martella, G.; Tscherter, A.; Platania, P.; Wu, B.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of Parkin-/- mice. J. Neurochem. 2009, 110, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Tran, T.; Ruhn, K.A.; Martinez, T.N.; Hong, J.; Marvin, M.; Hartley, M.; Treviño, I.; O’Brien, D.E.; Casey, B.; et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 2008, 28, 10825–10834. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in parkin-deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient Mice Exhibit Nigrostriatal Deficits but not Loss of Dopaminergic Neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef]
- Song, L.; McMackin, M.; Nguyen, A.; Cortopassi, G. Parkin deficiency accelerates consequences of mitochondrial DNA deletions and Parkinsonism. Neurobiol. Dis. 2017, 100, 30–38. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired mitophagy plays a role in denervation of neuromuscular junctions in ALS mice. Front. Neurosci. 2017, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Madeo, G.; Martella, G.; Schirinzi, T.; Ponterio, G.; Shen, J.; Bonsi, P.; Pisani, A. Aberrant striatal synaptic plasticity in monogenic parkinsonisms. Neuroscience 2012, 211, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xie, Z.; Turkson, S.; Zhuang, X. A53T human α-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci. 2015, 35, 890–905. [Google Scholar] [CrossRef]
- Pearlstein, E.; Michel, F.J.; Save, L.; Ferrari, D.C.; Hammond, C. Abnormal development of glutamatergic synapses afferent to dopaminergic neurons of the pink1–/– mouse model of Parkinson’s disease. Front. Cell. Neurosci. 2016, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Cortez, M.Q.; Moyzis, A.G.; Najor, R.H.; Lee, Y.; Gustafsson, Å.B. PINK1 is dispensable for mitochondrial recruitment of parkin and activation of mitophagy in cardiac myocytes. PLoS ONE 2015, 10, e0130707. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Pellegrini, L.; Hauser, D.N.; Li, Y.; Mamais, A.; Beilina, A.; Kumaran, R.; Wetzel, A.; Nixon-Abell, J.; Heaton, G.; Rudenko, I.; et al. Proteomic analysis reveals co-ordinated alterations in protein synthesis and degradation pathways in LRRK2 knockout mice. Hum. Mol. Genet. 2018, 27, 3257–3271. [Google Scholar] [CrossRef] [PubMed]
- Parisiadou, L.; Yu, J.; Sgobio, C.; Xie, C.; Liu, G.; Sun, L.; Gu, X.-L.; Lin, X.; Crowley, N.A.; Lovinger, D.M.; et al. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat. Neurosci. 2014, 17, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Yu, J.; Xie, C.; Rudrabhatla, P.; Chen, X.; Wu, J.; Parisiadou, L.; Liu, G.; Sun, L.; Ma, B.; et al. Leucine-rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER –Golgi export. EMBO J. 2014, 33, 2314–2331. [Google Scholar] [CrossRef]
- Sepulveda, B.; Mesias, R.; Li, X.; Yue, Z.; Benson, D.L. Short- and Long-Term Effects of LRRK2 on Axon and Dendrite Growth. PLoS ONE 2013, 8, e61986. [Google Scholar] [CrossRef]
- Dwyer, Z.; Rudyk, C.; Thompson, A.; Farmer, K.; Fenner, B.; Fortin, T.; Derksen, A.; Sun, H.; Hayley, S. Leucine-rich repeat kinase-2 (LRRK2) modulates microglial phenotype and dopaminergic neurodegeneration. Neurobiol. Aging 2020, 91, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Tsushima, H.; Kawakami, F.; Kawashima, R.; Kodo, M.; Imai, M.; Ichikawa, T. Leucine-rich repeat kinase 2 is associated with activation of the paraventricular nucleus of the hypothalamus and stress-related gastrointestinal dysmotility. Front. Neurosci. 2019, 13, 1–10. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Zuo, Z.; Zhang, Q.; Pan, Y.; Zeng, B.; Li, W.; Wei, H.; Liu, Z. Rip2 Is Required for Nod2-Mediated Lysozyme Sorting in Paneth Cells. J. Immunol. 2017, 198, 3729–3736. [Google Scholar] [CrossRef]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef]
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 2017, 96, 796–807.e6. [Google Scholar] [CrossRef]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J.; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Liu, J.; Cheng, S.; Vogel, P.; Mohan, S.; Brommage, R. Targeted disruption of leucine-rich repeat kinase 1 but not leucine-rich repeat kinase 2 in mice causes severe osteopetrosis. J. Bone Miner. Res. 2013, 28, 1962–1974. [Google Scholar] [CrossRef] [PubMed]
- Takagawa, T.; Kitani, A.; Fuss, I.; Levine, B.; Brant, S.R.; Tajima, M.; Nakamura, S.; Strober, W. Dectin-1–Induced immunity in a mouse model of colitis. Sci. Transl. Med. 2019, 10, 1–25. [Google Scholar]
- Beccano-kelly, D.A.; Volta, M.; Munsie, L.N.; Paschall, S.A. LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, behavioural hypoactivity and memory deficits. Hum. Mol. Genet. 2014, 24, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Parisiadou, L.; Xie, C.; Hyun, J.C.; Lin, X.; Gu, X.L.; Long, C.X.; Lobbestael, E.; Baekelandt, V.; Taymans, J.M.; Sun, L.; et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 2009, 29, 13971–13980. [Google Scholar] [CrossRef]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef]
- Beccano-Kelly, D.A.; Kuhlmann, N.; Tatarnikov, I.; Volta, M.; Munsie, L.N.; Chou, P.; Cao, L.P.; Han, H.; Tapia, L.; Farrer, M.J.; et al. Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front. Cell. Neurosci. 2014, 8, 1–11. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.-W.; Cooper, S.C.; Broxmeyer, H.E.; Cannon, J.R.; Kim, C.H. Parkinson disease–associated LRRK2 G2019S transgene disrupts marrow myelopoiesis and peripheral Th17 response. J. Leukoc. Biol. 2017, 102, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.P.L.; Abdelmotilib, H.A.; Hu, X.; Volpicelli-Daley, L.A.; Moehle, M.S.; Fraser, K.B.; Needle, E.; Chen, Y.; Steyn, S.J.; Galatsis, P.; et al. Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem. 2015, 290, 19433–19444. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.J.; Fuller, M.; Saville, J.T.; Cox, T.M. Reduced cerebral vascularization in experimental neuronopathic Gaucher disease. J. Pathol. 2018, 244, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Kelm-Nelson, C.A.; Brauer, A.F.L.; Barth, K.J.; Lake, J.M.; Sinnen, M.L.K.; Stehula, F.J.; Muslu, C.; Marongiu, R.; Kaplitt, M.G.; Ciucci, M.R. Characterization of early-onset motor deficits in the Pink1−/− mouse model of Parkinson disease. Brain Res. 2018, 1680, 1–12. [Google Scholar] [CrossRef]
- Das Banerjee, T.; Dagda, R.Y.; Dagda, M.; Chu, C.T.; Rice, M.; Vazquez-Mayorga, E.; Dagda, R.K. PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. J. Neurochem. 2017, 142, 545–559. [Google Scholar] [CrossRef]
- Haque, M.E.; Mount, M.P.; Safarpour, F.; Abdel-Messih, E.; Callaghan, S.; Mazerolle, C.; Kitada, T.; Slack, R.S.; Wallace, V.; Shen, J.; et al. Inactivation of Pink1 gene in Vivo sensitizes dopamine-producing neurons to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and can be rescued by autosomal recessive Parkinson disease genes, Parkin or DJ-1. J. Biol. Chem. 2012, 287, 23162–23170. [Google Scholar] [CrossRef] [PubMed]
- Martella, G.; Madeo, G.; Schirinzi, T.; Tassone, A.; Sciamanna, G.; Spadoni, F.; Stefani, A.; Shen, J.; Pisani, A.; Bonsi, P. Altered profile and D2-dopamine receptor modulation of high voltage-activated calcium current in striatal medium spiny neurons from animal models of Parkinson’s disease. Neuroscience 2011, 177, 240–251. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1 −/− mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Moisoi, N.; Fedele, V.; Edwards, J.; Martins, L.M. Loss of PINK1 enhances neurodegeneration in a mouse model of Parkinson’s disease triggered by mitochondrial stress. Neuropharmacology 2014, 77, 350–357. [Google Scholar] [CrossRef]
- Martella, G.; Platania, P.; Vita, D.; Sciamanna, G.; Cuomo, D.; Tassone, A.; Tscherter, A.; Kitada, T.; Bonsi, P.; Shen, J.; et al. Enhanced sensitivity to group II mGlu receptor activation at corticostriatal synapses in mice lacking the familial parkinsonism-linked genes PINK1 or Parkin. Exp. Neurol. 2009, 215, 388–396. [Google Scholar] [CrossRef]
- Bhatia, D.; Chung, K.P.; Nakahira, K.; Patino, E.; Rice, M.C.; Torres, L.K.; Muthukumar, T.; Choi, A.M.K.; Akchurin, O.M.; Choi, M.E. Mitophagy-dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 2019, 4, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Gispert, S.; Brehm, N. Methyl-arginine profile of brain from aged PINK1-KO+A53T-SNCA mice suggests altered mitochondrial biogenesis. Parkinsons. Dis. 2016, 2016, 4686185. [Google Scholar] [CrossRef]
