
On Iron Metabolism and Its Regulation




Abstract
1. Introduction
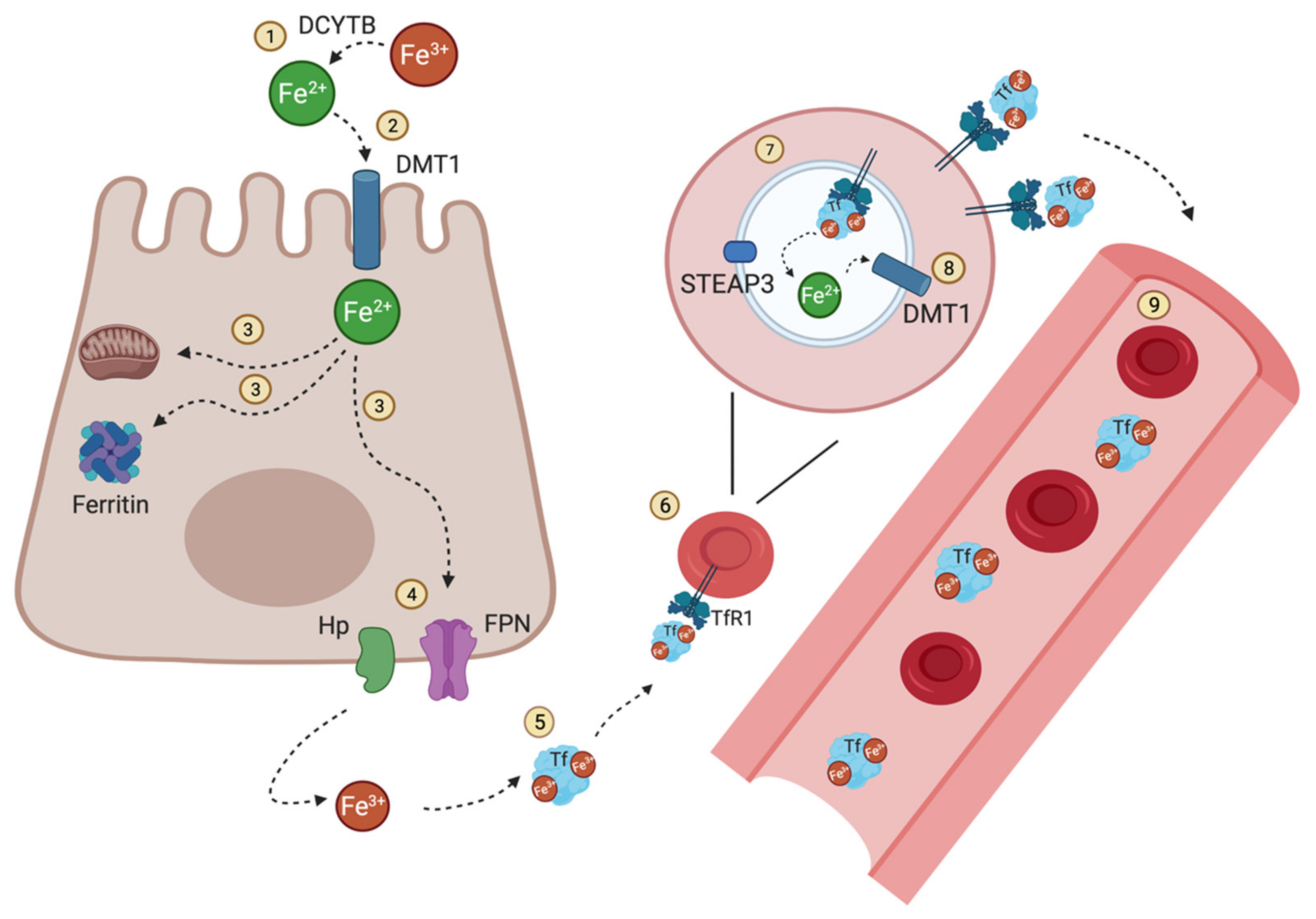
2. Iron Flow in the Human Body
3. Liver the Central Organ in Iron Homeostasis
4. Iron Regulation
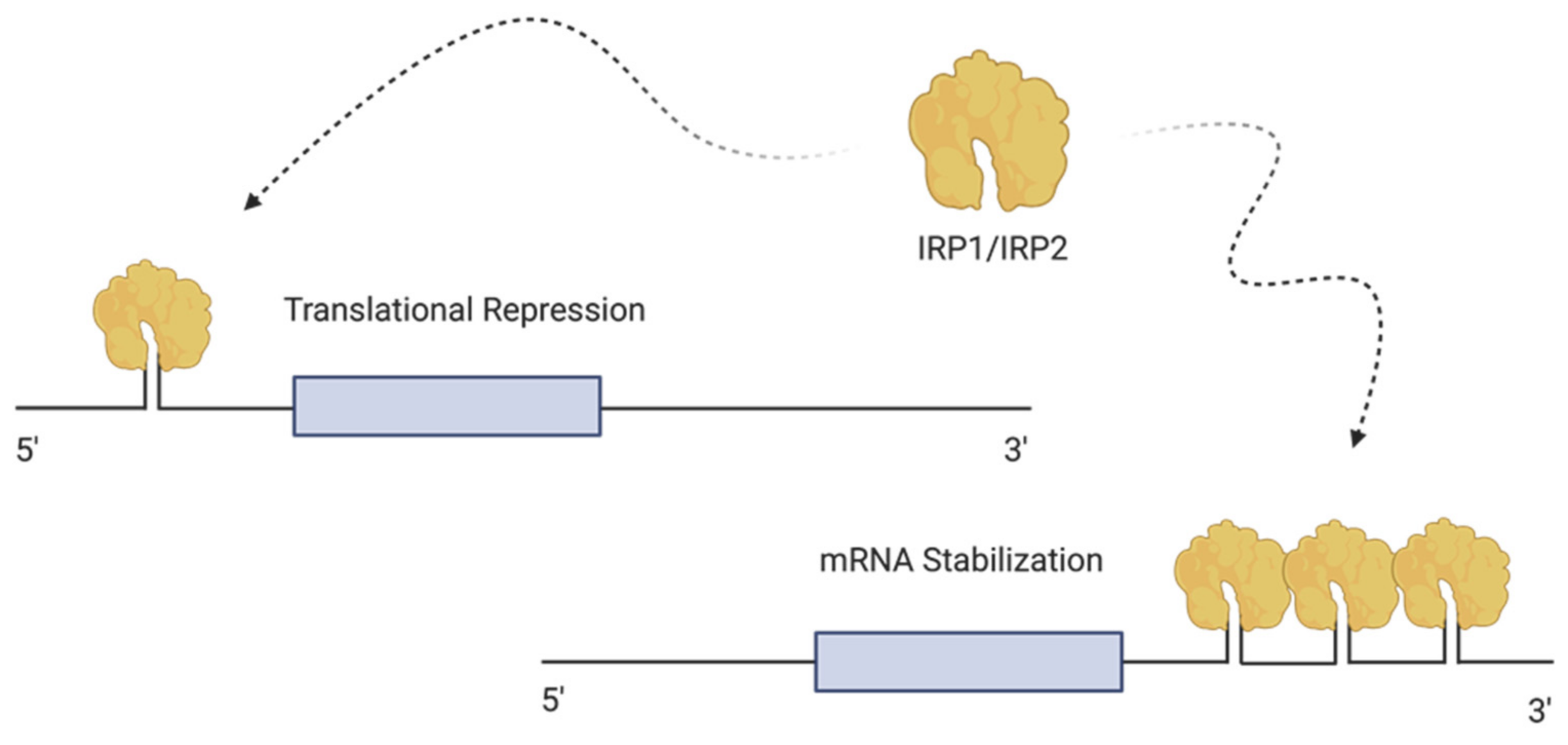
4.1. Cellular Regulation of Iron—The Iron Regulating Proteins (IRPs)
4.2. Sensing and Regulating Intracellular Iron by IRP1 and IRP2
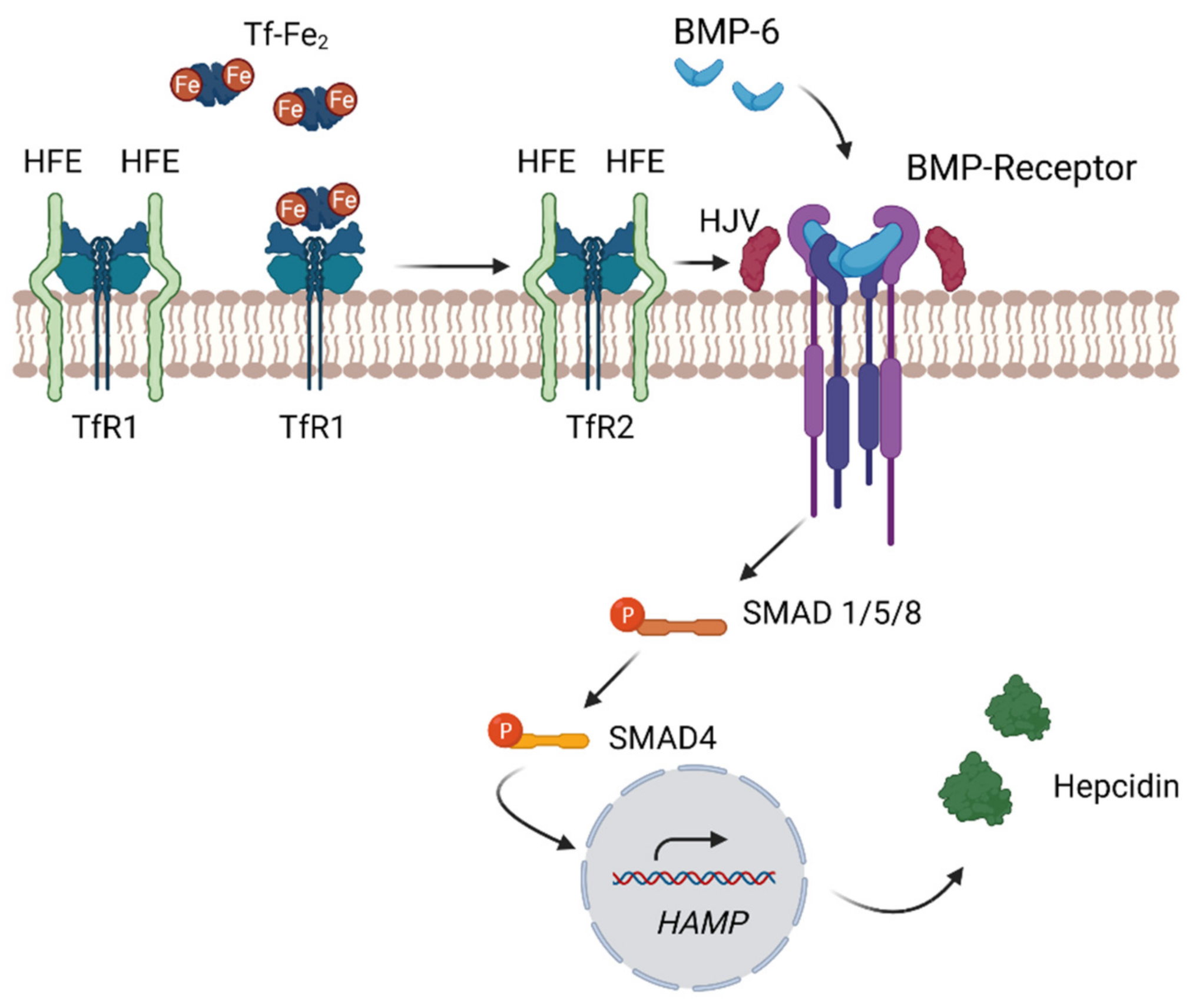
4.3. Systemic Regulation of Iron—The Hepcidin–Ferroportin Axis
4.4. Regulation of Hepcidin through the Bone Morphogenetic Protein
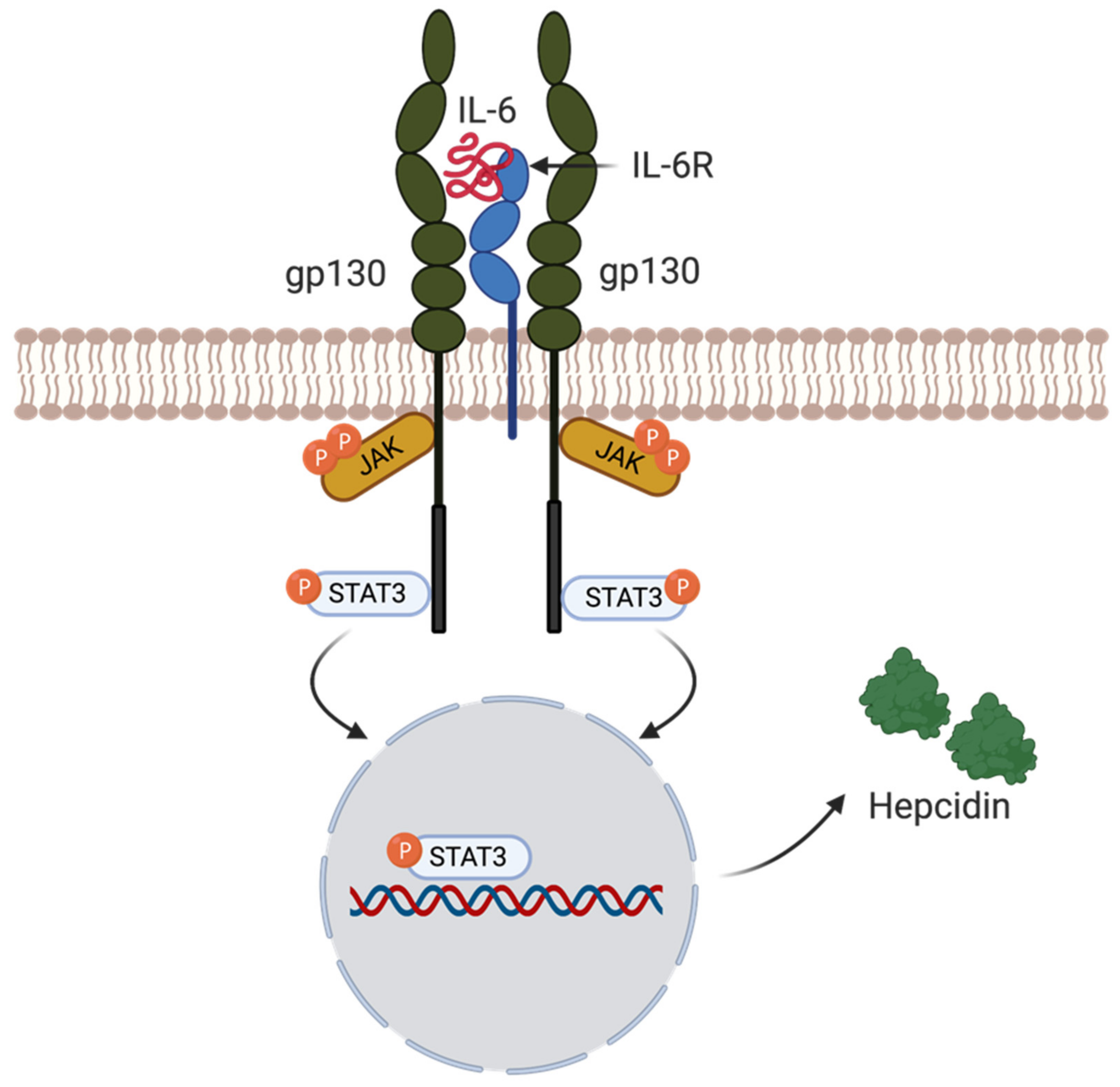
4.5. Hepcidin Regulation by Inflammation
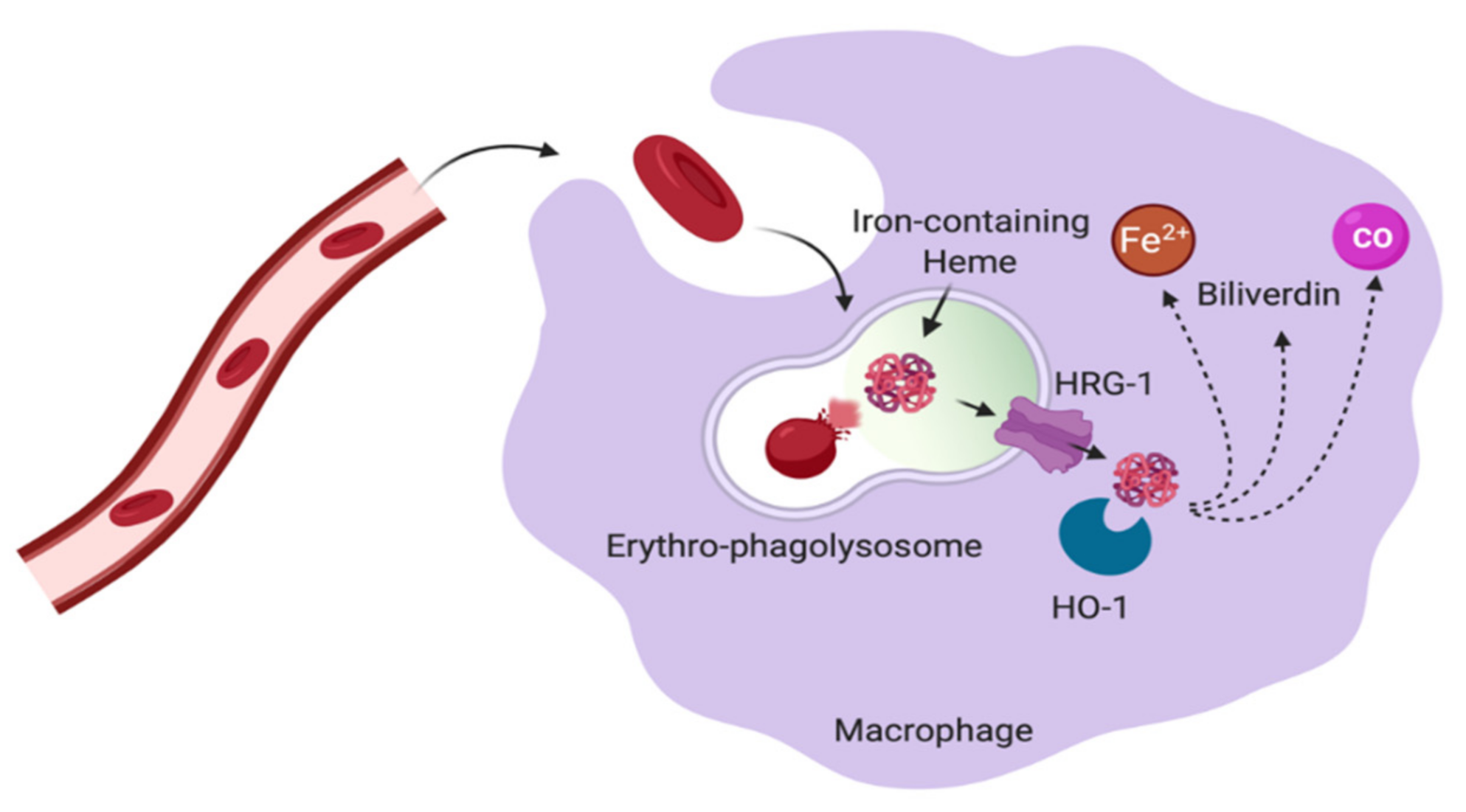
5. Macrophages in Control of Iron Homeostasis
5.1. Macrophage and Erythropoiesis
5.2. Red Pulp Macrophages in the Spleen
5.3. Liver Kupffer Cells
6. Monocytes and Their Contribution to Iron Metabolism
6.1. Monocyte Populations
6.2. Monocytes and Iron Handling
7. Iron, Immunity and Infection
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BM | Bone marrow |
| DMT1 | Divalent metal transporter 1 |
| FPN | Ferroportin |
| Ft | Ferritin |
| Hgb | Hemoglobin |
| HO-1 | Heme oxygenase |
| IFNγ | Interferon-gamma |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin 6 |
| IRE | Iron responsive elements |
| IRP1 | Iron regulating protein 1 |
