
Structural Fluctuations of the Human Proteasome α7 Homo-Tetradecamer Double Ring Imply the Proteasomal α-Ring Assembly Mechanism

 , , ,
, , ,  and
and
Abstract
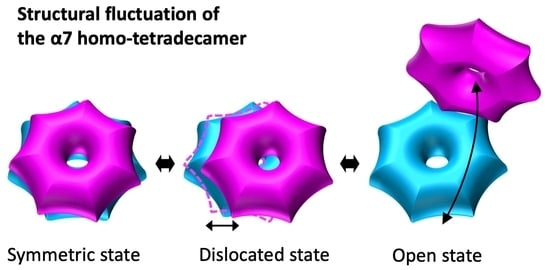
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The α7 Tetradecamer Double Ring Shows Three Different Structures in Solution
2.2. The α7 Subunit Models Were Fitted to the Densities in Each Cryo-EM Map
2.3. Cryo-EM Structures of the α7 Double Ring in Solution
3. Discussion
4. Materials and Methods
4.1. Sample Preparation for Cryo-EM
4.2. Image Processing for Cryo-EM
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 2017, 386, 463–471. [Google Scholar] [CrossRef]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef]
- Lowe, J.; Stock, D.; Jap, B.; Zwickl, P.; Baumeister, W.; Huber, R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. [Google Scholar] [CrossRef]
- Rabl, J.; Smith, D.M.; Yu, Y.; Chang, S.C.; Goldberg, A.L.; Cheng, Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell 2008, 30, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Kumoi, K.; Satoh, T.; Murata, K.; Hiromoto, T.; Mizushima, T.; Kamiya, Y.; Noda, M.; Uchiyama, S.; Yagi, H.; Kato, K. An archaeal homolog of proteasome assembly factor functions as a proteasome activator. PLoS ONE 2013, 8, e60294. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, J.; Lu, Y.; Ma, Y.B.; Lee, B.H.; Yu, Z.; Ouyang, Q.; Finley, D.J.; Kirschner, M.W.; Mao, Y. Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, 12991–12996. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, W.L.; Yu, D.; Ouyang, Q.; Lu, Y.; Mao, Y. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat. Commun. 2018, 9, 1360. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Yu, Z.; Yu, Y.; Wang, F.; Myasnikov, A.G.; Coffino, P.; Cheng, Y. Allosteric coupling between α-rings of the 20S proteasome. Nat. Commun. 2020, 11, 4580. [Google Scholar] [CrossRef]
- Dorn, I.T.; Eschrich, R.; Seemöller, E.; Guckenberger, R.; Tampé, R. High-resolution AFM-imaging and mechanistic analysis of the 20 S proteasome. J. Mol. Biol. 1999, 288, 1027–1036. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Hochstrasser, M.; Gaczynska, M. A tetrahedral transition state at the active sites of the 20S proteasome is coupled to opening of the alpha-ring channel. Structure 2009, 17, 1137–1147. [Google Scholar] [CrossRef]
- Yagi-Utsumi, M.; Sikdar, A.; Song, C.; Park, J.; Inoue, R.; Watanabe, H.; Burton-Smith, R.N.; Kozai, T.; Suzuki, T.; Kodama, A.; et al. Supramolecular tholos-like architecture constituted by archaeal proteins without functional annotation. Sci. Rep. 2020, 10, 1540. [Google Scholar] [CrossRef] [PubMed]
- Religa, T.L.; Sprangers, R.; Kay, L.E. Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science 2010, 328, 98–102. [Google Scholar] [CrossRef]
- Rennella, E.; Huang, R.; Yu, Z.; Kay, L.E. Exploring long-range cooperativity in the 20S proteasome core particle from Thermoplasma acidophilum using methyl-TROSY-based NMR. Proc. Natl. Acad. Sci. USA 2020, 117, 5298–5309. [Google Scholar] [CrossRef]
- Sprangers, R.; Kay, L.E. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature 2007, 445, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Satoh, T. Structural insights on the dynamics of proteasome formation. Biophys. Rev. 2018, 10, 597–604. [Google Scholar] [CrossRef]
