
Nuclear FGFR2 Interacts with the MLL-AF4 Oncogenic Chimera and Positively Regulates HOXA9 Gene Expression in t(4;11) Leukemia Cells


 , , ,
, , ,  ,
,  and
and 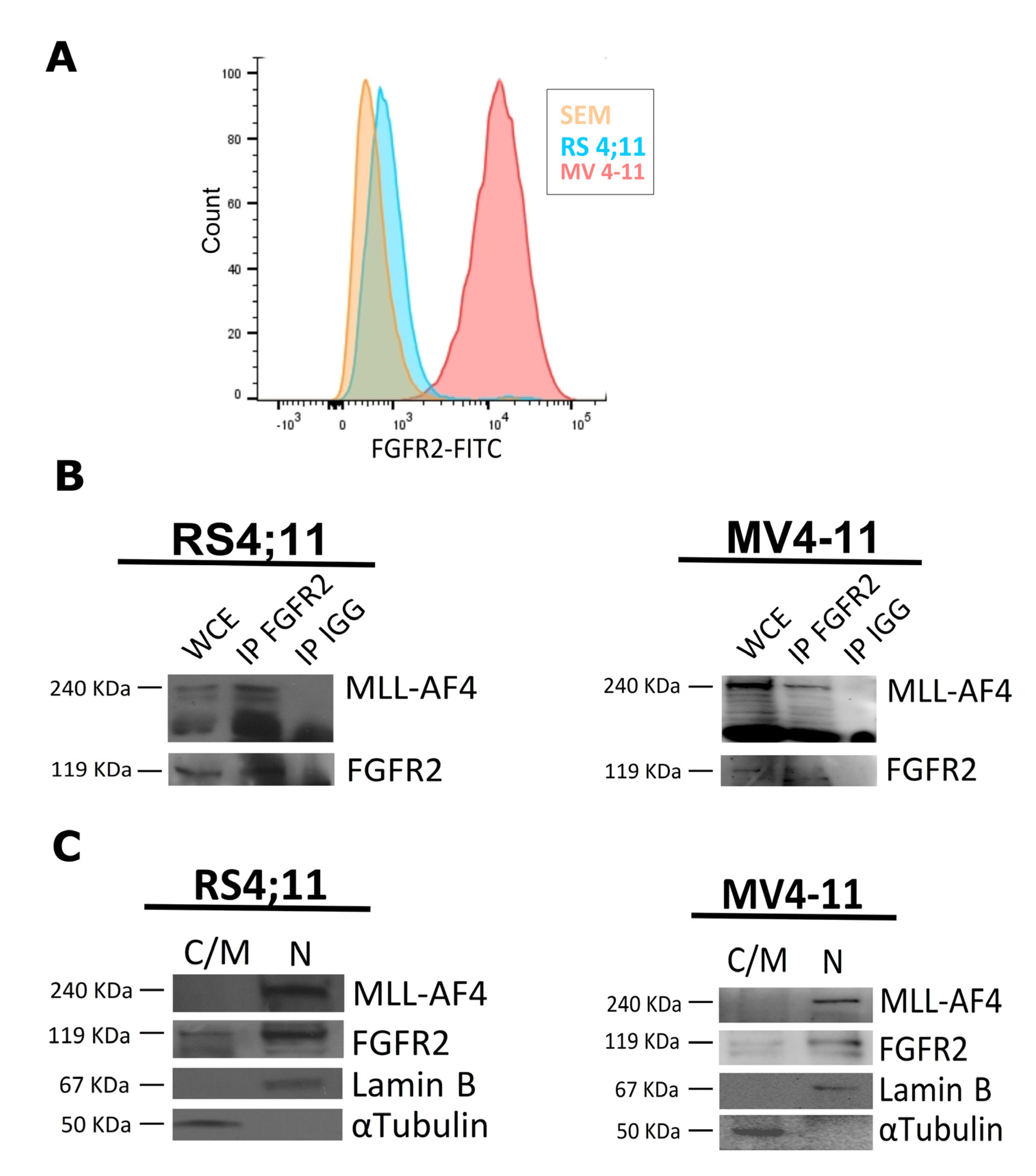{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
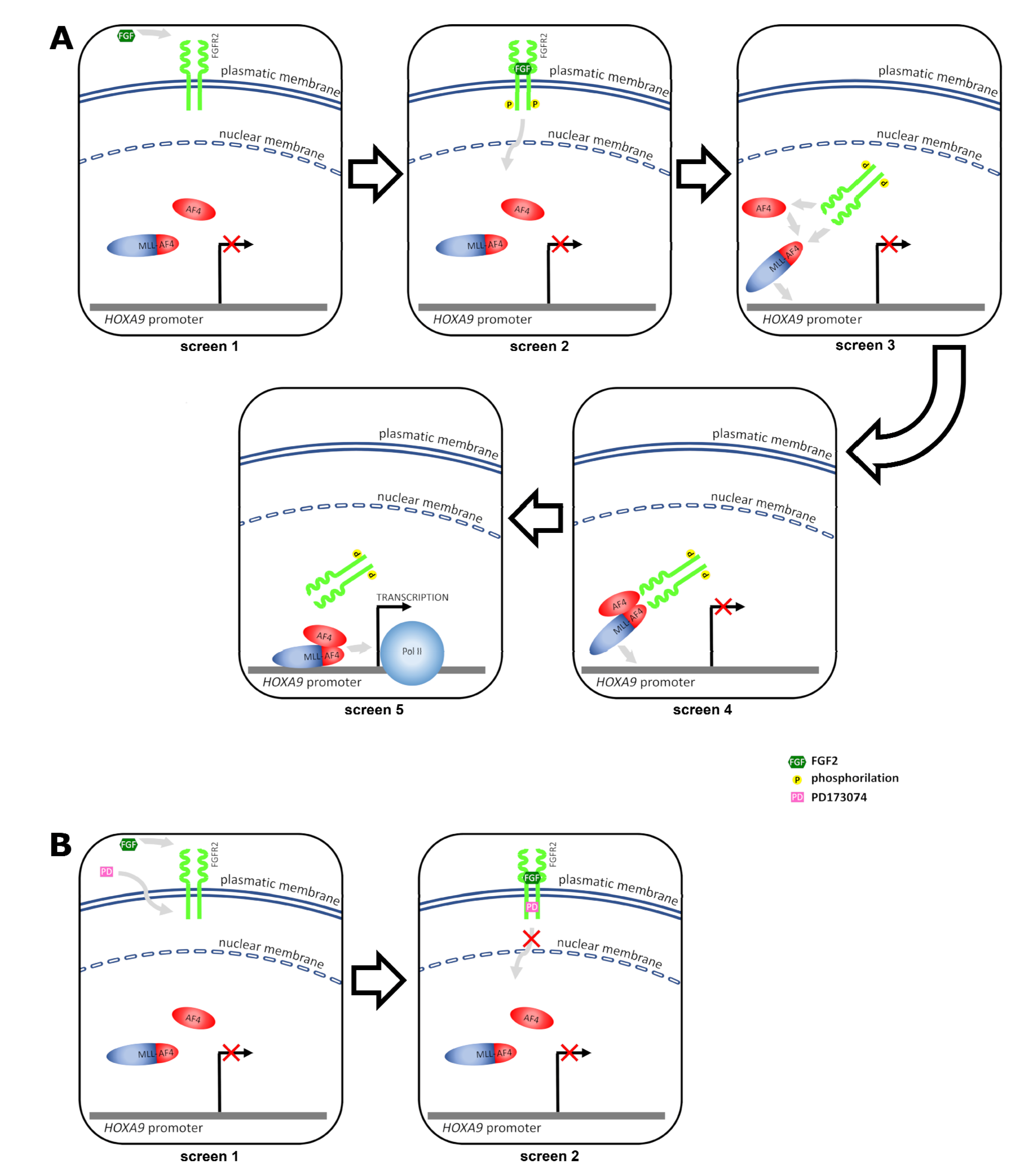
2.1. FGFR2 Is a Nuclear Interactor of MLL-AF4
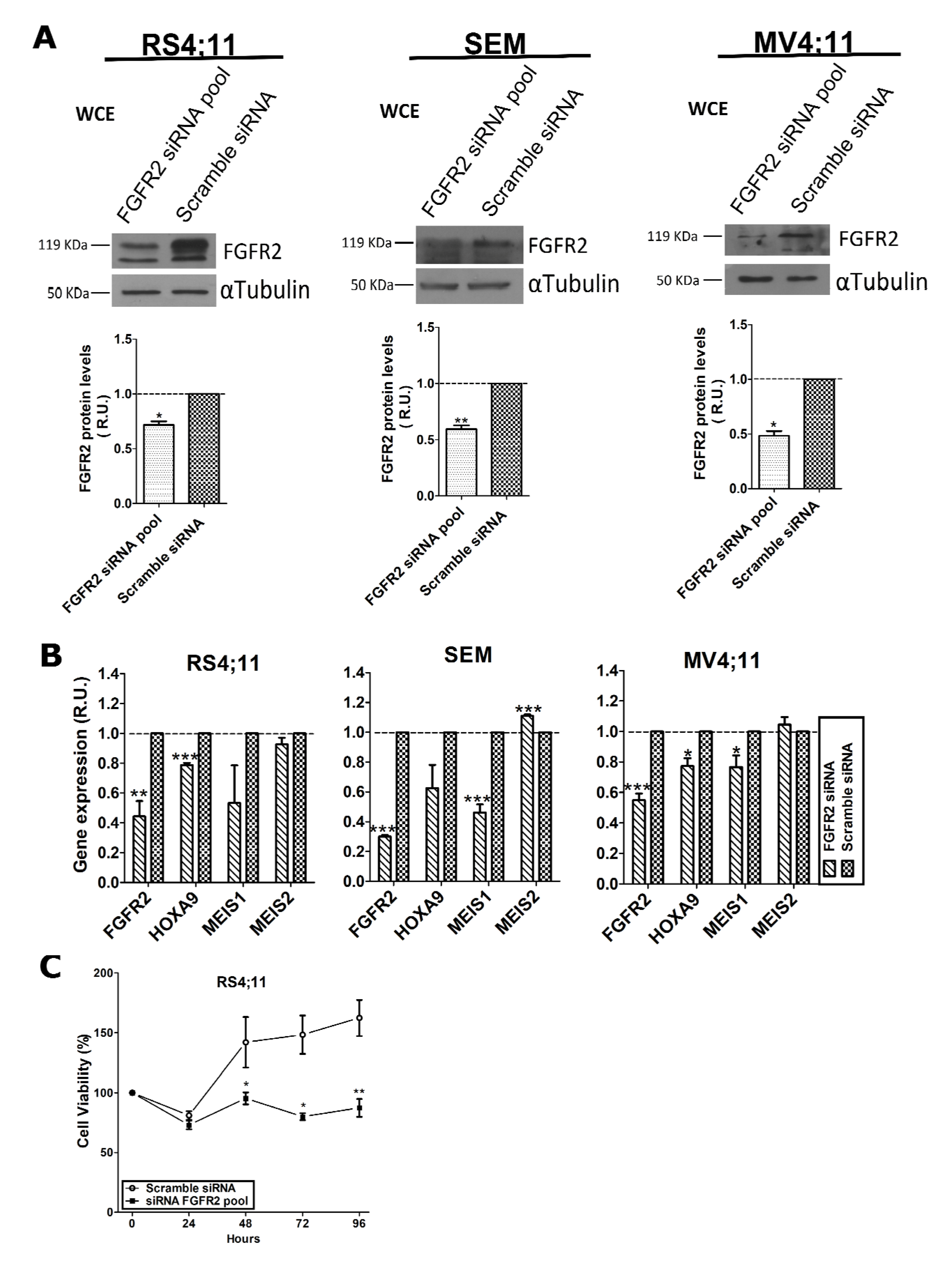
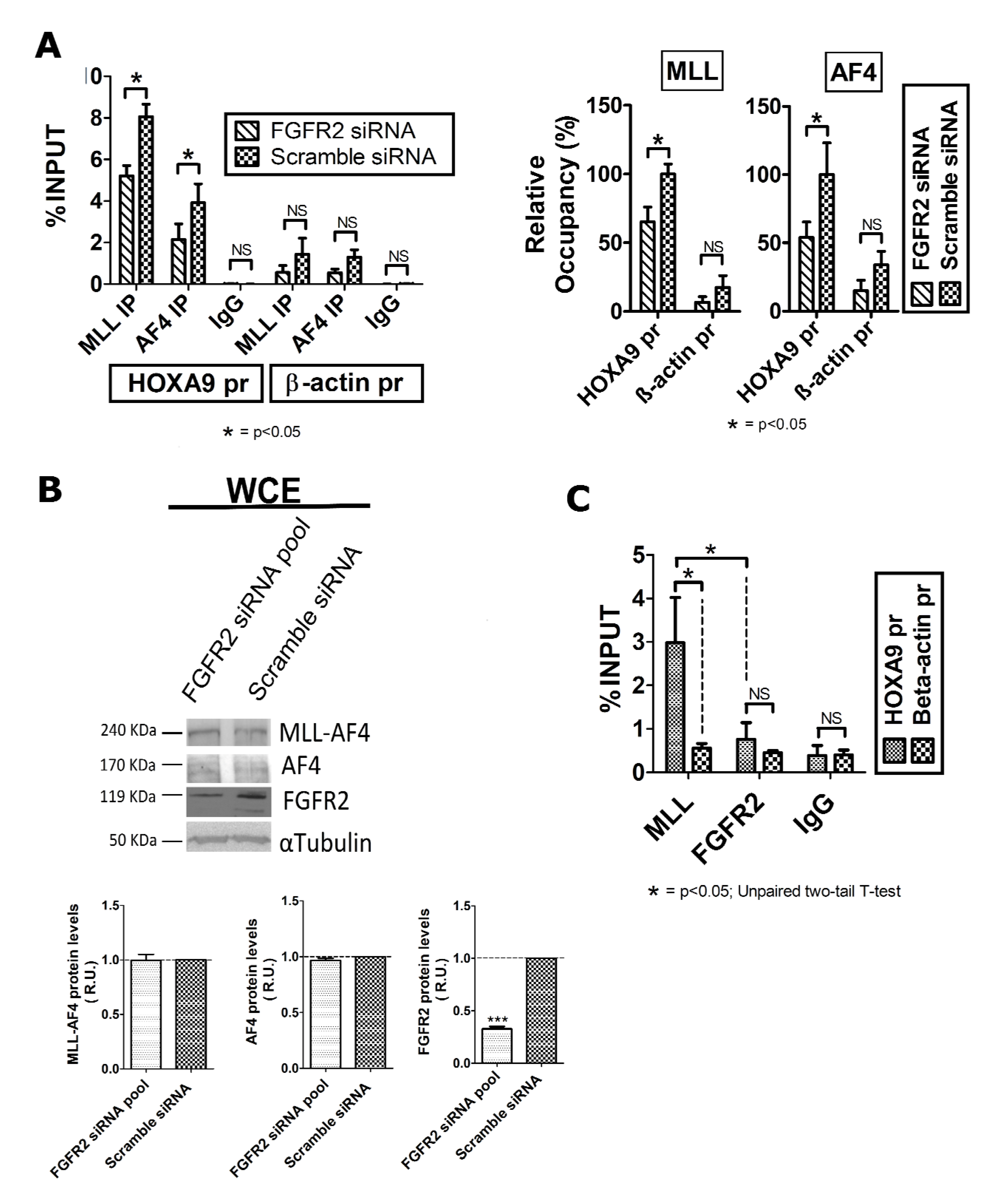
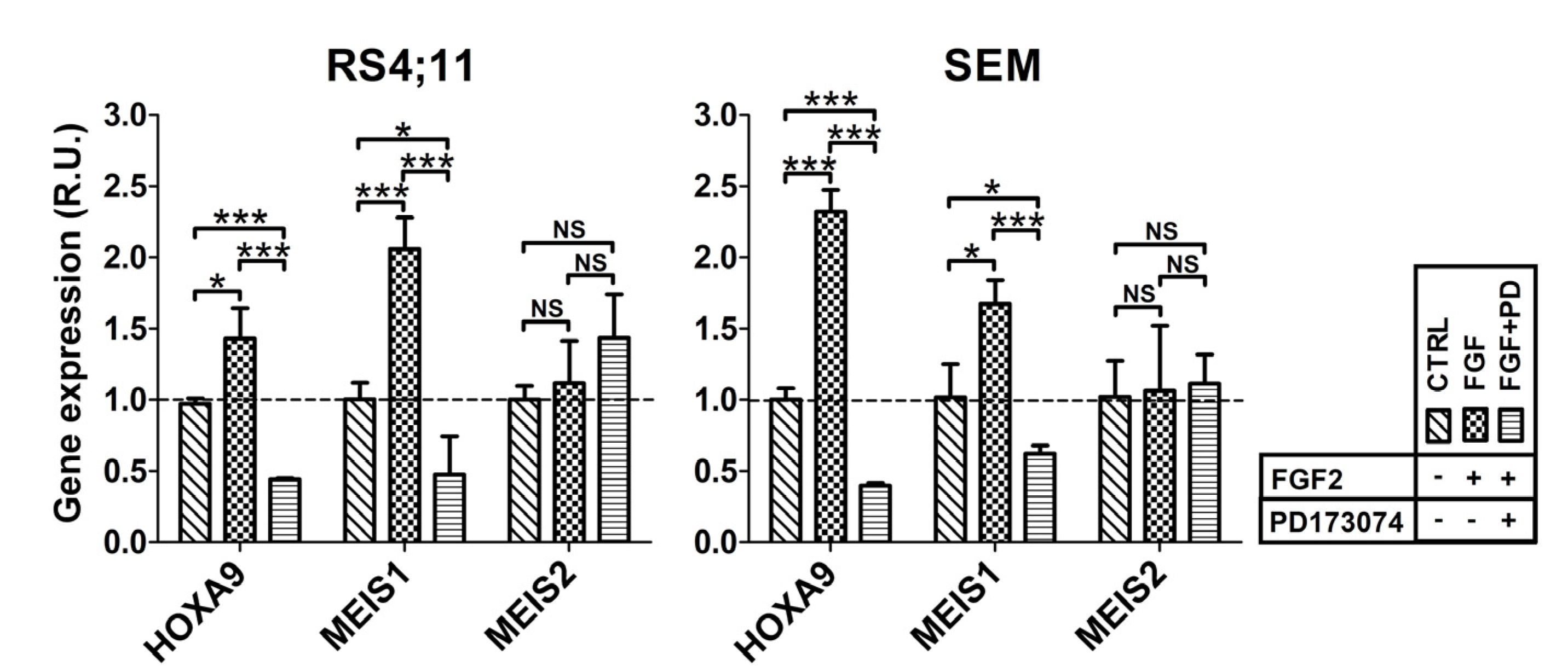
2.2. FGFR2 Silencing Affects Expression of MLL-AF4 Target Genes
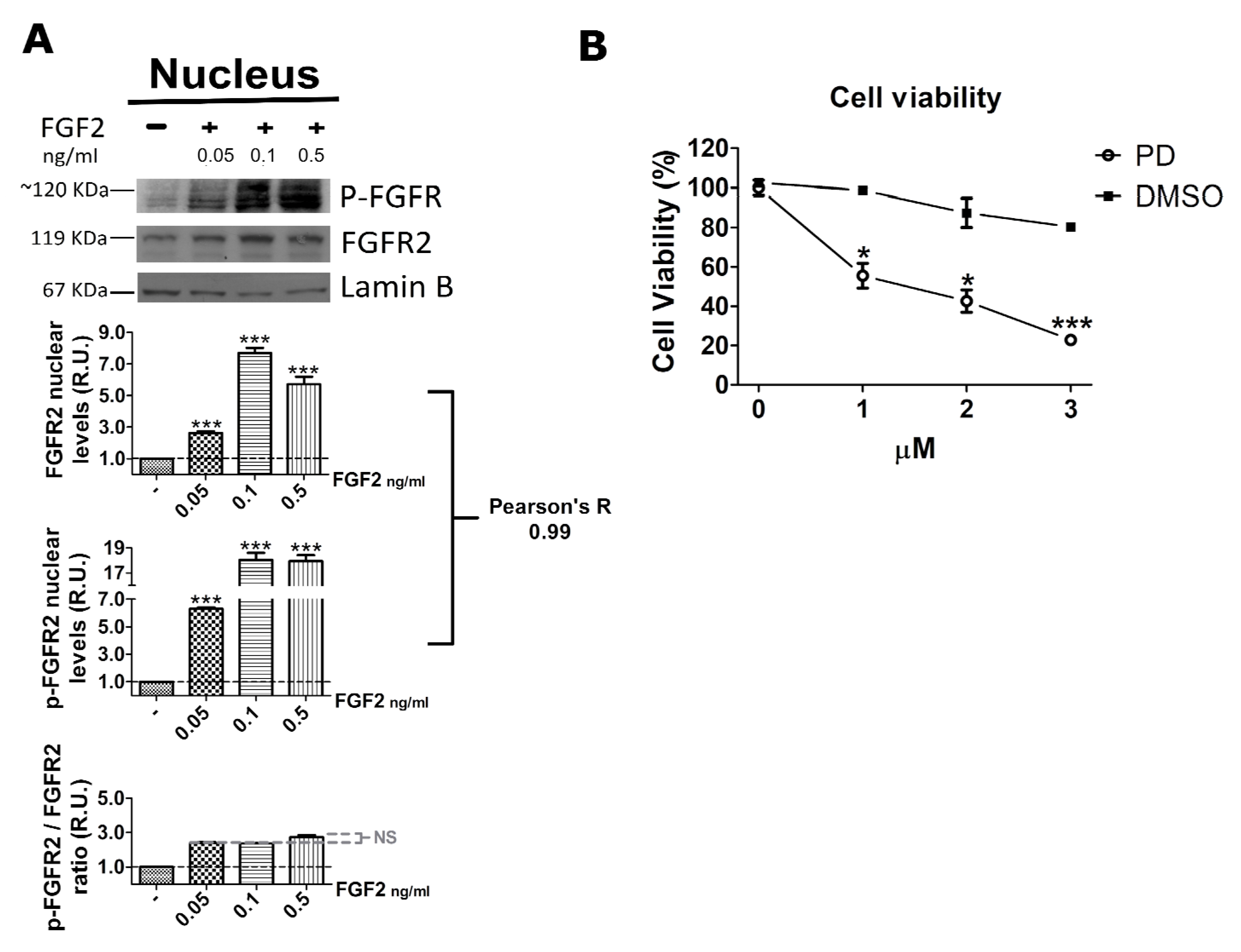
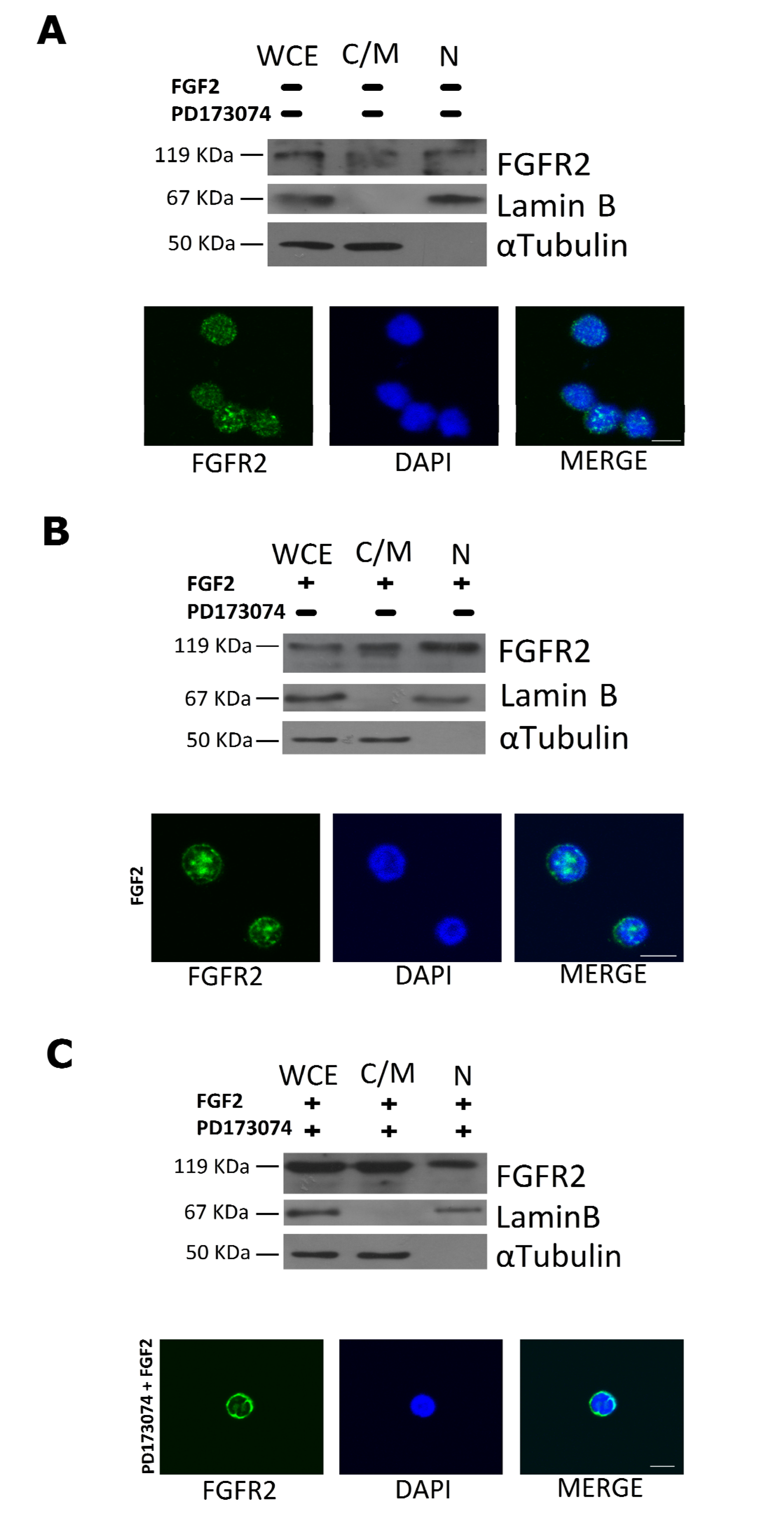
2.3. FGF2 Stimulates Nuclear Localization of FGFR2
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Lines
4.3. Flow Cytometry
4.4. Protein Extraction, Subcellular Fraction Isolation, Immunoprecipitation, and Western Blot Analysis
4.5. Small Interfering RNA (SiRNA)
4.6. Total RNA Isolation, Reverse Transcription (RT), and Real Time Polymerase Chain Reaction (PCR)
4.7. Cell Viability Assays
4.8. Chromatin Immunoprecipitation (ChIP) Assay
4.9. Treatment of t(4;11) Leukemia Cells with FGF2 and PD173074 Inhibitor
4.10. Immunofluorescence Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daser, A.; Rabbitts, T.H. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin. Cancer Biol. 2005, 15, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Gröger, D.; Park, T.S.; Emerenciano, M.; Pombo de Oliveira, M.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef]
- Bursen, A.; Schwabe, K.; Rüster, B.; Henschler, R.; Ruthardt, M.; Dingermann, T.; Marschalek, R. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood 2010, 115, 3570–3579. [Google Scholar] [CrossRef]
- Prieto, C.; Marschalek, R.; Kühn, A.; Bursen, A.; Bueno, C.; Menéndez, P. The AF4-MLL fusion transiently augments multilineage hematopoietic engraftment but is not sufficient to initiate leukemia in cord blood CD34+ cells. Oncotarget 2017, 8, 81936–81941. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wilkinson, A.C.; Ballabio, E.; Geng, H.; North, P.; Tapia, M.; Kerry, J.; Biswas, D.; Roeder, R.G.; Allis, C.D.; Melnick, A.; et al. RUNX1 Is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep. 2013, 3, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Bueno, C.; Calero-Nieto, F.J.; Wang, X.; Valdés-Mas, R.; Gutiérrez-Agüera, F.; Roca-Ho, H.; Ayllon, V.; Real, P.J.; Arambile, D.; Espinosa, L.; et al. Enhanced hemato-endothelial specification during human embryonic differentiation through developmental cooperation between AF4-MLL and MLL-AF4 fusions. Haematologica 2019, 104, 1189–1201. [Google Scholar] [CrossRef]
- Britten, O.; Ragusa, D.; Tosi, S.; Kamel, Y.M. MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)—Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells 2019, 8, 1341. [Google Scholar] [CrossRef]
- Kowarz, E.; Burmeister, T.; Lo Nigro, L.; Jansen, M.W.; Delabesse, E.; Klingebiel, T.; Dingermann, T.; Meyer, C.; Marschalek, R. Complex MLL rearrangements in t(4;11) leukemia patients with absent AF4.MLL fusion allele. Leukemia 2007, 21, 1232–1238. [Google Scholar] [CrossRef]
- Thomas, M.; Gessner, A.; Vornlocher, H.P.; Hadwiger, P.; Greil, J.; Heidenreich, O. Targeting MLL-AF4 with short interfering RNAs inhibits clonogenicity and engraftment of t(4;11)-positive human leukemic cells. Blood 2005, 106, 3559–3566. [Google Scholar] [CrossRef]
- Marschalek, R. Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol. 2011, 152, 141–154. [Google Scholar] [CrossRef]
- Godfrey, L.; Kerry, J.; Thorne, R.; Repapi, E.; Davies, J.O.; Tapia, M.; Ballabio, E.; Hughes, J.R.; Geng, H.; Konopleva, M.; et al. MLL-AF4 binds directly to a BCL-2 specific enhancer and modulates H3K27 acetylation. Exp. Hematol. 2016, 47, 64–75. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Collins, C.T.; Hess, J.L. Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr. Opin. Hematol. 2016, 23, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; Schneider, P.; Hagelstein, J.A.P.; van der Linden, M.H.; Stumpel, D.J.P.M.; de Menezes, R.X.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010, 115, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A. Transcriptional activation by MLL fusion proteins in leukemogenesis. Exp. Hematol. 2016, 46, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Orlovsky, K.; Kalinkovich, A.; Rozovskaia, T.; Shezen, E.; Itkin, T.; Alder, H.; Ozer, H.G.; Carramusa, L.; Avigdor, A.; Volinia, S.; et al. Down-regulation of homeobox genes MEIS1 and HOXA in MLL-rearranged acute leukemia impairs engraftment and reduces proliferation. Proc. Natl. Acad. Sci. USA 2011, 108, 7956–7961. [Google Scholar] [CrossRef]
- Yokoyama, A.; Lin, M.; Naresh, A.; Kitabayashi, I.; Cleary, M.L. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 2010, 17, 198–212. [Google Scholar] [CrossRef]
- Fioretti, T.; Cevenini, A.; Zanobio, M.; Raia, M.; Sarnataro, D.; Salvatore, F.; Esposito, G. Crosstalk between 14-3-3θ and AF4 enhances MLL-AF4 activity and promotes leukemia cell proliferation. Cell. Oncol. 2019, 42, 829–845. [Google Scholar] [CrossRef]
- Okuda, H.; Stanojevic, B.; Kanai, A.; Kawamura, T.; Takahashi, S.; Matsui, H.; Takaori-Kondo, A.; Yokoyama, A. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Investig. 2017, 127, 1918–1931. [Google Scholar] [CrossRef]
- Esposito, G.; Cevenini, A.; Cuomo, A.; de Falco, F.; Sabbatino, D.; Pane, F.; Ruoppolo, M.; Salvatore, F. Protein network study of human AF4 reveals its central role in RNA Pol II-mediated transcription and in phosphorylation-dependent regulatory mechanisms. Biochem. J. 2011, 438, 121–131. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Lonic, A.; Barry, E.; Quach, C.; Kobe, B.; Saunders, N.; Guthridge, M.A. Fibroblast Growth Factor Receptor 2 Phosphorylation on Serine 779 Couples to 14-3-3 and Regulates Cell Survival and Proliferation. Mol. Cell. Biol. 2008, 28, 3372–3385. [Google Scholar] [CrossRef][Green Version]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Montor, W.R.; Salas, A.R.O.S.E.; Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 55. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed]
- Korc, M.; Friesel, R.E. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targets 2009, 9, 639–651. [Google Scholar] [CrossRef]
- Esposito, M.T. The Impact of PI3-kinase/RAS Pathway Cooperating Mutations in the Evolution of KMT2A-rearranged Leukemia. HemaSphere 2019, 3, e195. [Google Scholar] [CrossRef]
- Whelan, J.T.; Ludwig, D.L.; Bertrand, F.E. HoxA9 induces insulin-like growth factor-1 receptor expression in B-lineage acute lymphoblastic leukemia. Leukemia 2008, 22, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Nakamura, T.; Canaani, E.; Croce, C.M. ALL1 fusion proteins induce deregulation of EphA7 and ERK phosphorylation in human acute leukemias. Proc. Natl. Acad. Sci. USA 2007, 104, 14442–14447. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, M.K.; Maher, P.A.; Stachowiak, E.K. Integrative nuclear signaling in cell development—A role for FGF receptor-1. DNA Cell Biol. 2007, 26, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Cerliani, J.P.; Guillardoy, T.; Giulianelli, S.; Vaque, J.P.; Gutkind, J.S.; Vanzulli, S.I.; Martins, R.; Zeitlin, E.; Lamb, C.A.; Lanari, C. Interaction between FGFR-2, STAT5, and progesterone receptors in breast cancer. Cancer Res. 2011, 71, 3720–3731. [Google Scholar] [CrossRef] [PubMed]
- Tuzon, C.T.; Rigueur, D.; Merrill, A.E. Nuclear Fibroblast Growth Factor Receptor Signaling in Skeletal Development and Disease. Curr. Osteoporos. Rep. 2019, 17, 138–146. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Fioretti, T.; Sarnataro, D.; Esposito, G.; Ammendola, R. Nuclear localization of Formyl-Peptide Receptor 2 in human cancer cells. Arch. Biochem. Biophys. 2016, 603, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Liao, H.J. Receptor tyrosine kinases in the nucleus. Cold Spring Harb. Perspect. Biol. 2013, 5, a008979. [Google Scholar] [CrossRef]
- Porębska, N.; Latko, M.; Kucińska, M.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Targeting cellular trafficking of fibroblast growth factor receptors as a strategy for selective cancer treatment. J. Clin. Med. 2018, 8, 7. [Google Scholar] [CrossRef]
- Kerry, J.; Godfrey, L.; Repapi, E.; Tapia, M.; Blackledge, N.P.; Ma, H.; Ballabio, E.; O’Byrne, S.; Ponthan, F.; Heidenreich, O.; et al. MLL-AF4 spreading identifies binding sites that are distinct from super-enhancers and that govern sensitivity to DOT1L inhibition in leukemia. Cell Rep. 2017, 18, 482–495. [Google Scholar] [CrossRef]
- Martin, A.J.; Grant, A.; Ashfield, A.M.; Palmer, C.N.; Baker, L.; Quinlan, P.R.; Purdie, C.A.; Thompson, A.M.; Jordan, L.B.; Berg, J.B. FGFR2 protein expression in breast cancer: Nuclear localisation and correlation with patient genotype. BMC Res. Notes 2011, 4, 72. [Google Scholar] [CrossRef]
- Schmahl, J.; Kim, Y.; Colvin, J.S.; Ornitz, D.M.; Capel, B. Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 2004, 131, 3627–3636. [Google Scholar] [CrossRef]
- Bansal, R.; Magge, S.; Winkler, S. Specific inhibitor of FGF receptor signaling: FGF-2-mediated effects on proliferation, differentiation, and MAPK activation are inhibited by PD173074 in oligodendrocyte-lineage cells. J. Neurosci. Res. 2003, 74, 486–493. [Google Scholar] [CrossRef]
- Pardo, O.E.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.R.; Stamp, G.W.; Aboagye, E.O.; Seckl, M.J. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [PubMed]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Boer, J.M.; Besselink, N.J.M.; Koudijs, M.J.; Pieters, R.; den Boer, M.L. Fibroblast growth factor receptor signaling in pediatric B-cell precursor acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 1875. [Google Scholar] [CrossRef]
- Kasbekar, M.; Nardi, V.; Dal Cin, P.; Brunner, A.M.; Burke, M.; Chen, Y.B.; Connolly, C.; Fathi, A.T.; Foster, J.; Macrae, M.; et al. Targeted FGFR inhibition results in a durable remission in an FGFR1-driven myeloid neoplasm with eosinophilia. Blood Adv. 2020, 4, 3136–3140. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, M.K.; Birkaya, B.; Aletta, J.M.; Narla, S.T.; Benson, C.A.; Decker, B.; Stachowiak, E.K. Nuclear FGF receptor-1 and CREB binding protein: An integrative signaling module. J. Cell. Physiol. 2015, 230, 989–1002. [Google Scholar] [CrossRef]
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; van den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009, 113, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Castaldo, M.; Parisi, M.; Faraonio, R.; Esposito, G.; Ammendola, R. Formyl Peptide Receptor 1 Modulates Endothelial Cell Functions by NADPH Oxidase-Dependent VEGFR2 Transactivation. Oxid. Med. Cell. Longev. 2018, 2018, 2609847. [Google Scholar] [CrossRef]
- Castaldo, M.; Zollo, C.; Esposito, G.; Ammendola, R.; Cattaneo, F. NOX2-Dependent Reactive Oxygen Species Regulate Formyl-Peptide Receptor 1-Mediated TrkA Transactivation in SH-SY5Y Cells. Oxid. Med. Cell. Longev. 2019, 2019, 2051235. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Russo, R.; Castaldo, M.; Chambery, A.; Zollo, C.; Esposito, G.; Pedone, P.V.; Ammendola, R. Phosphoproteomic analysis sheds light on intracellular signaling cascades triggered by Formyl-Peptide Receptor 2. Sci. Rep. 2019, 9, 17894. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fioretti, T.; Cevenini, A.; Zanobio, M.; Raia, M.; Sarnataro, D.; Cattaneo, F.; Ammendola, R.; Esposito, G. Nuclear FGFR2 Interacts with the MLL-AF4 Oncogenic Chimera and Positively Regulates HOXA9 Gene Expression in t(4;11) Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 4623. https://doi.org/10.3390/ijms22094623
Fioretti T, Cevenini A, Zanobio M, Raia M, Sarnataro D, Cattaneo F, Ammendola R, Esposito G. Nuclear FGFR2 Interacts with the MLL-AF4 Oncogenic Chimera and Positively Regulates HOXA9 Gene Expression in t(4;11) Leukemia Cells. International Journal of Molecular Sciences. 2021; 22(9):4623. https://doi.org/10.3390/ijms22094623
Chicago/Turabian StyleFioretti, Tiziana, Armando Cevenini, Mariateresa Zanobio, Maddalena Raia, Daniela Sarnataro, Fabio Cattaneo, Rosario Ammendola, and Gabriella Esposito. 2021. "Nuclear FGFR2 Interacts with the MLL-AF4 Oncogenic Chimera and Positively Regulates HOXA9 Gene Expression in t(4;11) Leukemia Cells" International Journal of Molecular Sciences 22, no. 9: 4623. https://doi.org/10.3390/ijms22094623
APA StyleFioretti, T., Cevenini, A., Zanobio, M., Raia, M., Sarnataro, D., Cattaneo, F., Ammendola, R., & Esposito, G. (2021). Nuclear FGFR2 Interacts with the MLL-AF4 Oncogenic Chimera and Positively Regulates HOXA9 Gene Expression in t(4;11) Leukemia Cells. International Journal of Molecular Sciences, 22(9), 4623. https://doi.org/10.3390/ijms22094623