| IRP2 | iron regulating protein 2 |
| KC’s | Kupffer cells |
| MΦ | Macrophage |
| NTBI | Non transferrin-bound iron |
| RBC | Red blood cell |
| RPM | Red pulp macrophages |
| TBI | Transferrin-bound iron |
| Tf | Transferrin |
| TfR1 | Transferrin receptor 1 |
| TfR2 | Transferrin receptor 2 |
References
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of Iron Metabolism in Health and Disease. Hemodial. Int. 2017, 21, S6–S20. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An Overview of Molecular Basis of Iron Metabolism Regulation and the Associated Pathologies. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1347–1359. [Google Scholar] [CrossRef]
- Schwartz, A.J.; Converso-Baran, K.; Michele, D.E.; Shah, Y.M. A Genetic Mouse Model of Severe Iron Deficiency Anemia Reveals Tissue-Specific Transcriptional Stress Responses and Cardiac Remodeling. J. Biol. Chem. 2019, 294, 14991–15002. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in Metal-Induced Oxidative Stress and Human Disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Zhao, Z. Iron and Oxidizing Species in Oxidative Stress and Alzheimer’s Disease. Aging Med. 2019, 2, 82–87. [Google Scholar] [CrossRef]
- Scindia, Y.; Leeds, J.; Swaminathan, S. Iron Homeostasis in Healthy Kidney and Its Role in Acute Kidney Injury. Semin. Nephrol. 2019, 39, 76–84. [Google Scholar] [CrossRef]
- Johnson Wimbley, T.D.; Graham, D.Y. Diagnosis and Management of Iron Deficiency Anemia in the 21st Century. Therap. Adv. Gastroenterol. 2011, 4, 177–184. [Google Scholar] [CrossRef]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62. [Google Scholar]
- Daher, R.; Karim, Z. Iron Metabolism: State of the Art. Transfus. Clin. Biol. 2017, 24, 115–119. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Musallam, K.M.; Taher, A.T. Iron Deficiency Anaemia Revisited. J. Intern. Med. 2020, 287, 153–170. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.H.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular Iron Uptake, Trafficking and Metabolism: Key Molecules and Mechanisms and Their Roles in Disease. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef]
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on Iron and Its Importance for Human Health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Fleming, R.E.; Ponka, P. Iron Overload in Human Disease. N. Engl. J. Med. 2012, 366, 1549. [Google Scholar] [CrossRef]
- Wang, L.; Cherayil, B.J. Ironing out the Wrinkles in Host Defense: Interactions between Iron Homeostasis and Innate Immunity. J. Innate Immun. 2009, 1, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Theurl, I.; Swirski, F.K.; Weiss, G. “Pumping Iron”—How Macrophages Handle Iron at the Systemic, Microenvironmental, and Cellular Levels. Pflugers Arch. Eur. J. Physiol. 2017, 469, 397–418. [Google Scholar] [CrossRef]
- Yeo, J.H.; Colonne, C.K.; Tasneem, N.; Cosgriff, M.P.; Fraser, S.T. The Iron Islands: Erythroblastic Islands and Iron Metabolism. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 466–471. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Hepatic Iron Metabolism. Semin. Liver Dis. 2005, 25, 420–432. [Google Scholar] [CrossRef]
- Ginzburg, Y.Z. Hepcidin-Ferroportin Axis in Health and Disease, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 110. [Google Scholar] [CrossRef]
- Tissot, J.-D.; Gassner, C.; Favrat, B.; Waeber, G.; Buser, A.; Frey, B.M.; Waldvogel-Abramowski, S. Physiology of Iron Metabolism. Transfus. Med. Hemother. 2017, 41, 213–221. [Google Scholar] [CrossRef]
- Kawabata, H. Transferrin and Transferrin Receptors Update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-Transferrin Bound Iron: A Key Role in Iron Overload and Iron Toxicity. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia Inhibits Ferritinophagy, Increases Mitochondrial Ferritin, and Protects from Ferroptosis. Redox Biol. 2020, 36. [Google Scholar] [CrossRef] [PubMed]
- Rishi, G.; Subramaniam, V.N. The Liver in Regulation of Iron Homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G157–G165. [Google Scholar] [CrossRef]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-Induced Endocytosis of Ferroportin Is Dependent on Ferroportin Ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE System in Vivo: Insights from Mouse Models. Front. Pharmacol. 2014, 5, 1–15. [Google Scholar] [CrossRef]
- Rouault, T.A.; Klausner, R.D. Post-Transcriptional Regulation of Genes of Iron Metabolism in Mammalian Cells. JBIC J. Biol. Inorg. Chem. 1996, 494–499. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing Acts: Molecular Control of Mammalian Iron Metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- Chen, C.; Paw, B.H. Cellular and Mitochondrial Iron Homeostasis in Vertebrates. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.S.; Morán, E.; Sánchez, M. Cellular Iron Metabolism—The IRP/IRE Regulatory Network. 2012. Available online: https://books.google.com.hk/books?id=41WECgAAQBAJ&pg=PA67&dq=Cellular+Iron+Metabolism+%E2%80%93+The+IRP+/+IRE+Regulatory+Network.+2012&hl=zh-CN&sa=X&ved=2ahUKEwipzeaUgp7wAhXCxYsBHc7YBrkQ6AEwAHoECAEQAg#v=onepage&q=Cellular%20Iron%20Metabolism%20%E2%80%93%20The%20IRP%20%2F%20IRE%20Regulatory%20Network.%202012&f=false (accessed on 20 December 2020).
- Rouault, T.A. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Tan, E.K. Iron Regulatory Protein (IRP)-Iron Responsive Element (IRE) Signaling Pathway in Human Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 1–12. [Google Scholar] [CrossRef]
- Sebastiani, G.; Wilkinson, N.; Pantopoulos, K. Pharmacological Targeting of the Hepcidin/Ferroportin Axis. Front. Pharmacol. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Crichton, R.R.; Taylor, D.L.; Della Corte, L.; Srai, S.K.; Dexter, D.T. Iron and the Immune System. J. Neural Transm. 2011, 118, 315–328. [Google Scholar] [CrossRef]
- Pantopoulos, K. Inherited Disorders of Iron Overload. Front. Nutr. 2018, 5, 1–11. [Google Scholar] [CrossRef]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. Int. Rev. J. 2017, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J. Regulation of Iron Metabolism by Hepcidin under Conditions of Inflammation. J. Biol. Chem. 2015, 290, 18975–18983. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Muckenthaler, M.U. Out of Balance-Systemic Iron Homeostasis in Iron-Related Disorders. Nutrients 2013, 5, 3034–3061. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of Erythroferrone as an Erythroid Regulator of Iron Metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, A.M.; Björkman, P.J. HFE and Transferrin Directly Compete for Transferrin Receptor in Solution and at the Cell Surface. J. Biol. Chem. 2004, 279, 25866–25875. [Google Scholar] [CrossRef] [PubMed]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.P.; Townsend, A.R.M.; Drakesmith, H. Hepcidin Regulation by Innate Immune and Infectious Stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Singh, P.; Srivastava, A. Synthesis, Nature and Utility of Universal Iron Chelator—Siderophore: A Review. Microbiol. Res. 2018, 212–213, 103–111. [Google Scholar] [CrossRef]
- Soares, M.P.; Hamza, I. Macrophages and Iron Metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef]
- Heideveld, E.; van den Akker, E. Digesting the Role of Bone Marrow Macrophages on Hematopoiesis. Immunobiology 2017, 222, 814–822. [Google Scholar] [CrossRef]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of Tissue Iron Homeostasis: The Macrophage “Ferrostat”. JCI Insight 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Philpott, C.C.; Jadhav, S. The Ins and Outs of Iron: Escorting Iron through the Mammalian Cytosol. Free Radic. Biol. Med. 2019, 133, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Shvartsman, M.; Cabantchik, Z.I. Intracellular Iron Trafficking: Role of Cytosolic Ligands. BioMetals 2012, 25, 711–723. [Google Scholar] [CrossRef]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A Family of Molecules for Iron Storage, Antioxidation and More. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Rishi, G.; Subramaniam, V.N. The Relationship between Systemic Iron Homeostasis and Erythropoiesis. Biosci. Rep. 2017, 37, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Pagani, A.; Nai, A.; Silvestri, L. The Mutual Control of Iron and Erythropoiesis. Int. J. Lab. Hematol. 2016, 38, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Weed, R.I.; Reed, C.F.; Berg, G. Is Hemoglobin An Essential Structural Component Of Human Erythrocyte Membranes? J. Clin. Investig. 1963, 42, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Sandeep Prabhu, K.; Paulson, R.F. Monocyte-Derived Macrophages Expand the Murine Stress Erythropoietic Niche during the Recovery from Anemia. Blood 2018, 132, 2580–2593. [Google Scholar] [CrossRef]
- Ganz, T. Erythropoietic Regulators of Iron Metabolism. Free Radic. Biol. Med. 2019, 133, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Gifford, S.C.; Derganc, J.; Shevkoplyas, S.S.; Yoshida, T.; Bitensky, M.W. A Detailed Study of Time-Dependent Changes in Human Red Blood Cells: From Reticulocyte Maturation to Erythrocyte Senescence. Br. J. Haematol. 2006, 135, 395–404. [Google Scholar] [CrossRef]
- Haldar, M.; Kohyama, M.; So, A.Y.L.; Kc, W.; Wu, X.; Briseño, C.G.; Satpathy, A.T.; Kretzer, N.M.; Arase, H.; Rajasekaran, N.S.; et al. Heme-Mediated SPI-C Induction Promotes Monocyte Differentiation into Iron-Recycling Macrophages. Cell 2014, 156, 1223–1234. [Google Scholar] [CrossRef]
- Nai, A.; Lidonnici, M.R.; Federico, G.; Pettinato, M.; Olivari, V.; Carrillo, F.; Geninatti Crich, S.; Ferrari, G.; Camaschella, C.; Silvestri, L.; et al. NCOA4-Mediated Ferritinophagy in Macrophages Is Crucial to Sustain Erythropoiesis in Mice. Haematologica 2020. [Google Scholar] [CrossRef]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017. [Google Scholar] [CrossRef]
- Borges Da Silva, H.; Fonseca, R.; Pereira, R.M.; Cassado, A.A.; Álvarez, J.M.; D’Império Lima, M.R. Splenic Macrophage Subsets and Their Function during Blood-Borne Infections. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Klei, T.R.; Dalimot, J.; Nota, B.; Veldthuis, M.; Mul, F.P.; Rademakers, T.; Hoogenboezem, M.; van Zwieten, R.; van Burgen, R. Hemolysis in the Spleen Drives Erythrocyte Turnover. Blood 2020, 136, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Buffet, P.A.; Milon, G.; Brousse, V.; Correas, J.M.; Dousset, B.; Couvelard, A.; Kianmanesh, R.; Farges, O.; Sauvanet, A.; Paye, F.; et al. Ex Vivo Perfusion of Human Spleens Maintains Clearing and Processing Functions. Blood 2006, 107, 3745–3752. [Google Scholar] [CrossRef]
- Duez, J.; Holleran, J.P.; Ndour, P.A.; Pionneau, C.; Diakité, S.; Roussel, C.; Dussiot, M.; Amireault, P.; Avery, V.M.; Buffet, P.A. Mechanical Clearance of Red Blood Cells by the Human Spleen: Potential Therapeutic Applications of a Biomimetic RBC Filtration Method. Transfus. Clin. Biol. 2015, 22, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Fens, M.H.A.M.; Storm, G.; Pelgrim, R.C.M.; Ultee, A.; Byrne, A.T.; Gaillard, C.A.; van Solinge, W.W.; Schiffelers, R.M. Erythrophagocytosis by Angiogenic Endothelial Cells Is Enhanced by Loss of Erythrocyte Deformability. Exp. Hematol. 2010, 38, 282–291. [Google Scholar] [CrossRef]
- Nagelkerke, S.Q.; Bruggeman, C.W.; Den Haan, J.M.M.; Mul, E.P.J.; Van Den Berg, T.K.; Van Bruggen, R.; Kuijpers, T.W. Red Pulp Macrophages in the Human Spleen Are a Distinct Cell Population with a Unique Expression of Fc-g Receptors. Blood Adv. 2018, 2, 941–963. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 Is Essential for Heme Transport from the Phagolysosome of Macrophages during Erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Korolnek, T.; Hamza, I. Macrophages and Iron Trafficking at the Birth and Death of Red Cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the Heme Recycling System in Heme Oxygenase 1-Deficient Mice: Effects on Macrophage Viability and Tissue Iron Distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed]
- Kovtunovych, G.; Ghosh, M.C.; Ollivierre, W.; Weitzel, R.P.; Eckhaus, M.A.; Tisdale, J.F.; Yachie, A.; Rouault, T.A. Wild-Type Macrophages Reverse Disease in Heme Oxygenase 1-Deficient Mice. Blood 2014, 124, 1522–1530. [Google Scholar] [CrossRef]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Bouwens, L.; Baekeland, M.; de Zanger, R.; Wisse, E. Quantitation, Tissue Distribution and Proliferation Kinetics of Kupffer Cells in Normal Rat Liver. Hepatology 1986, 6, 718–722. [Google Scholar] [CrossRef]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Zhong, W.; Zhang, J.; Zhang, X.; Wang, Y.; McClain, C.J. Kupffer Cell Depletion Protects against the Steatosis, but Not the Liver Damage, Induced by Marginal-Copper, High-Fructose Diet in Male Rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G934–G945. [Google Scholar] [CrossRef] [PubMed]
- Theurl, M.; Theurl, I.; Hochegger, K.; Obrist, P.; Subramaniam, N.; van Rooijen, N.; Schuemann, K.; Weiss, G. Kupffer Cells Modulate Iron Homeostasis in Mice via Regulation of Hepcidin Expression. J. Mol. Med. 2008, 86, 825–835. [Google Scholar] [CrossRef]
- Sprangers, S.; Vries, T.J.D.; Everts, V. Monocyte Heterogeneity: Consequences for Monocyte-Derived Immune Cells. J. Immunol. Res. 2016, 2016. [Google Scholar] [CrossRef]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion during Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef]
- Houthuys, E.; Movahedi, K.; De Baetselier, P.; Van Ginderachter, J.A.; Brouckaert, P. A Method for the Isolation and Purification of Mouse Peripheral Blood Monocytes. J. Immunol. Methods 2010, 359, 1–10. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal Differentiation of Classical Monocytes as They Survey Steady-State Tissues and Transport Antigen to Lymph Nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Ohteki, T. Monopoiesis in Humans and Mice. Int. Immunol. 2018, 30, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Haschka, D.; Petzer, V.; Kocher, F.; Tschurtschenthaler, C.; Schaefer, B.; Seifert, M.; Sopper, S.; Sonnweber, T.; Feistritzer, C.; Arvedson, T.L.; et al. Classical and Intermediate Monocytes Scavenge Non-Transferrin-Bound Iron and Damaged Erythrocytes. JCI Insight 2019, 4, 1–23. [Google Scholar] [CrossRef]
- Pinto, J.P.; Arezes, J.; Dias, V.; Oliveira, S.; Vieira, I.; Costa, M.; Vos, M.; Carlsson, A.; Rikers, Y.; Rangel, M.; et al. Physiological Implications of NTBI Uptake by T Lymphocytes. Front. Pharmacol. 2014, 5 FEB, 1–14. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The Mononuclear Phagocyte System: A New Classification of Macrophages, Monocytes, and Their Precursor Cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- Schaible, U.E.; Kaufmann, S.H.E. Iron and Microbial Infection. Nat. Rev. Microbiol. 2004, 2, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Weiss, G. The Iron Age of Host—Microbe Interactions. EMBO Rep. 2015, 16, 1482–1500. [Google Scholar] [CrossRef]
- Hamad, M.; Bajbouj, K. The Re-Emerging Role of Iron in Infection and Immunity. Integr. Mol. Med. 2016, 3, 807–810. [Google Scholar] [CrossRef]
- Skaar, E.P.; Raffatellu, M. Metals in infectious diseases and nutritional immunity. Metallomics 2015, 7, 926–928. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Iron and Infection. Int. J. Hematol. 2018, 107, 7–15. [Google Scholar] [CrossRef]
- Nairz, M.; Haschka, D.; Demetz, E.; Weiss, G. Iron at the Interface of Immunity and Infection. Front. Pharmacol. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Gwamaka, M.; Kurtis, J.D.; Sorensen, B.E.; Holte, S.; Morrison, R.; Mutabingwa, T.K.; Fried, M.; Duffy, P.E. Iron Deficiency Protects against Severe Plasmodium Falciparum Malaria and Death in Young Children. Clin. Infect. Dis. 2012, 54, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Núñez, G.; Sakamoto, K.; Soares, M.P. Innate Nutritional Immunity. J. Immunol. 2018, 201, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Anzaldi, L.L.; Skaar, E.P. Overcoming the Heme Paradox: Heme Toxicity and Tolerance in Bacterial Pathogens. Infect. Immun. 2010, 78, 4977–4989. [Google Scholar] [CrossRef] [PubMed]
- Richard, K.L.; Kelley, B.R.; Johnson, J.G. Heme Uptake and Utilization by Gram-Negative Bacterial Pathogens. Front. Cell. Infect. Microbiol. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subset | Marker | Function |
|---|---|---|
| Mouse | ||
| Classical | Ly6ChighCD11b+ | Proinflammatory |
| Non-classical | Ly6ClowCD11b+ | Patrolling |
| Human Classical | CD15+CD16− | Immune response |
| Intermediate | CD14+CD16+ | Proinflammatory |
| Non-classical | CD14+CD16++ | Patrolling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogt, A.-C.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. https://doi.org/10.3390/ijms22094591
Vogt A-CS, Arsiwala T, Mohsen M, Vogel M, Manolova V, Bachmann MF. On Iron Metabolism and Its Regulation. International Journal of Molecular Sciences. 2021; 22(9):4591. https://doi.org/10.3390/ijms22094591
Chicago/Turabian StyleVogt, Anne-Cathrine S., Tasneem Arsiwala, Mona Mohsen, Monique Vogel, Vania Manolova, and Martin F. Bachmann. 2021. "On Iron Metabolism and Its Regulation" International Journal of Molecular Sciences 22, no. 9: 4591. https://doi.org/10.3390/ijms22094591
APA StyleVogt, A.-C. S., Arsiwala, T., Mohsen, M., Vogel, M., Manolova, V., & Bachmann, M. F. (2021). On Iron Metabolism and Its Regulation. International Journal of Molecular Sciences, 22(9), 4591. https://doi.org/10.3390/ijms22094591