
Methods for Identifying Microbial Natural Product Compounds that Target Kinetoplastid RNA Structural Motifs by Homology and De Novo Modeled 18S rRNA

 ,
,
Abstract
1. Introduction
2. RNA as a Drug Target
3. Results and Discussion
3.1. Sequence selection Homology and De Novo Modeling Structure Prediction and Analysis of the 18S rRNA of the Selected Kinetoplastids Leishmania Major, Trypanosoma brucei, and Trypanosoma cruzi
3.2. 18S rRNA Secondary Structure
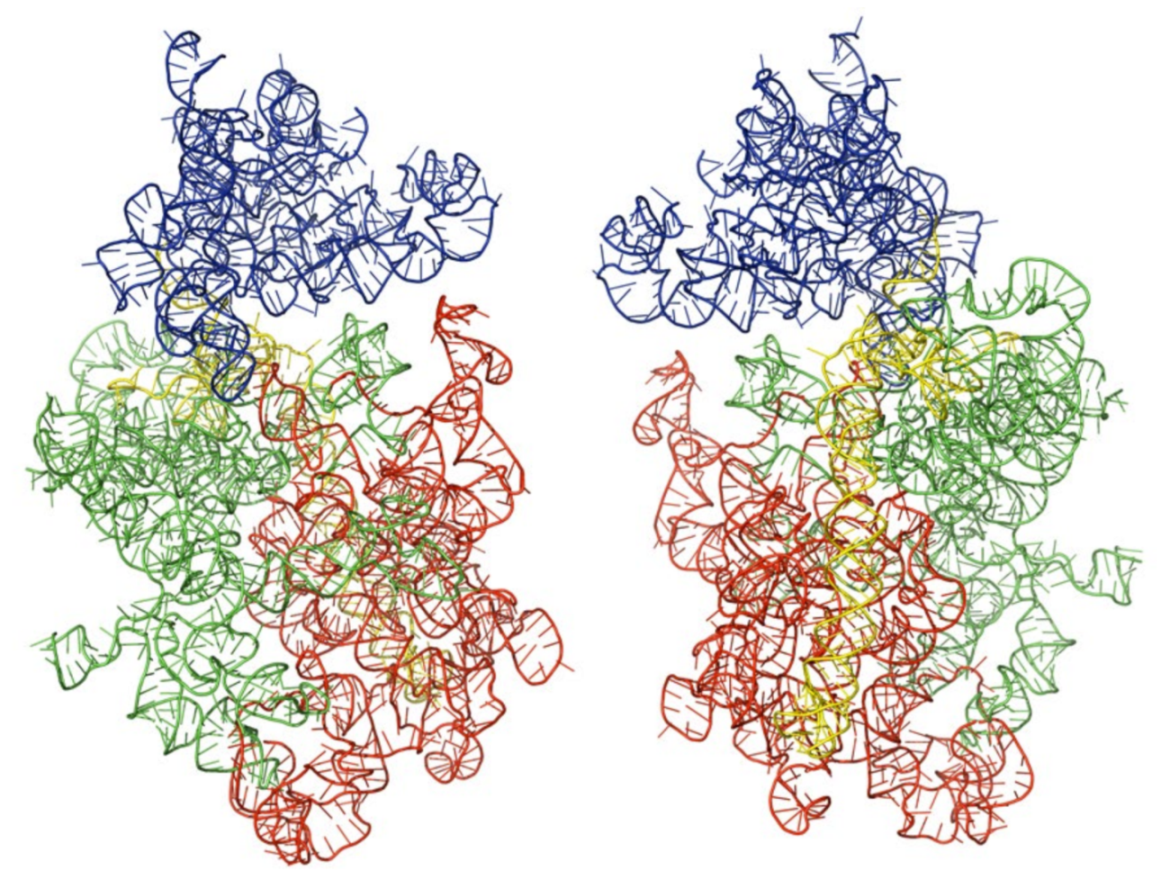
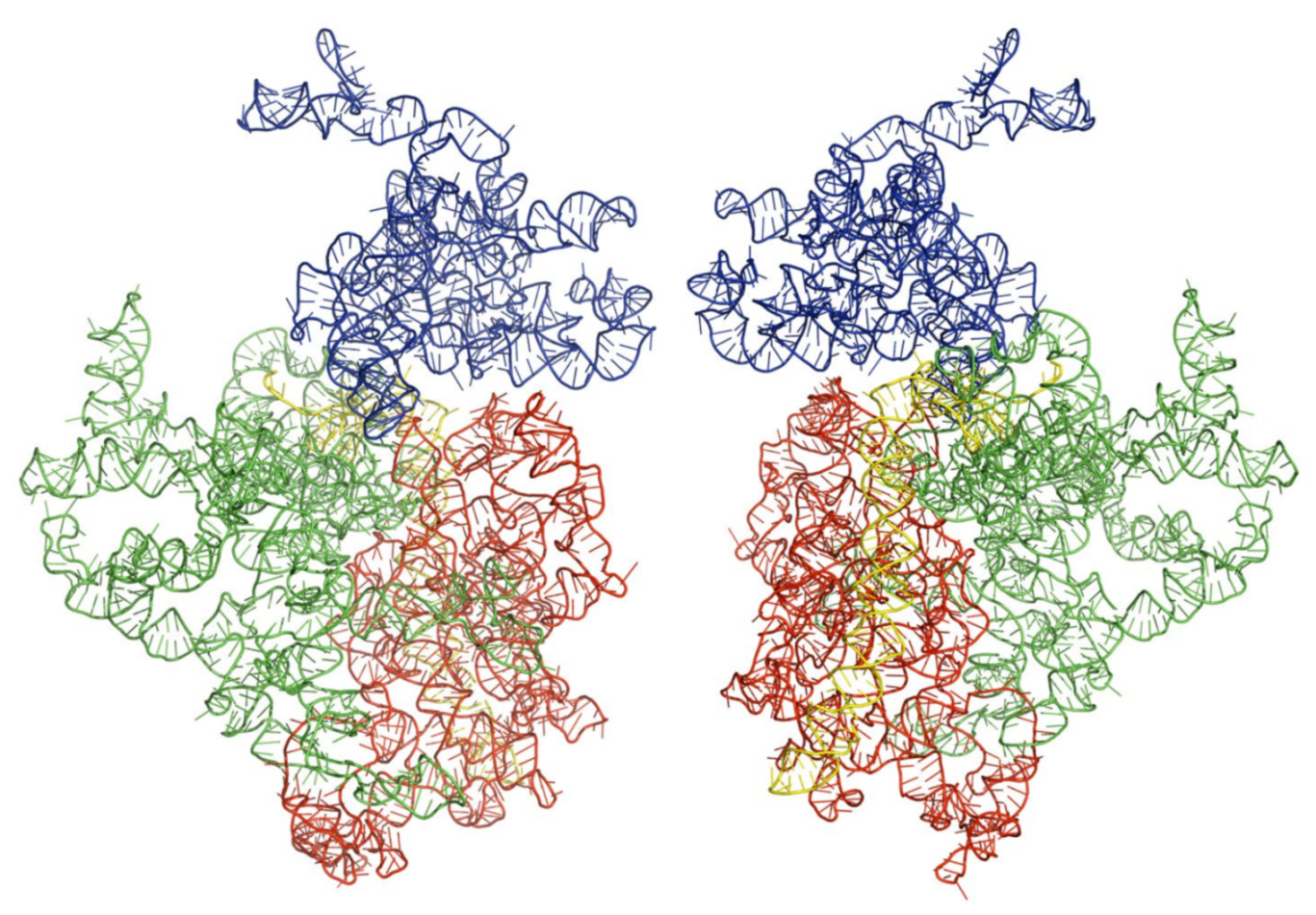
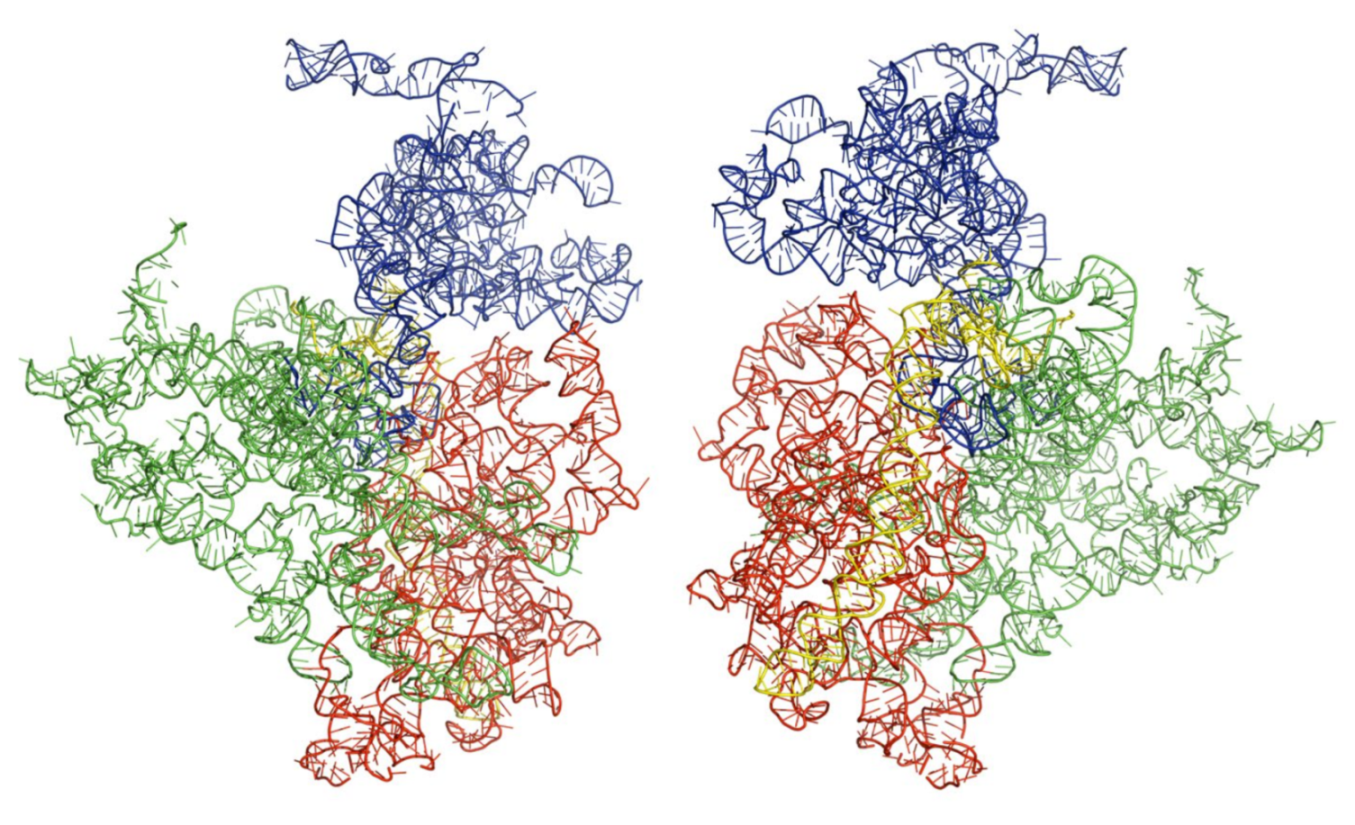
3.3. Three-Dimensional Structures of the Modeled Kinetoplastids
3.4. Myxobacterial Secondary Metabolites Databases
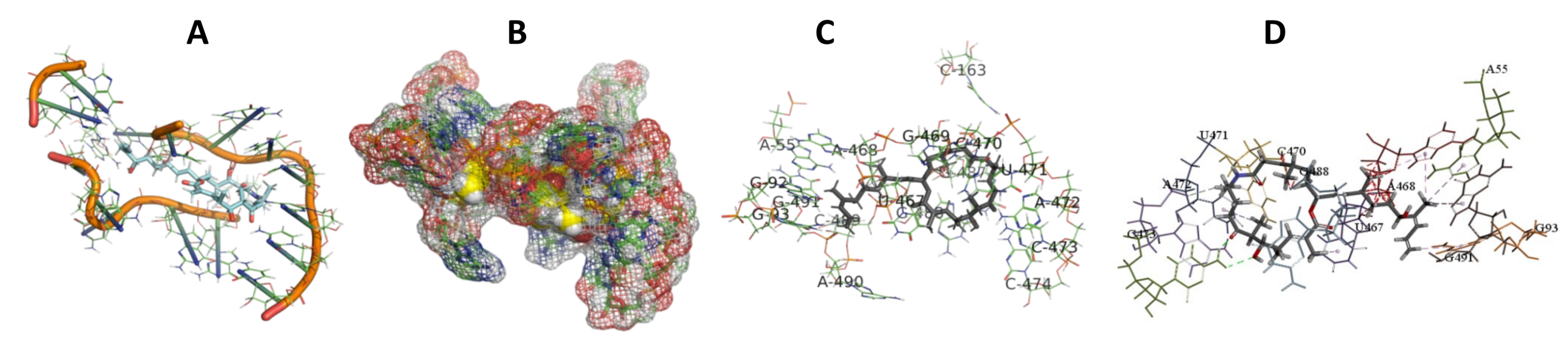
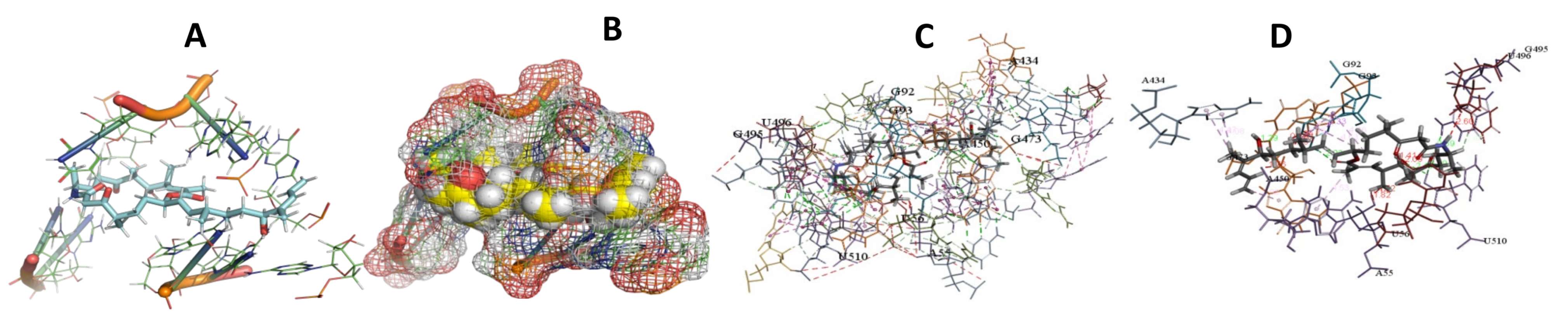
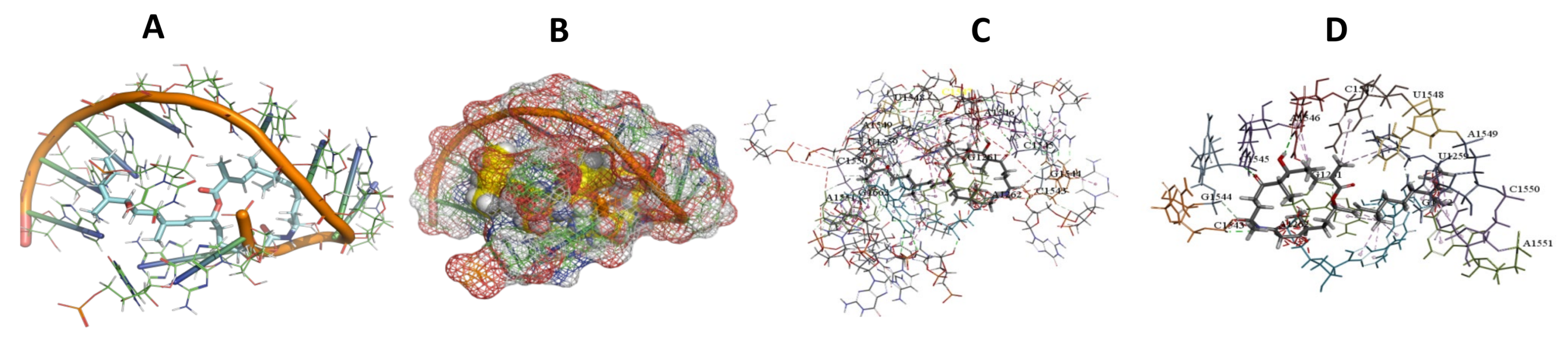
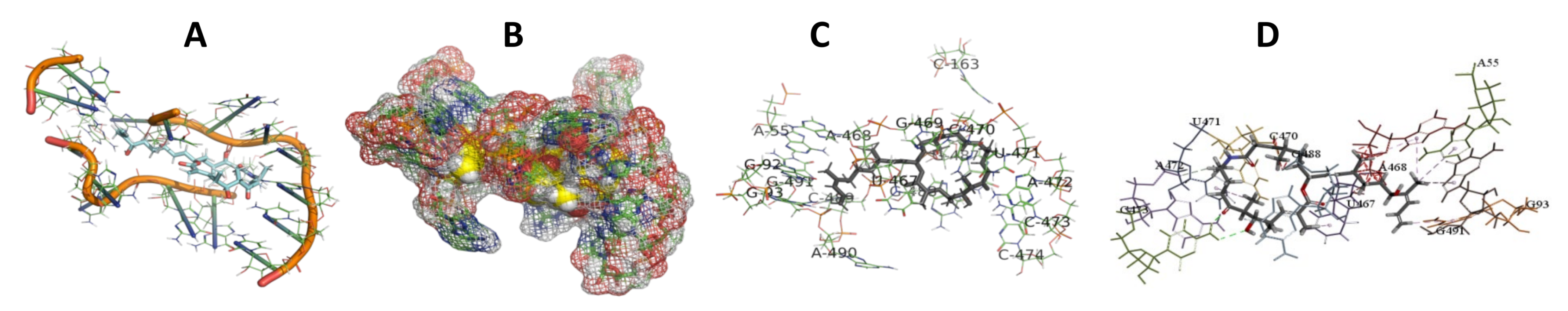
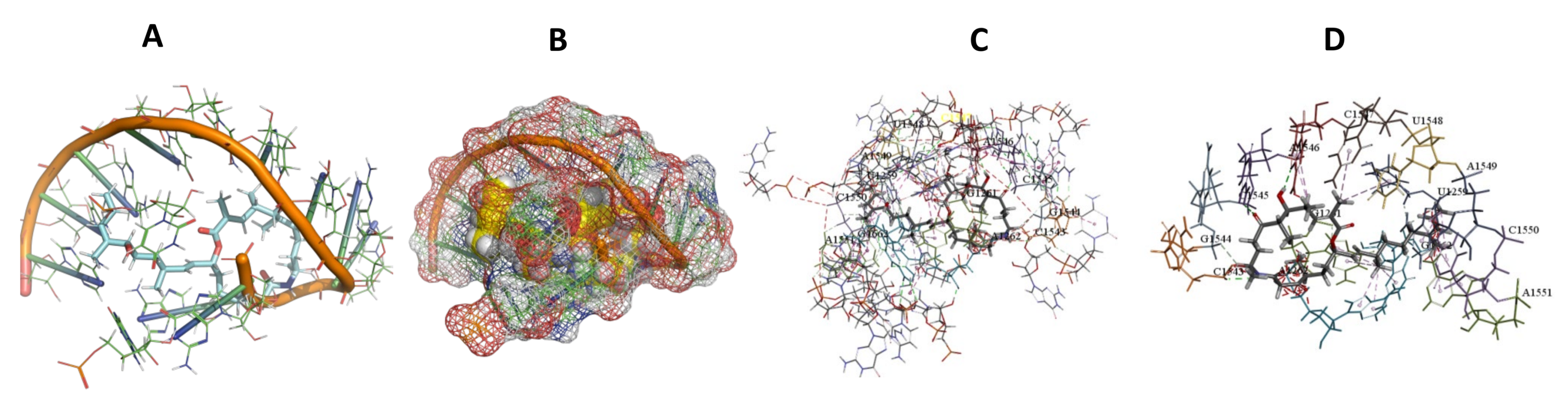
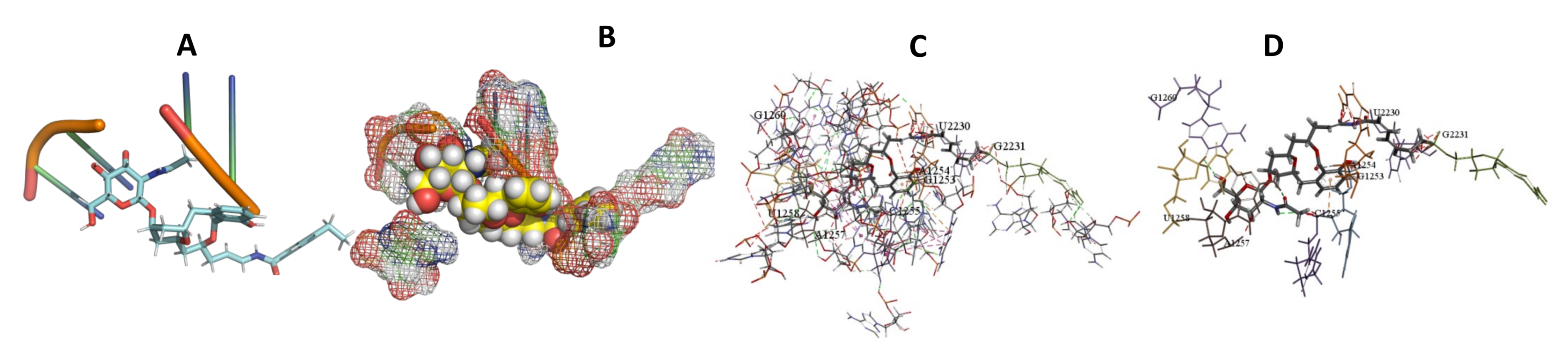
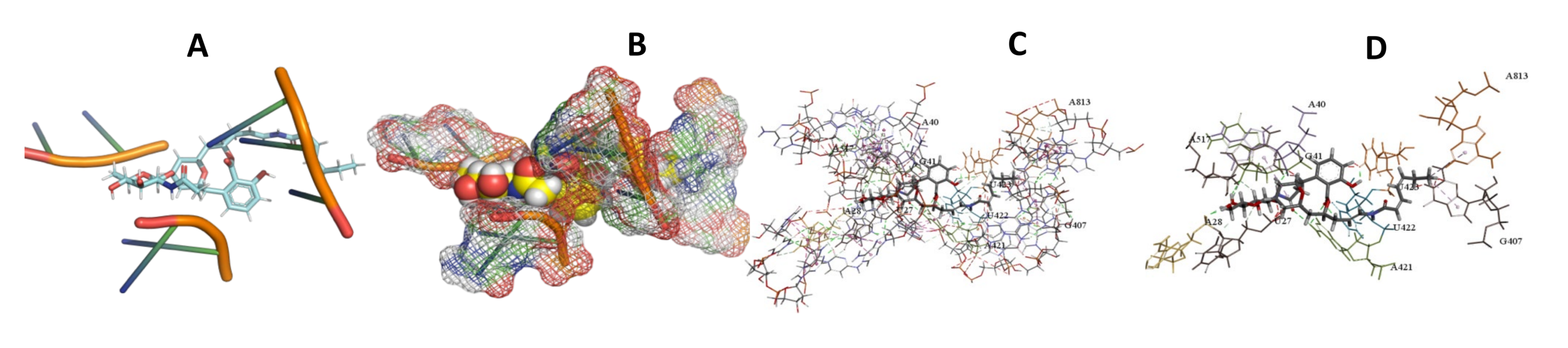
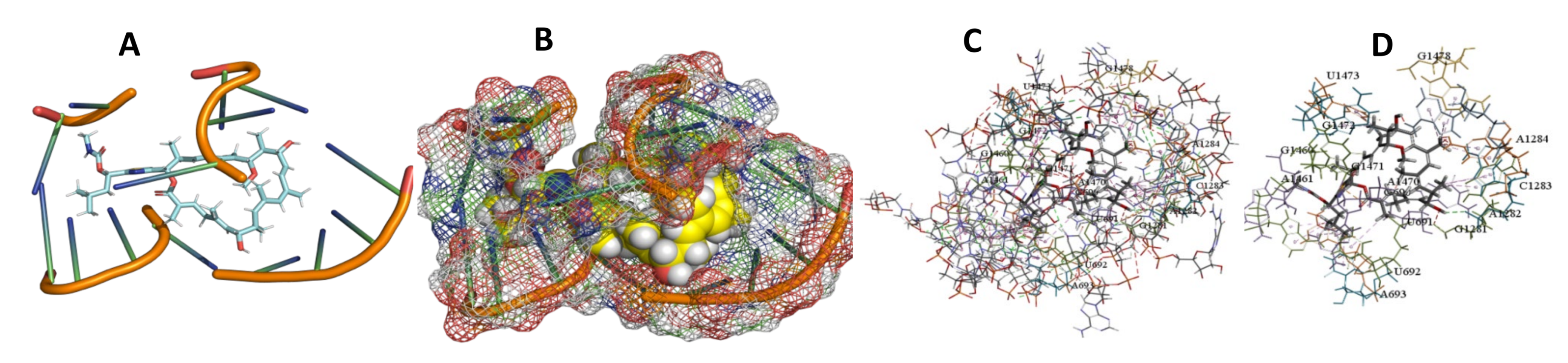
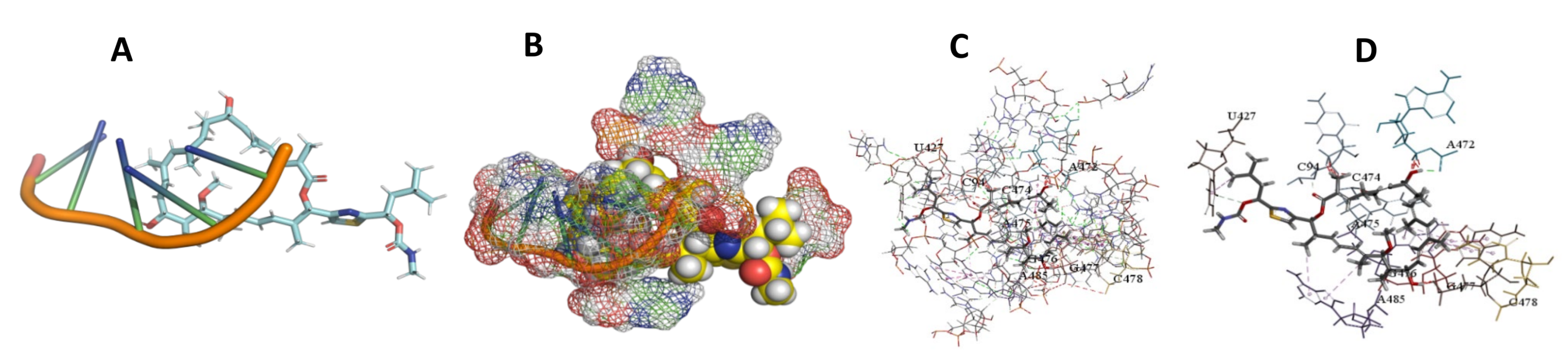
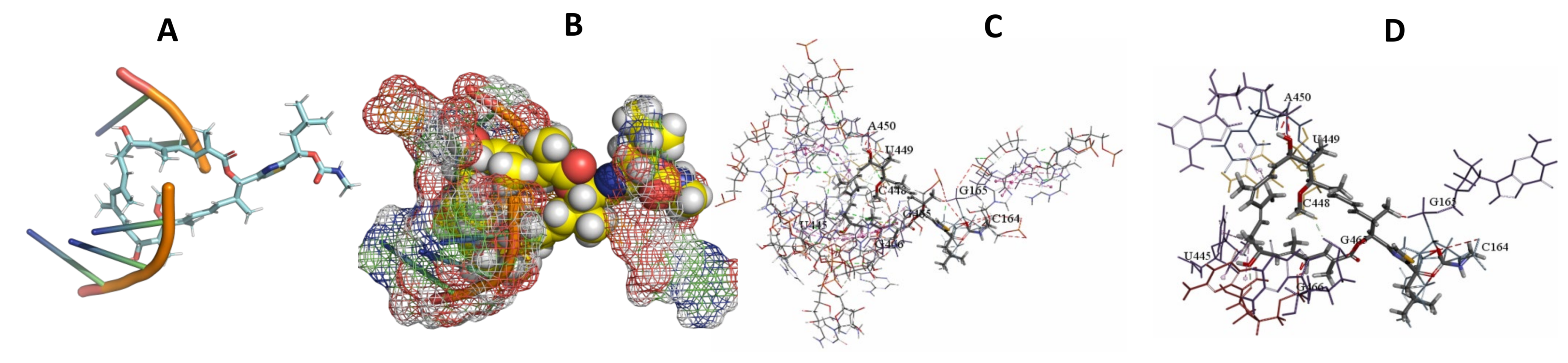
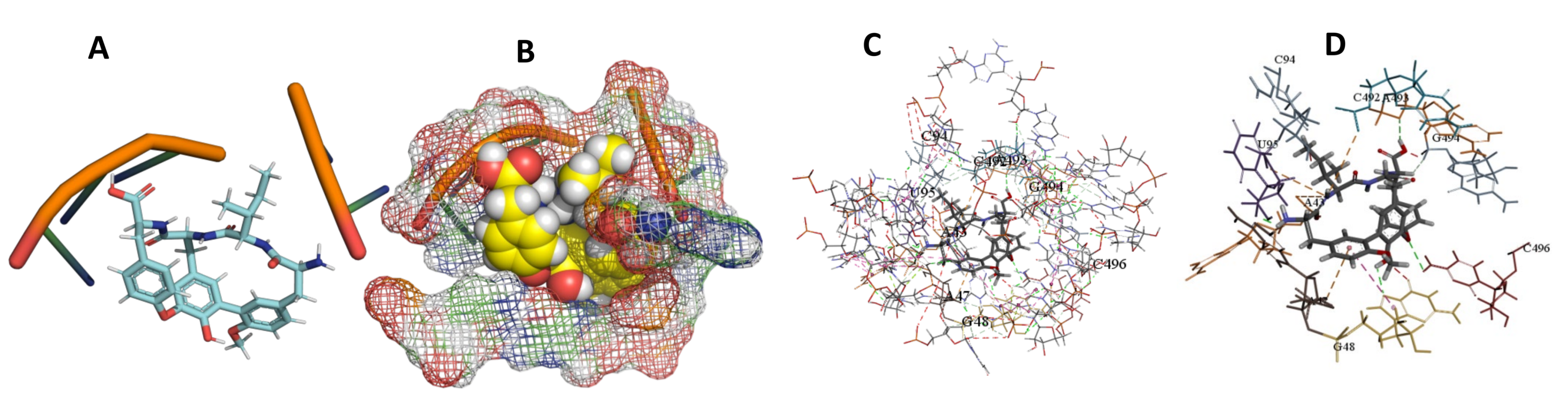
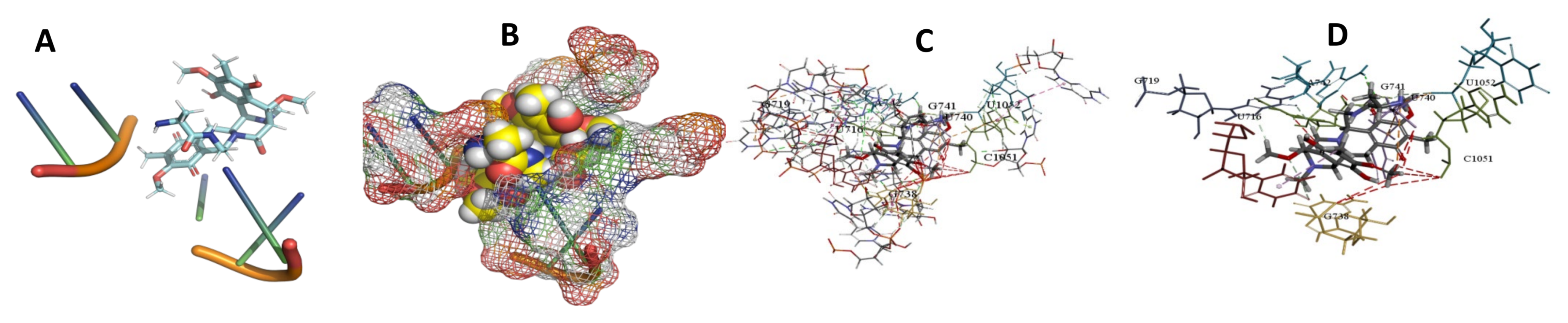
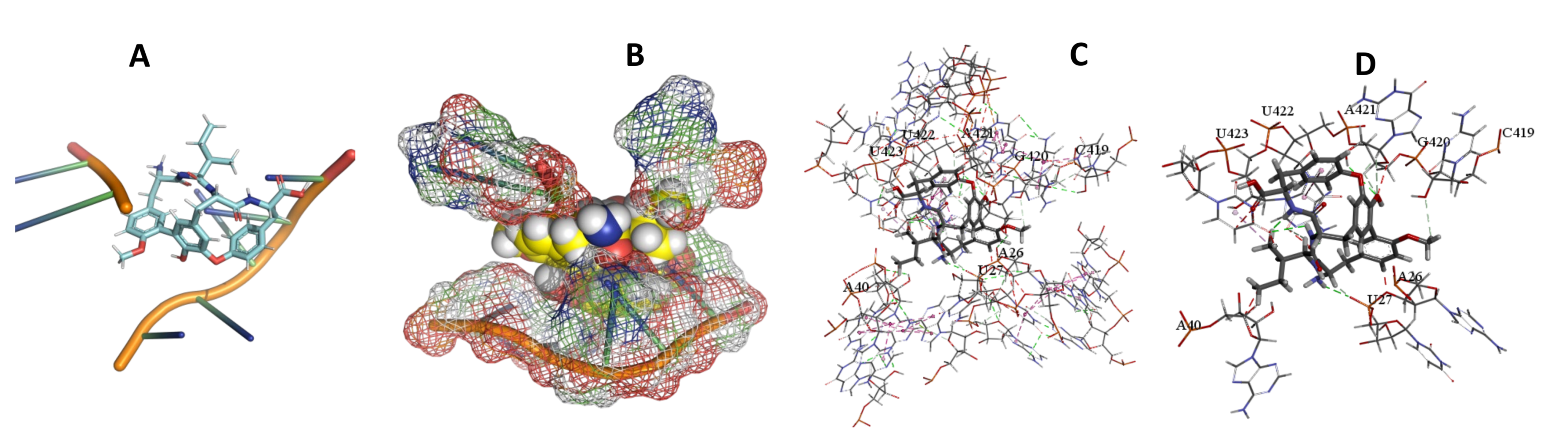
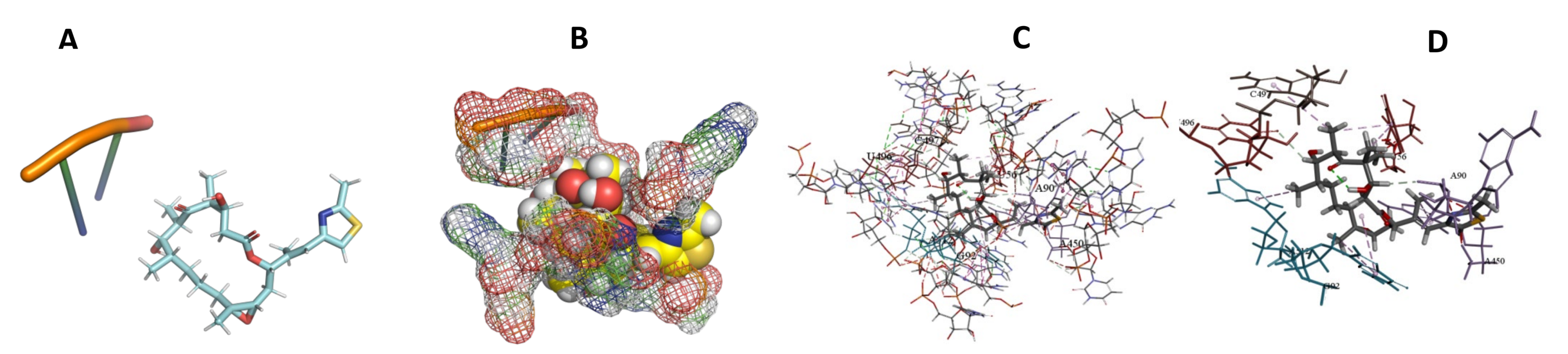
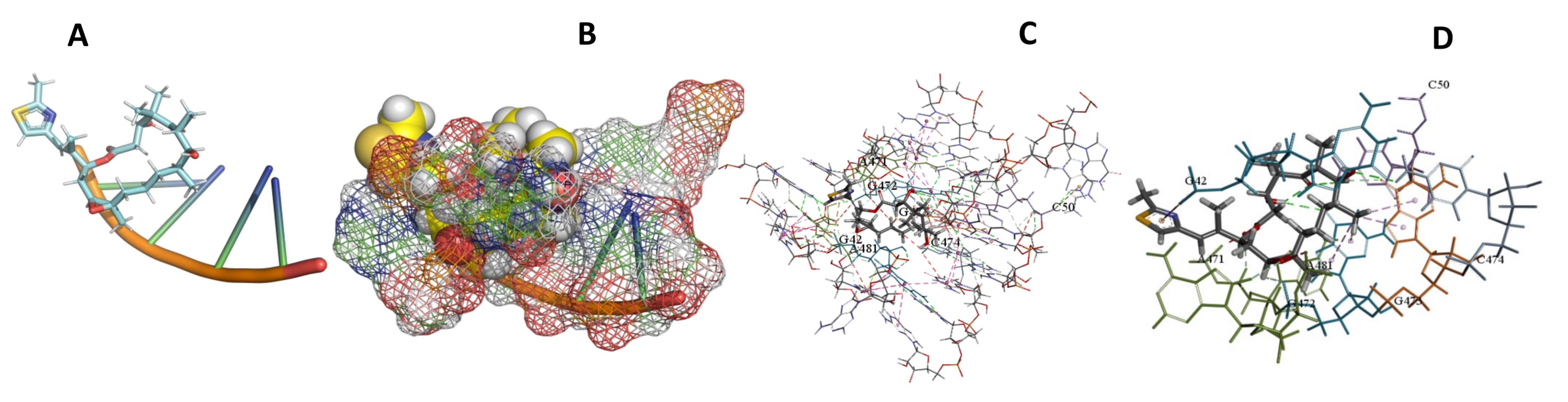
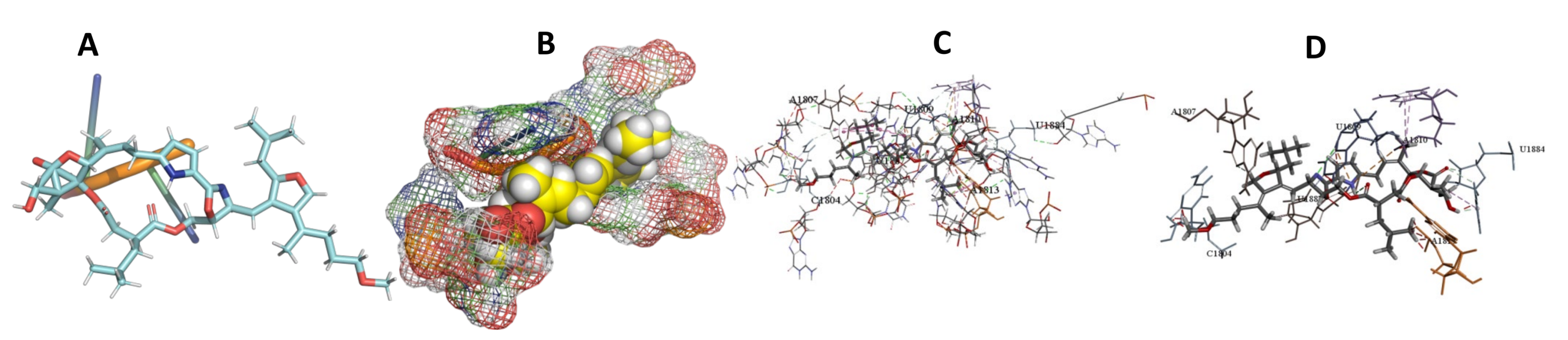
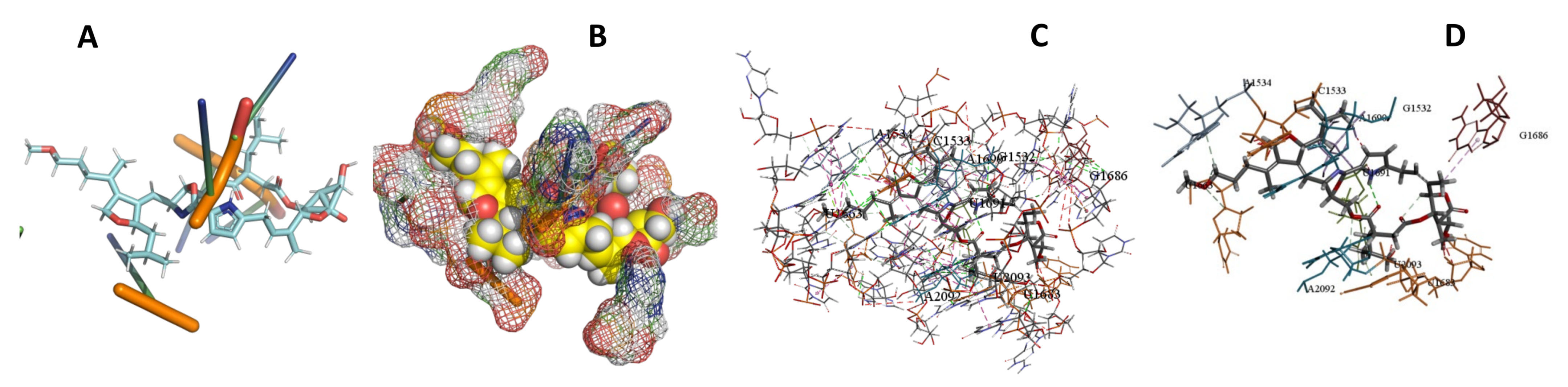
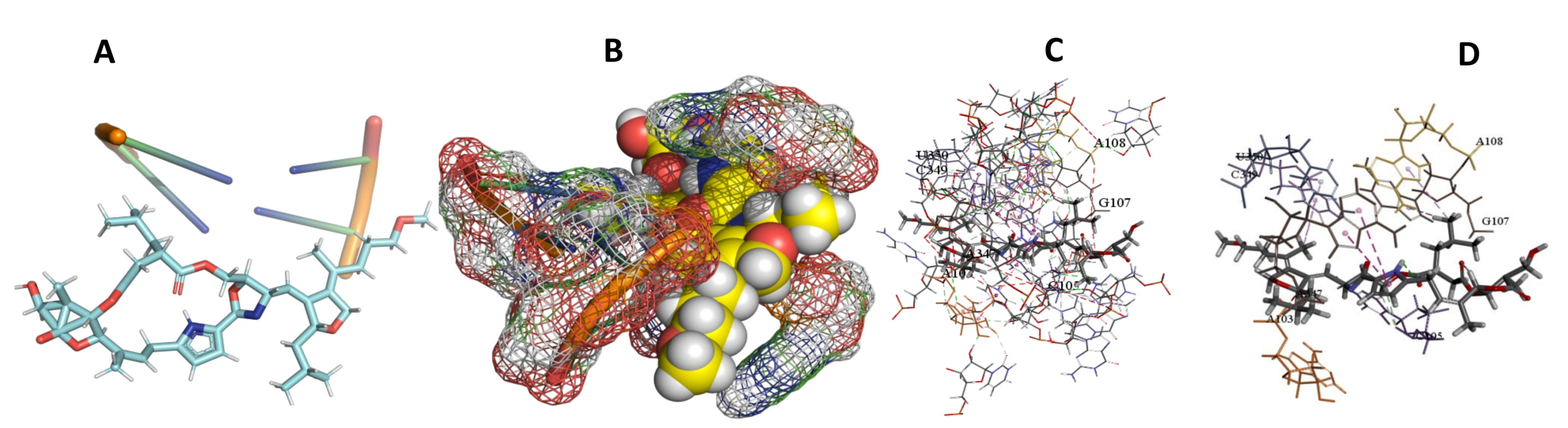
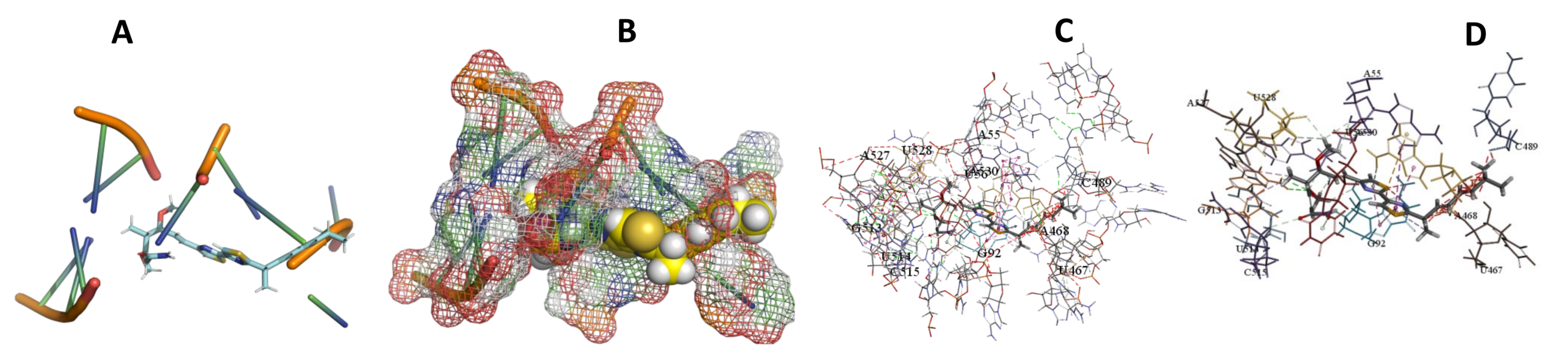
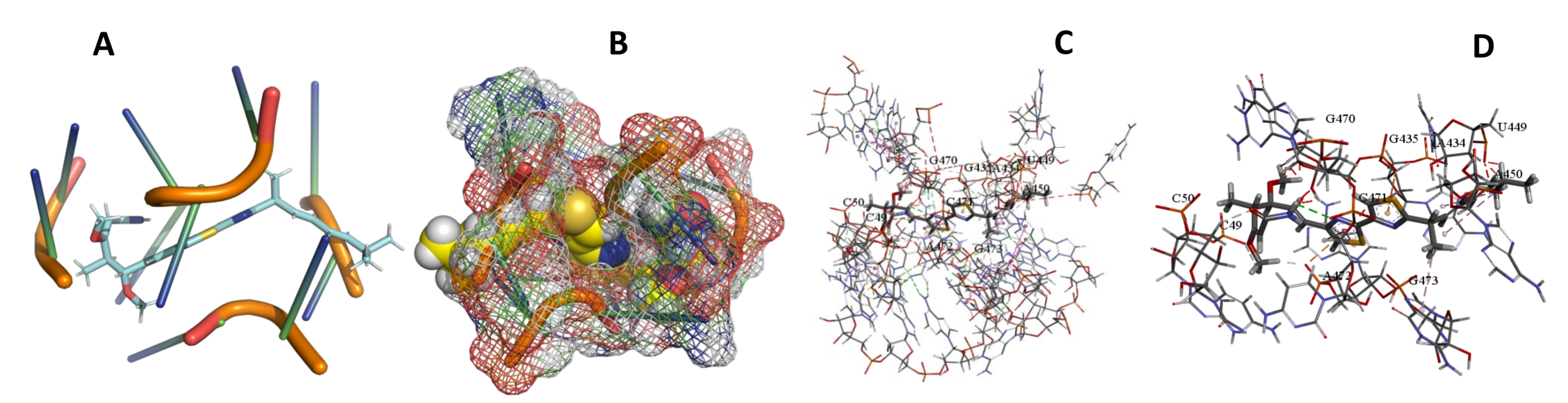
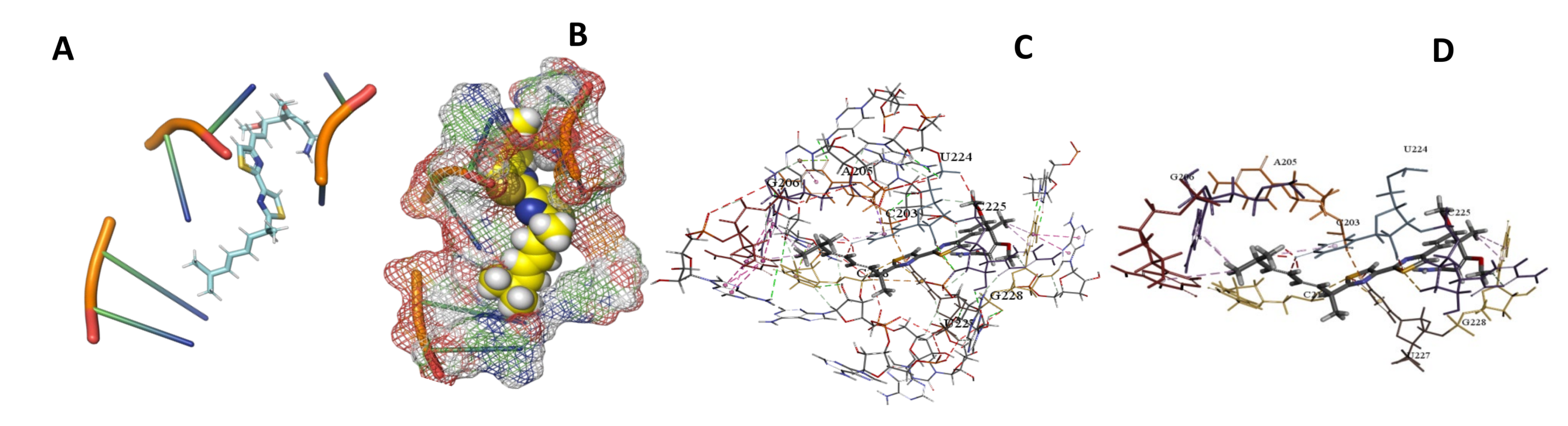
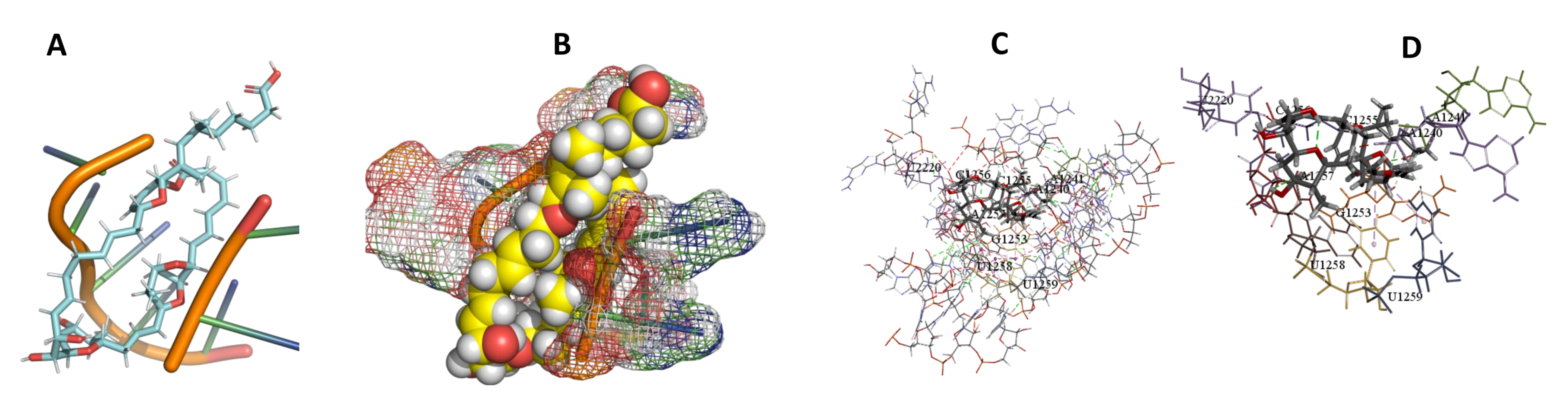
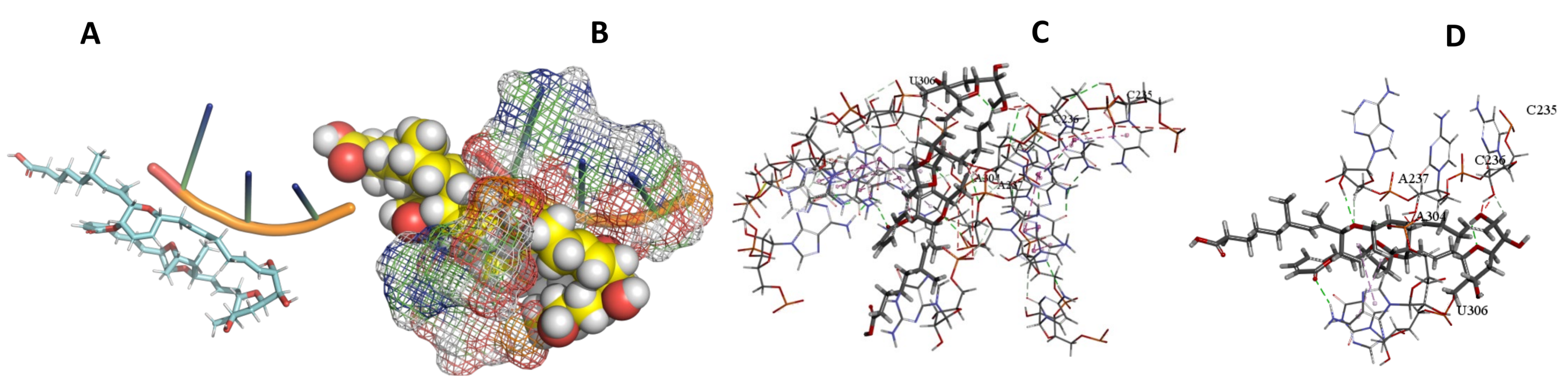
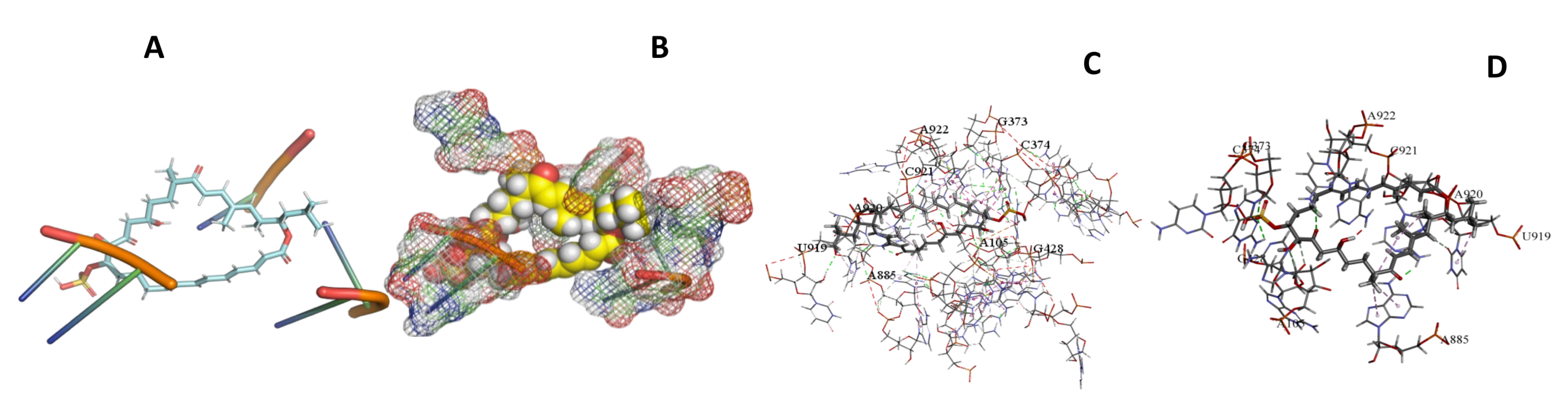
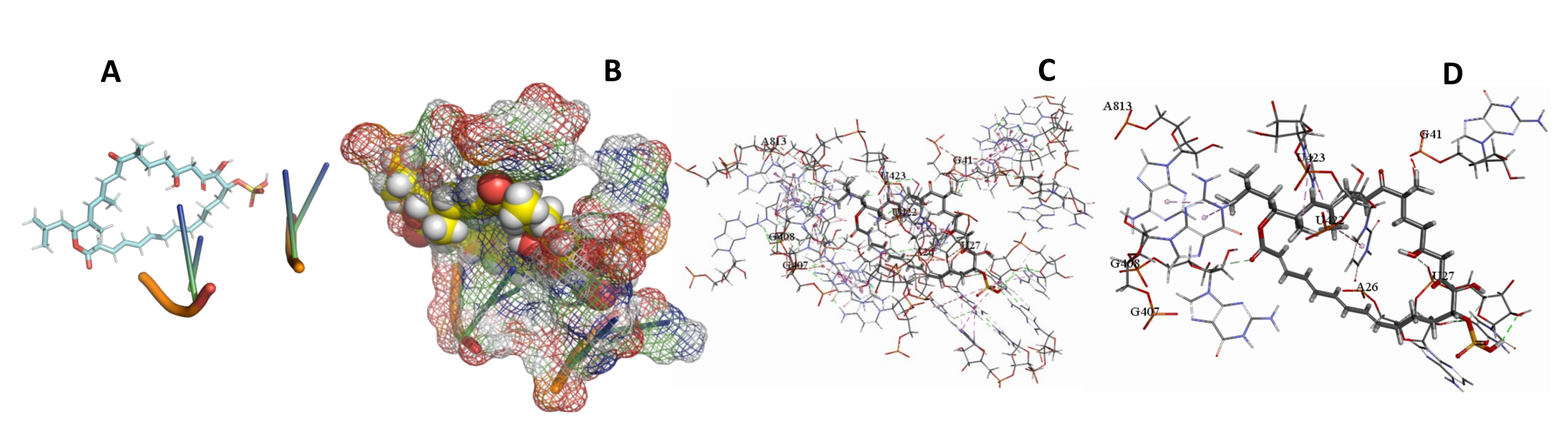
3.5. Binding and Docking Results
Selected Kinetoplastid 18S rRNA Structure Preparation
3.6. Binding Site Identification
- (1)
- Structure-based drug design: this method assumes that the structure of a biological target is known (the protein/DNA has been crystallized or a 3D model of the target is built). Compounds are then designed/screened to fit the structural characteristics of the target, thus rendering strong molecular interactions that stabilize the compound at the targets binding site. This technique is the most widely used in computational chemistry and yields a plethora of potential compounds that may then be screened for activity.
- (2)
- Ligand-based drug design: this method assumes that only the structure of the drug is known and that there is an absence of the 3D biological target. Optimized compounds are then designed based on the knowledge of the drug’s chemical analogs and their biological activity. Quantitative structure activity relationship (QSAR) features are designed based on physiochemical attributes of a set of chosen analogs and their biological activity with a target molecule. These QSAR features are then used as a template to screen for potential compounds with more favorable characteristics. Computational tools are now also available to predict potential targets of a compound prior to QSAR analysis.
3.7. Conclusion and Recommendation
4. Materials and Methods
4.1. Selection and Three-Dimentional Modeling of Kinetioplastids 18S rRNA
4.2. Selecting a Template
4.3. Obtaining and Verifying Template Sequences and Three-Dimensional Coordinate Files
4.4. Homology and De Novo Modeling
4.5. Ligand Docking Simulation
4.5.1. Active Site Prediction
4.5.2. Preparation of 18S rRNA and Ligand Molecules for Docking
4.6. