
Combination of Niraparib, Cisplatin and Twist Knockdown in Cisplatin-Resistant Ovarian Cancer Cells Potentially Enhances Synthetic Lethality through ER-Stress Mediated Mitochondrial Apoptosis Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Twist Expression Is Strongly Correlated with Expression of PARP1 Involved in DNA Damage Response and Repair in CisR OC Cells
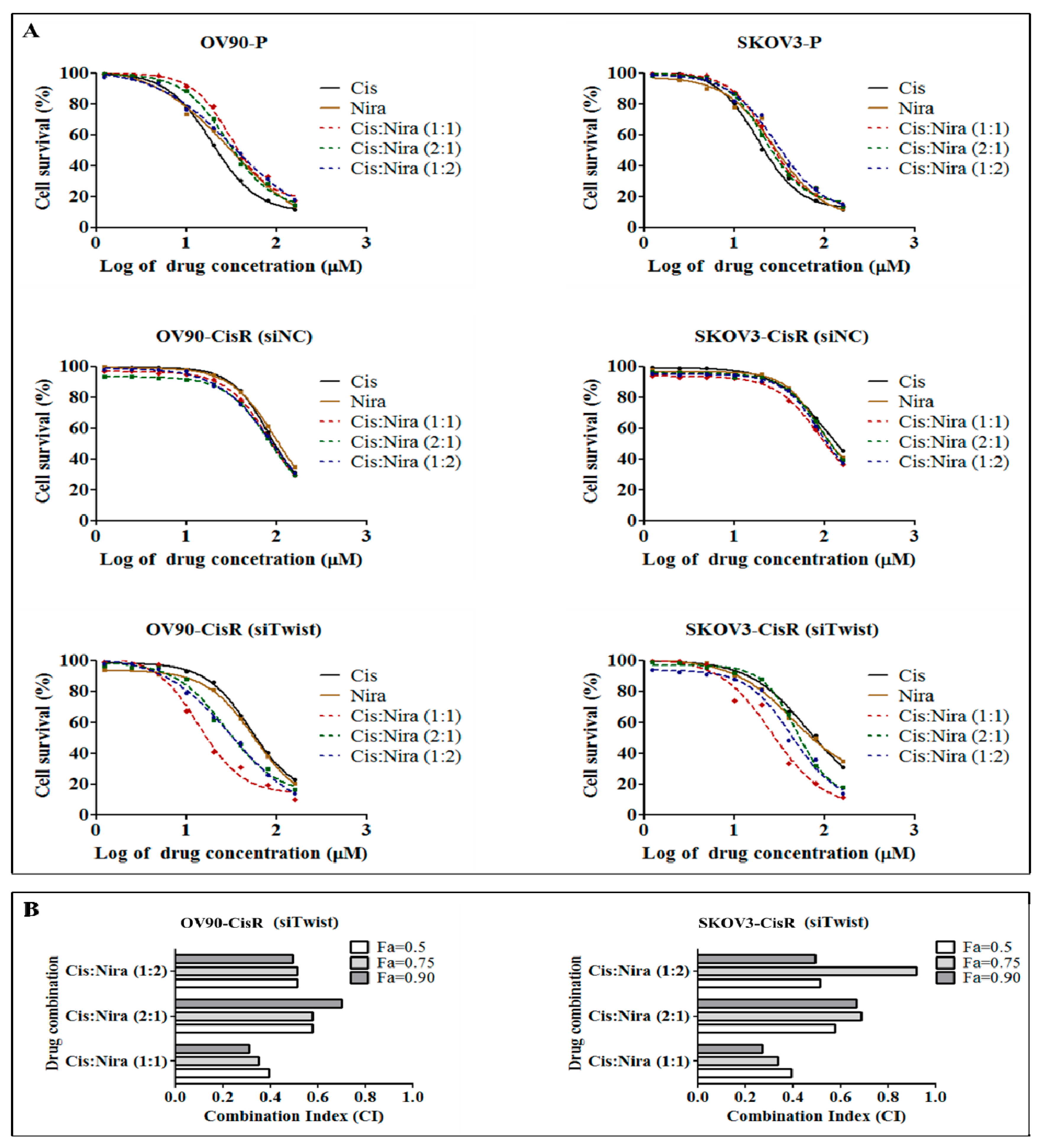
2.2. The Twist Deficient Cisplatin-Resistant OC Cells Were More Susceptible to Cisplatin and PARPi
2.3. Combination Therapy Exhibits Synergetic Effect with Suppression of DNA Damage Response Capacity and Inducing Apoptosis on Twist Deficient Cisplatin-Resistant OC Cells
2.4. Combination Therapy Is Considerably Effective against Three-Dimensional Cultured of Twist Knockdown CisR OC Cells
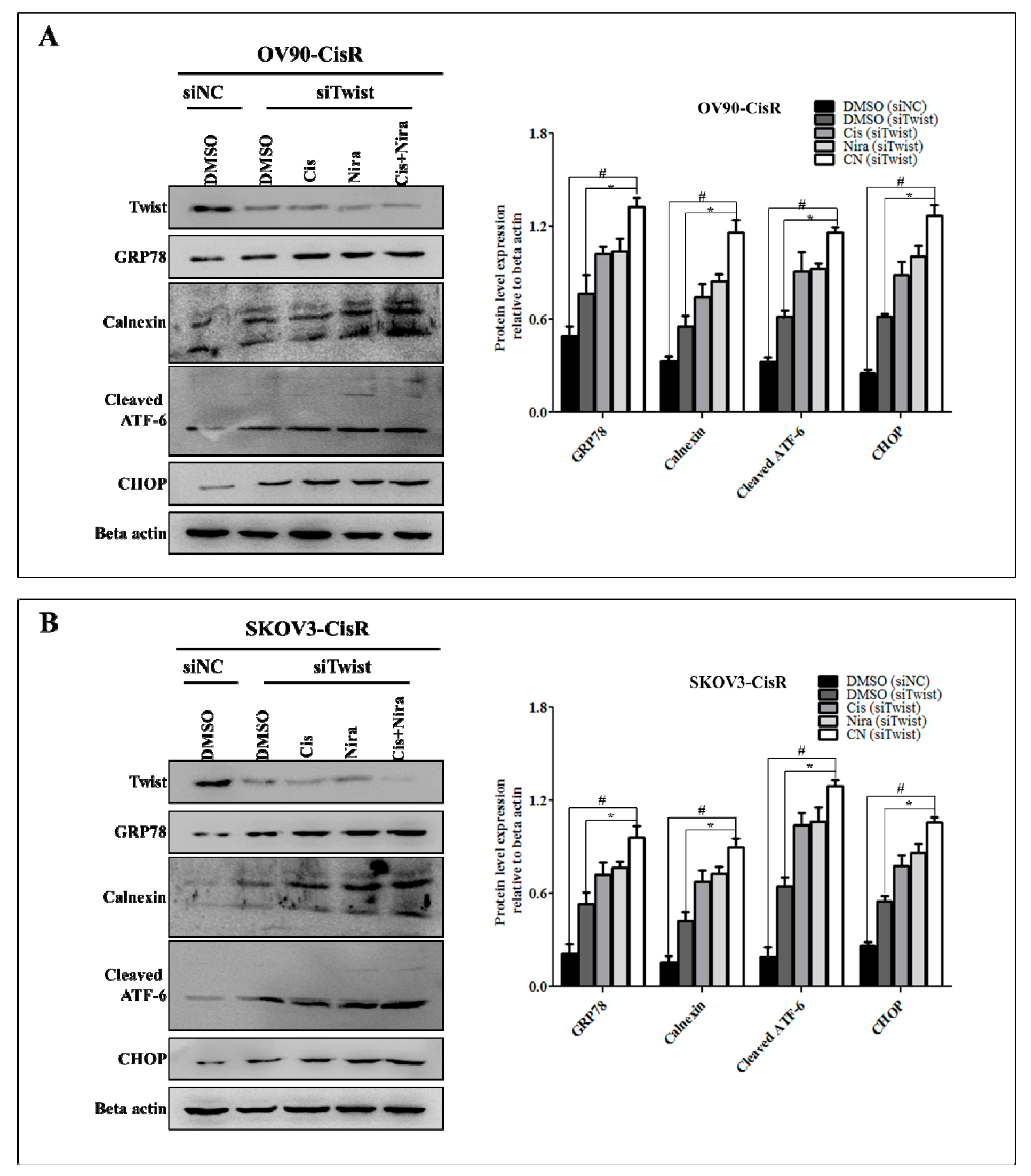
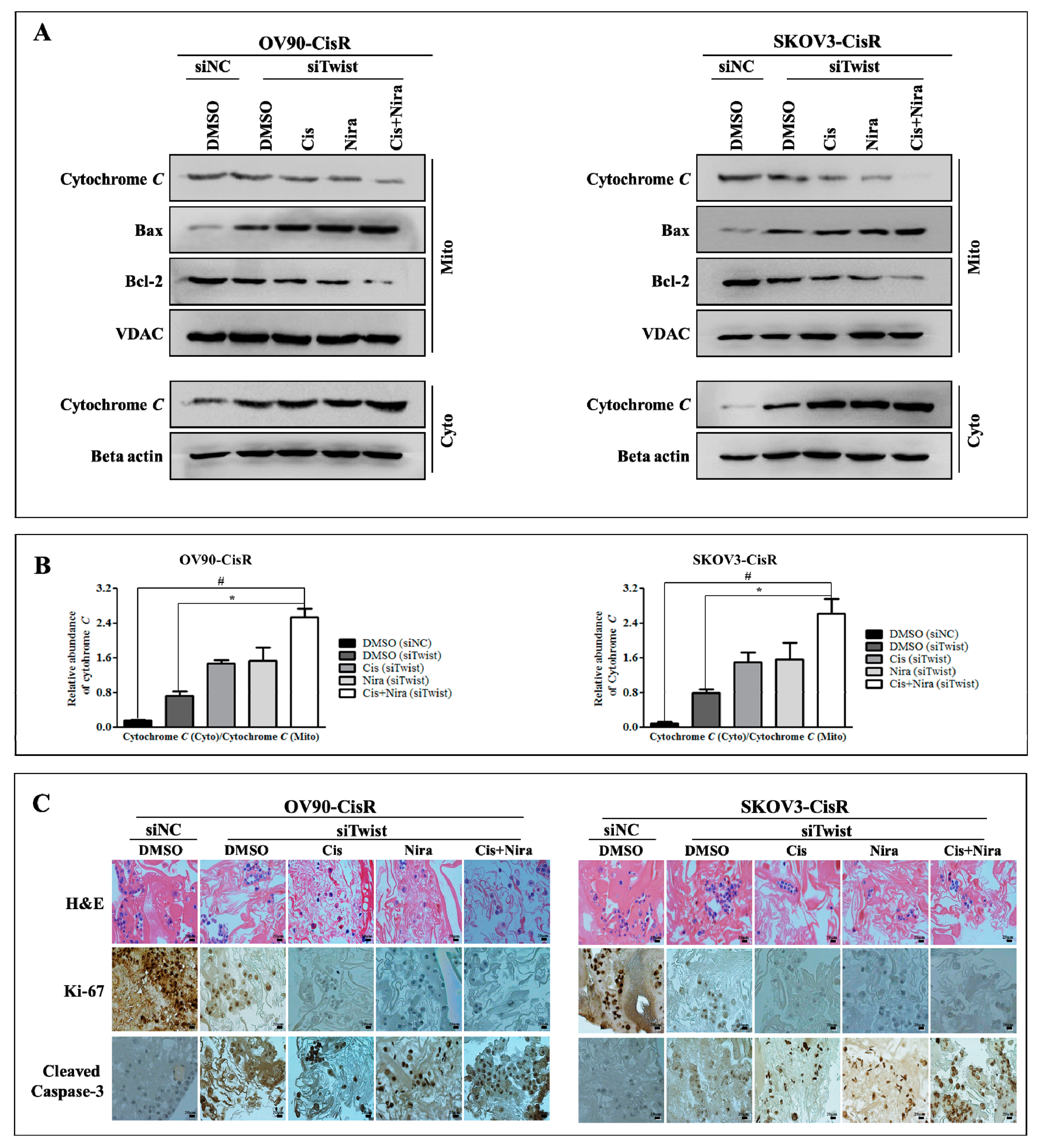
2.5. Cancer Cell-Death by Cisplatin and Niraparib Accomplished through Boosting ER Stress-Mediated Mitochondrial-Depended Apoptosis on Three-Dimensional Cultured of Twist Knockdown Cisplatin-Resistant OC Cells
2.6. The Synthetic Lethality of Cisplatin and Niraparib Consists of Expressing Apoptotic Marker and Suppressing Cell Proliferative Marker on Three-Dimensional Cultured of Twist Knockdown Cisplatin-Resistant OC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. siRNA Transfection
4.3. Measurement of Cell Viability and IC50
4.4. Colony Formation Assay
4.5. Generation of Three Dimensional (3D) Spheroidal Model
4.6. Mitochondrial Apoptosis Staining
4.7. Mitochondrial and Cytosolic Separation
4.8. Western Blotting
4.9. Histology and Immunohistochemistry
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef]
- Kawahara, N.; Ogawa, K.; Nagayasu, M.; Kimura, M.; Sasaki, Y.; Kobayashi, H. Candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer. Biomed. Rep. 2017, 7, 391–399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2012, 31, 14. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; Adams, D.J.; Ranzani, M. Synthetic lethality: Emerging targets and opportunities in melanoma. Pigment Cell Melanoma Res. 2017, 30, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Megchelenbrink, W.; Notebaart, R.A.; Huynen, M.A. Predicting human genetic interactions from cancer genome evolution. PLoS ONE 2015, 10, e0125795. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]
- Possik, P.A.; Muller, J.; Gerlach, C.; Kenski, J.C.; Huang, X.; Shahrabi, A.; Krijgsman, O.; Song, J.Y.; Smit, M.A.; Gerritsen, B.; et al. Parallel in vivo and in vitro melanoma RNAi dropout screens reveal synthetic lethality between hypoxia and DNA damage response inhibition. Cell Rep. 2014, 9, 1375–1386. [Google Scholar] [CrossRef]
- Bonanno, L. Predictive models for customizing chemotherapy in advanced non-small cell lung cancer (NSCLC). Transl. Lung Cancer Res. 2013, 2, 160–171. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Jiang, H.; Hemann, M.T.; Yaffe, M.B. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle 2009, 8, 3112–3119. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Li, M.; Li, H.; Liu, F.; Bi, R.; Tu, X.; Chen, L.; Ye, S.; Cheng, X. Characterization of ovarian clear cell carcinoma using target drug-based molecular biomarkers: Implications for personalized cancer therapy. J. Ovarian Res. 2017, 10, 9. [Google Scholar] [CrossRef]
- Friedlander, M.; Gebski, V.; Gibbs, E.; Davies, L.; Bloomfield, R.; Hilpert, F.; Wenzel, L.B.; Eek, D.; Rodrigues, M.; Clamp, A.; et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): A placebo-controlled, phase 3 randomised trial. Lancet Oncol. 2018, 19, 1126–1134. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: An updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Ru, G.Q.; Wang, H.J.; Xu, W.J.; Zhao, Z.S. Upregulation of Twist in gastric carcinoma associated with tumor invasion and poor prognosis. Pathol. Oncol. Res. 2011, 17, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Ohuchida, K.; Mizumoto, K.; Ohhashi, S.; Yamaguchi, H.; Konomi, H.; Nagai, E.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M. Twist, a novel oncogene, is upregulated in pancreatic cancer: Clinical implication of Twist expression in pancreatic juice. Int. J. Cancer 2007, 120, 1634–1640. [Google Scholar] [CrossRef]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xie, F.; Bao, X.; Chen, W.; Xu, Q. miR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol. Cancer 2014, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Park, H.J.; Choi, Y.; Won, Y.J.; Lee, S.J.; Park, D.Y. TWIST mediates resistance to paclitaxel by regulating Akt and Bcl-2 expression in gastric cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317722070. [Google Scholar] [CrossRef]
- Ji, H.; Lu, H.W.; Li, Y.M.; Lu, L.; Wang, J.L.; Zhang, Y.F.; Shang, H. Twist promotes invasion and cisplatin resistance in pancreatic cancer cells through growth differentiation factor 15. Mol. Med. Rep. 2015, 12, 3841–3848. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ling, M.T.; Guan, X.Y.; Tsao, S.W.; Cheung, H.W.; Lee, D.T.; Wong, Y.C. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 2004, 23, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, J.Y.; Kim, H.S.; Yoon, H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. Int. J. Mol. Sci. 2020, 21, 7613. [Google Scholar] [CrossRef]
- Maestro, R.; Dei Tos, A.P.; Hamamori, Y.; Krasnokutsky, S.; Sartorelli, V.; Kedes, L.; Doglioni, C.; Beach, D.H.; Hannon, G.J. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999, 13, 2207–2217. [Google Scholar] [CrossRef]
- Valsesia-Wittmann, S.; Magdeleine, M.; Dupasquier, S.; Garin, E.; Jallas, A.C.; Combaret, V.; Krause, A.; Leissner, P.; Puisieux, A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 2004, 6, 625–630. [Google Scholar] [CrossRef]
- Roberts, C.M.; Tran, M.A.; Pitruzzello, M.C.; Wen, W.; Loeza, J.; Dellinger, T.H.; Mor, G.; Glackin, C.A. TWIST1 drives cisplatin resistance and cell survival in an ovarian cancer model, via upregulation of GAS6, L1CAM, and Akt signalling. Sci. Rep. 2016, 6, 37652. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liao, Q.; He, H.; Zhong, D.; Yin, K. TWIST interacts with beta-catenin signaling on osteosarcoma cell survival against cisplatin. Mol. Carcinog. 2014, 53, 440–446. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef]
- Song, Y.H.; Shiota, M.; Yokomizo, A.; Uchiumi, T.; Kiyoshima, K.; Kuroiwa, K.; Oda, Y.; Naito, S. Twist1 and Y-box-binding protein-1 are potential prognostic factors in bladder cancer. Urol. Oncol. 2014, 32, 31.e31–31.e37. [Google Scholar] [CrossRef]