- Gerards, W.L.; Enzlin, J.; Haner, M.; Hendriks, I.L.; Aebi, U.; Bloemendal, H.; Boelens, W. The human alpha-type proteasomal subunit HsC8 forms a double ringlike structure, but does not assemble into proteasome-like particles with the beta-type subunits HsDelta or HsBPROS26. J. Biol. Chem. 2017, 272, 10080–10086. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Noda, M.; Yagi, H.; Thammaporn, R.; Seetaha, S.; Satoh, T.; Kato, K.; Uchiyama, S. Disassembly of the self-assembled, double-ring structure of proteasome alpha7 homo-tetradecamer by alpha6. Sci. Rep. 2015, 5, 18167. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Hamada, K.; Kato, K.; Kurimoto, E.; Okamoto, K.; Morimoto, Y.; Ikeda, S.; Naito, S.; Furusaka, M.; Itoh, K.; et al. SANS simulation of aggregated protein in aqueous solution. Nucl. Instrum. Methods. Phys. Res. A 2009, 600, 272–274. [Google Scholar] [CrossRef]
- Sugiyama, M.; Kurimoto, E.; Yagi, H.; Mori, K.; Fukunaga, T.; Hirai, M.; Zaccai, G.; Kato, K. Kinetic asymmetry of subunit exchange of homooligomeric protein as revealed by deuteration-assisted small-angle neutron scattering. Biophys. J. 2011, 101, 2037–2042. [Google Scholar] [CrossRef]
- Sekiguchi, T.; Satoh, T.; Kurimoto, E.; Song, C.; Kozai, T.; Watanabe, H.; Ishii, K.; Yagi, H.; Yanaka, S.; Uchiyama, S.; et al. Mutational and Combinatorial Control of Self-Assembling and Disassembling of Human Proteasome alpha Subunits. Int. J. Mol. Sci. 2019, 20, 2308. [Google Scholar] [CrossRef]
- Kozai, T.; Sekiguchi, T.; Satoh, T.; Yagi, H.; Kato, K.; Uchihashi, T. Two-step process for disassembly mechanism of proteasome alpha7 homo-tetradecamer by alpha6 revealed by high-speed atomic force microscopy. Sci. Rep. 2017, 7, 15373. [Google Scholar] [CrossRef] [PubMed]
- Kimanius, D.; Forsberg, B.O.; Scheres, S.; Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 2016, 5, e18722. [Google Scholar] [CrossRef]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comp. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Satoh, T.; Sekiguchi, T.; Kato, K.; Murata, K. Structural Fluctuations of the Human Proteasome α7 Homo-Tetradecamer Double Ring Imply the Proteasomal α-Ring Assembly Mechanism. Int. J. Mol. Sci. 2021, 22, 4519. https://doi.org/10.3390/ijms22094519
Song C, Satoh T, Sekiguchi T, Kato K, Murata K. Structural Fluctuations of the Human Proteasome α7 Homo-Tetradecamer Double Ring Imply the Proteasomal α-Ring Assembly Mechanism. International Journal of Molecular Sciences. 2021; 22(9):4519. https://doi.org/10.3390/ijms22094519
Chicago/Turabian StyleSong, Chihong, Tadashi Satoh, Taichiro Sekiguchi, Koichi Kato, and Kazuyoshi Murata. 2021. "Structural Fluctuations of the Human Proteasome α7 Homo-Tetradecamer Double Ring Imply the Proteasomal α-Ring Assembly Mechanism" International Journal of Molecular Sciences 22, no. 9: 4519. https://doi.org/10.3390/ijms22094519
APA StyleSong, C., Satoh, T., Sekiguchi, T., Kato, K., & Murata, K. (2021). Structural Fluctuations of the Human Proteasome α7 Homo-Tetradecamer Double Ring Imply the Proteasomal α-Ring Assembly Mechanism. International Journal of Molecular Sciences, 22(9), 4519. https://doi.org/10.3390/ijms22094519