- Cabin, D.E.; Gispert-Sanchez, S.; Murphy, D.; Auburger, G.; Myers, R.R.; Nussbaum, R.L. Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiol. Aging 2005, 26, 25–35. [Google Scholar] [CrossRef]
- Gispert, S.; Brehm, N.; Weil, J.; Seidel, K.; Rüb, U.; Kern, B.; Walter, M.; Roeper, J.; Auburger, G. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 2015, 24, 1061–1076. [Google Scholar] [CrossRef]
- Jin, G.; Xu, C.; Zhang, X.; Long, J.; Rezaeian, A.H.; Liu, C.; Furth, M.E.; Kridel, S.; Pasche, B.; Bian, X.-W.; et al. Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol. 2018, 19, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome–lysosome fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Seven, Y.B.; Howard, J.; Duffy, C.; Altshuler, M.; Moloney, C.; Giasson, B.I.; Lewis, J. Partial loss of ATP13A2 causes selective gliosis independent of robust lipofuscinosis. Mol. Cell. Neurosci. 2018, 92, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, P.J.; Fleming, S.M.; Clippinger, A.K.; Lewis, J.; Tsunemi, T.; Giasson, B.; Dickson, D.W.; Mazzulli, J.R.; Bardgett, M.E.; Haik, K.L.; et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet 2013, 22, 2067–2082. [Google Scholar] [CrossRef] [PubMed]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Harvey, K.; Dominguez-Meijide, A.; Gerhardt, E. LRRK2, alpha-synuclein, and tau: Partners in crime or unfortunate bystanders? Biochem. Soc. Trans. 2019, 47, 827–838. [Google Scholar] [CrossRef]
- Cresto, N.; Gardier, C.; Gubinelli, F.; Gaillard, M.C.; Liot, G.; West, A.B.; Brouillet, E. The unlikely partnership between LRRK2 and α-synuclein in Parkinson’s disease. Eur. J. Neurosci. 2019, 49, 339–363. [Google Scholar] [CrossRef]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK 2 regulates endoplasmic reticulum–mitochondrial tethering through the PERK -mediated ubiquitination pathway. EMBO J. 2020, 39, e100875. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Galter, D. The MitoPark Mouse—An animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S185–S188. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, R.; Wang, G. Impact of Dopamine Oxidation on Dopaminergic Neurodegeneration. ACS Chem. Neurosci. 2019, 10, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine Oxidation and Autophagy. Parkinsons. Dis. 2012, 2012, 920953. [Google Scholar] [CrossRef] [PubMed]
- Kanďár, R. The ratio of oxidized and reduced forms of selected antioxidants as a possible marker of oxidative stress in humans. Biomed. Chromatogr. 2016, 30, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Yamamoto, Y.; Matsumoto, N.; Arai, Y.; Hirose, N. Increased oxidative stress and coenzyme Q10 deficiency in centenarians. J. Clin. Biochem. Nutr. 2018, 63, 129–136. [Google Scholar] [CrossRef]
- Sourris, K.C.; Harcourt, B.E.; Tang, P.H.; Morley, A.L.; Huynh, K.; Penfold, S.A.; Coughlan, M.T.; Cooper, M.E.; Nguyen, T.-V.; Ritchie, R.H.; et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic. Biol. Med. 2012, 52, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Melser, S.; Zottola, A.C.P.; Serrat, R.; Puente, N.; Grandes, P.; Marsicano, G.; Hebert-Chatelain, E. Functional Analysis of Mitochondrial CB1 Cannabinoid Receptors (mtCB1) in the Brain. Methods Enzymol. 2017, 593, 143–174. [Google Scholar]
- Valenzuela, R.; Costa-Besada, M.A.; Iglesias-Gonzalez, J.; Perez-Costas, E.; Villar-Cheda, B.; Garrido-Gil, P.; Melendez-Ferro, M.; Soto-Otero, R.; Lanciego, J.L.; Henrion, D.; et al. Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 2016, 7, e2427. [Google Scholar] [CrossRef]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciúncula, L.O. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J. Alzheimer’s Dis. JAD 2013, 34, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. Cafeína, receptores de adenosina, memoria y enfermedad de Alzheimer. Med. Clin. 2008, 131, 790–795. [Google Scholar] [CrossRef]
- Canas, P.M.; Porciuncula, L.O.; Cunha, G.M.A.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.A.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A Receptor Blockade Prevents Synaptotoxicity and Memory Dysfunction Caused by -Amyloid Peptides via p38 Mitogen-Activated Protein Kinase Pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S167–S174. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife coffee and tea drinking and the risk of late-life dementia: A population-based CAIDE study. J. Alzheimer’s Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Pedata, F.; Pugliese, A.M.; Melani, A.; Gianfriddo, M. A2A receptors in neuroprotection of dopaminergic neurons. Neurology 2003, 61, S49–S50. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Navarro, G. Adenosine A2A receptor antagonists in neurodegenerative diseases: Huge potential and huge challenges. Front. Psychiatry 2018, 9, 1–5. [Google Scholar] [CrossRef]
- Franco, R. Café y salud mental. Aten. Primaria 2009, 41, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.M.; Xu, Z.; Tan, L.C.S. Epidemiology of Parkinson’s Disease-East Versus West. Mov. Disord. Clin. Pract. 2018, 5, 14–28. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Ribeiro, J.A.; Sebastiao, A.M. Caffeine and adenosine. J. Alzheimer’s Dis. 2010, 20, S3–S15. [Google Scholar] [CrossRef]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr. Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Liu, R.; Guo, X.; Park, Y.; Huang, X.; Sinha, R.; Freedman, N.D.; Hollenbeck, A.R.; Blair, A.; Chen, H. Caffeine intake, smoking, and risk of parkinson disease in men and women. Am. J. Epidemiol. 2012, 175, 1200–1207. [Google Scholar] [CrossRef]



| Gene | Inheritance | Proposed Disease Mechanism | Disease Onset | Mutation | Frequency | Confidence as Actual PD Gene | Year of Discovery | Animal Model | |
|---|---|---|---|---|---|---|---|---|---|
| Early | Late | ||||||||
| SNCA | D | GoF or overexpression | Often with dementia | Missense or multiplication | Very rare | Very high | 1997,2003 | + | |
| LRRK2 | D | GoF | X | Missense | common | Very high | 2004 | + | |
| GBA | D | Likely LoF | X | Missense or LoF | common | Very high | 2009 | + | |
| UCHL1 | D | LoF | NA | Missense | unclear | Low | 1998 | + | |
| VPS35 | D | LoF | X | Missense | Very rare | Very high | 2011 | + | |
| PRKN | R | LoF | X | Missense or LoF | rare | Very high | 1998 | + | |
| PINK1 | R | LoF | X | Missense or loss of function | rare | Very high | 2004 | + | |
| ATP13A2 | R | LoF | Atypical PD | Missense or LoF | Very rare | Very high | 2006 | + | |
| PLA2G6 | R | LoF | X | Missense or LoF | rare | Very high | 2009 | NA | |
| DNAJC6 | R | LoF | X | Missense o LoF | Very rare | High | 2012 | NA | |
| SYNJ1 | R | LoF | (often) Atypical PD | Missense or loss of function | Very rare | Very high | 2013 | NA | |
| DJ-1/PARK7 | R | LoF | X | Missense | Very rare | Very High | 2003 | + | |
| FBXO7 | R | LoF | X | Missense | Very rare | Very High | 2008 | NA | |
| Gene | Ensembl Identifier | Protein | Function (Information Obtained from STRING Data Base) |
|---|---|---|---|
| SNCA | ENSP00000338345 | α-synuclein | Involved in regulating dopamine release and transport. Induces the fibrillization of tau protein. Reduces neuronal responsiveness to different apoptotic stimuli, thus promoting a decreased caspase-3 activation. |
| LRRK2 | ENSP00000298910 | Leucine-rich repeat serine/threonine-protein kinase 2 | Regulates autophagy in a positive way by means of the calcium- dependent activation of the CaMKK/AMPK pathway, also involving the activation of nicotinic acid adenine dinucleotide phosphate (NAADP) receptors, increases in lysosomal pH, and release of Ca++ from lysosomes. |
| GBA | ENSP00000314508 | Glucosylceramidase beta | Involved in the hydrolization of glucocerebroside. Localized in lysosomes. |
| UCHL1 | ENSP00000284440 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | Deubiquitinating enzyme, generates ubiquitin monomers. Might prevent the degradation of monoubiquitin in lysosomes. Its expression is highly specific to neurons and to cells of the diffuse neuroendocrine system and their tumors. |
| VPS35 | ENSP00000299138 | Vacuolar protein sorting-associated protein 35 | Involved in autophagy. Is part of the retromer cargo-selective complex (CSC), which is responsible for transporting select cargo proteins between vesicular structures (e.g., endosomes, lysosomes, vacuoles) and the Golgi apparatus. |
| PRKN | ENSP00000355865 | E3 ubiquitin-protein ligase parkin | Ubiquitin ligase; covalently binds ubiquitin residues onto proteins. Involved in the removal of abnormally folded or damaged proteins thanks to ‘Lys-63’-linked polyubiquitination of misfolded proteins. |
| PINK1 | ENSP00000364204 | Serine/threonine-protein kinase PINK1 | Localized in mitochondria. Protects cells against stress-induced mitochondrial dysfunction by phosphorylating mitochondrial proteins. By means of activation and translocation of PRKN participates in the clearance of damaged mitochondria via selective autophagy (mitophagy). |
| ATP13A2 | ENSP00000327214 | Cation-transporting ATPase 13A2 | ATPase involved in the transport of divalent transition metal cations and the maintenance of neuronal integrity. It is necessary for a correct lysosomal and mitochondrial maintenance. |
| PLA2G6 | ENSP00000333142 | 85/88 kDa calcium-independent phospholipase A2 | Involved in the release of fatty acids from phospholipids. Implicated in normal phospholipid remodeling. It has also been involved in NO- or vasopressin-induced arachidonic acid release and in leukotriene and prostaglandin production. |
| DNAJC6 | ENSP00000360108 | Putative tyrosine-protein phosphatase auxilin | Promotes uncoating of clathrin-coated vesicles by recruiting HSPA8/HSC70 to clathrin-coated vesicles. Involved in clathrin-mediated endocytosis in neurons. |
| SYNJ1 | ENSP00000409667 | Synaptojanin-1 | Phosphoinositide phosphatase, regulates levels of membrane phosphatidylinositol-4,5-bisphosphate (PIP2). Involved in the rearrangement of actin filaments downstream of tyrosine kinase and ASH/GRB2 by means of hydrolyzing PIP2 bound to actin regulatory proteins. |
| DJ-1/ PARK7 | ENSP00000418770 | Protein/nucleic acid deglycase DJ-1 | Under an oxidative condition, via its chaperone activity, inhibits the aggregation of α-synuclein, thus functioning as a redox-sensitive chaperone and as a sensor for oxidative stress. Deglycates proteins and nucleotides, and the Maillard adducts formed between amino groups of proteins or nucleotides and reactive carbonyl groups of glyoxals. |
| FBXO7 | ENSP00000266087 | F-box only protein 7 | Part of a SCF (SKP1-CUL1-F- box protein) E3 ubiquitin-protein ligase complex involved in protein ubiquitination. Role in the clearance of damaged mitochondria (mitophagy). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, R.; Rivas-Santisteban, R.; Navarro, G.; Pinna, A.; Reyes-Resina, I. Genes Implicated in Familial Parkinson’s Disease Provide a Dual Picture of Nigral Dopaminergic Neurodegeneration with Mitochondria Taking Center Stage. Int. J. Mol. Sci. 2021, 22, 4643. https://doi.org/10.3390/ijms22094643
Franco R, Rivas-Santisteban R, Navarro G, Pinna A, Reyes-Resina I. Genes Implicated in Familial Parkinson’s Disease Provide a Dual Picture of Nigral Dopaminergic Neurodegeneration with Mitochondria Taking Center Stage. International Journal of Molecular Sciences. 2021; 22(9):4643. https://doi.org/10.3390/ijms22094643
Chicago/Turabian StyleFranco, Rafael, Rafael Rivas-Santisteban, Gemma Navarro, Annalisa Pinna, and Irene Reyes-Resina. 2021. "Genes Implicated in Familial Parkinson’s Disease Provide a Dual Picture of Nigral Dopaminergic Neurodegeneration with Mitochondria Taking Center Stage" International Journal of Molecular Sciences 22, no. 9: 4643. https://doi.org/10.3390/ijms22094643
APA StyleFranco, R., Rivas-Santisteban, R., Navarro, G., Pinna, A., & Reyes-Resina, I. (2021). Genes Implicated in Familial Parkinson’s Disease Provide a Dual Picture of Nigral Dopaminergic Neurodegeneration with Mitochondria Taking Center Stage. International Journal of Molecular Sciences, 22(9), 4643. https://doi.org/10.3390/ijms22094643