- Shiota, M.; Song, Y.; Yokomizo, A.; Kiyoshima, K.; Tada, Y.; Uchino, H.; Uchiumi, T.; Inokuchi, J.; Oda, Y.; Kuroiwa, K.; et al. Foxo3a suppression of urothelial cancer invasiveness through Twist1, Y-box-binding protein 1, and E-cadherin regulation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5654–5663. [Google Scholar] [CrossRef]
- Hung, J.J.; Yang, M.H.; Hsu, H.S.; Hsu, W.H.; Liu, J.S.; Wu, K.J. Prognostic significance of hypoxia-inducible factor-1alpha, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax 2009, 64, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, J.; Kim, Y.J.; Wong, M.K.; Wang, P. Codelivery of doxorubicin and paclitaxel by cross-linked multilamellar liposome enables synergistic antitumor activity. Mol. Pharm. 2014, 11, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Tardi, P.; Johnstone, S.; Harasym, N.; Xie, S.; Harasym, T.; Zisman, N.; Harvie, P.; Bermudes, D.; Mayer, L. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk. Res. 2009, 33, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Harasym, T.O.; Tardi, P.G.; Harasym, N.L.; Shew, C.R.; Johnstone, S.A.; Ramsay, E.C.; Bally, M.B.; Janoff, A.S. Ratiometric dosing of anticancer drug combinations: Controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol. Cancer Ther. 2006, 5, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Lee, G.Y.; Kenny, P.A.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4, 359–365. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Jiang, Q.; Ding, X.; Xu, W.; Wang, D.W.; Chen, M. The ER stress-mediated mitochondrial apoptotic pathway and MAPKs modulate tachypacing-induced apoptosis in HL-1 atrial myocytes. PLoS ONE 2015, 10, e0117567. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling. Cancers 2019, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Denning, M.F.; Wang, Y.; Tibudan, S.; Alkan, S.; Nickoloff, B.J.; Qin, J.Z. Caspase activation and disruption of mitochondrial membrane potential during UV radiation-induced apoptosis of human keratinocytes requires activation of protein kinase C. Cell Death Differ. 2002, 9, 40–52. [Google Scholar] [CrossRef]
- Reers, M.; Smiley, S.T.; Mottola-Hartshorn, C.; Chen, A.; Lin, M.; Chen, L.B. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995, 260, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhan, Y.; Chen, B.; Lu, Y.; Yin, T.; Zhou, S.; Zhang, W.; Liu, X.; Du, B.; Wei, X.; et al. Tubeimoside I-induced lung cancer cell death and the underlying crosstalk between lysosomes and mitochondria. Cell Death Dis. 2020, 11, 708. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Lin, X.H.; Liu, H.H.; Zhang, R.; Chen, R.X. Suppression of DRP1mediated mitophagy increases the apoptosis of hepatocellular carcinoma cells in the setting of chemotherapy. Oncol. Rep. 2020, 43, 1010–1018. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, M. Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci. 2020, 111, 3111–3121. [Google Scholar] [CrossRef]
- Lord, R.V.; Brabender, J.; Gandara, D.; Alberola, V.; Camps, C.; Domine, M.; Cardenal, F.; Sanchez, J.M.; Gumerlock, P.H.; Taron, M.; et al. Low ERCC1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 2286–2291. [Google Scholar]
- Cabelof, D.C.; Guo, Z.; Raffoul, J.J.; Sobol, R.W.; Wilson, S.H.; Richardson, A.; Heydari, A.R. Base excision repair deficiency caused by polymerase beta haploinsufficiency: Accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003, 63, 5799–5807. [Google Scholar]
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Hilton, J.F.; Hadfield, M.J.; Tran, M.T.; Shapiro, G.I. Poly(ADP-ribose) polymerase inhibitors as cancer therapy. Front. Biosci. 2013, 18, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Gourley, C.; Balmana, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving from Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Carey, J.P.W.; Karakas, C.; Bui, T.; Chen, X.; Vijayaraghavan, S.; Zhao, Y.; Wang, J.; Mikule, K.; Litton, J.K.; Hunt, K.K.; et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 742–757. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA Mutation-Associated Advanced Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 7165–7170. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Grzegrzolka, J.; Biala, M.; Wojtyra, P.; Kobierzycki, C.; Olbromski, M.; Gomulkiewicz, A.; Piotrowska, A.; Rys, J.; Podhorska-Okolow, M.; Dziegiel, P. Expression of EMT Markers SLUG and TWIST in Breast Cancer. Anticancer Res. 2015, 35, 3961–3968. [Google Scholar] [PubMed]
- Nuti, S.V.; Mor, G.; Li, P.; Yin, G. TWIST and ovarian cancer stem cells: Implications for chemoresistance and metastasis. Oncotarget 2014, 5, 7260–7271. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Hosono, S.; Terauchi, M.; Shibata, K.; Ino, K.; Yamamoto, E.; Nomura, S.; Nawa, A.; Kikkawa, F. Twist expression predicts poor clinical outcome of patients with clear cell carcinoma of the ovary. Oncology 2006, 71, 394–401. [Google Scholar] [CrossRef]
- Elloul, S.; Vaksman, O.; Stavnes, H.T.; Trope, C.G.; Davidson, B.; Reich, R. Mesenchymal-to-epithelial transition determinants as characteristics of ovarian carcinoma effusions. Clin. Exp. Metastasis 2010, 27, 161–172. [Google Scholar] [CrossRef]
- Caruso, D.; Papa, A.; Tomao, S.; Vici, P.; Panici, P.B.; Tomao, F. Niraparib in ovarian cancer: Results to date and clinical potential. Ther. Adv. Med. Oncol. 2017, 9, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Mirza, M.R.; Matulonis, U.A. The poly (ADP ribose) polymerase inhibitor niraparib: Management of toxicities. Gynecol. Oncol. 2018, 149, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Vidi, P.A.; Bissell, M.J.; Lelievre, S.A. Three-dimensional culture of human breast epithelial cells: The how and the why. Methods Mol. Biol. 2013, 945, 193–219. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Hirai, S.; Yamaguchi, M.; Tanaka, Y.; Tada, M.; Yamada, G.; Hasegawa, T.; Miyagi, Y.; Niki, T.; Watanabe, A.; et al. Survivin knockdown induces senescence in TTF1-expressing, KRAS-mutant lung adenocarcinomas. Int. J. Oncol. 2018, 53, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kraskiewicz, H.; FitzGerald, U. InterfERing with endoplasmic reticulum stress. Trends Pharmacol. Sci. 2012, 33, 53–63. [Google Scholar] [CrossRef]
- Schleicher, S.M.; Moretti, L.; Varki, V.; Lu, B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: Implications for future therapeutic approaches. Drug Resist. Updat. 2010, 13, 79–86. [Google Scholar] [CrossRef]
- Rosati, E.; Sabatini, R.; Rampino, G.; De Falco, F.; Di Ianni, M.; Falzetti, F.; Fettucciari, K.; Bartoli, A.; Screpanti, I.; Marconi, P. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood 2010, 116, 2713–2723. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Dalton, L.E.; Clarke, H.J.; Knight, J.; Lawson, M.H.; Wason, J.; Lomas, D.A.; Howat, W.J.; Rintoul, R.C.; Rassl, D.M.; Marciniak, S.J. The endoplasmic reticulum stress marker CHOP predicts survival in malignant mesothelioma. Br. J. Cancer 2013, 108, 1340–1347. [Google Scholar] [CrossRef]
- Huber, A.L.; Lebeau, J.; Guillaumot, P.; Petrilli, V.; Malek, M.; Chilloux, J.; Fauvet, F.; Payen, L.; Kfoury, A.; Renno, T.; et al. p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Mol. Cell 2013, 49, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harbor Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Foty, R. A simple hanging drop cell culture protocol for generation of 3D spheroids. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahar, E.; Kim, J.-Y.; Kim, D.-C.; Kim, H.-S.; Yoon, H. Combination of Niraparib, Cisplatin and Twist Knockdown in Cisplatin-Resistant Ovarian Cancer Cells Potentially Enhances Synthetic Lethality through ER-Stress Mediated Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2021, 22, 3916. https://doi.org/10.3390/ijms22083916
Bahar E, Kim J-Y, Kim D-C, Kim H-S, Yoon H. Combination of Niraparib, Cisplatin and Twist Knockdown in Cisplatin-Resistant Ovarian Cancer Cells Potentially Enhances Synthetic Lethality through ER-Stress Mediated Mitochondrial Apoptosis Pathway. International Journal of Molecular Sciences. 2021; 22(8):3916. https://doi.org/10.3390/ijms22083916
Chicago/Turabian StyleBahar, Entaz, Ji-Ye Kim, Dong-Chul Kim, Hyun-Soo Kim, and Hyonok Yoon. 2021. "Combination of Niraparib, Cisplatin and Twist Knockdown in Cisplatin-Resistant Ovarian Cancer Cells Potentially Enhances Synthetic Lethality through ER-Stress Mediated Mitochondrial Apoptosis Pathway" International Journal of Molecular Sciences 22, no. 8: 3916. https://doi.org/10.3390/ijms22083916
APA StyleBahar, E., Kim, J.-Y., Kim, D.-C., Kim, H.-S., & Yoon, H. (2021). Combination of Niraparib, Cisplatin and Twist Knockdown in Cisplatin-Resistant Ovarian Cancer Cells Potentially Enhances Synthetic Lethality through ER-Stress Mediated Mitochondrial Apoptosis Pathway. International Journal of Molecular Sciences, 22(8), 3916. https://doi.org/10.3390/ijms22083916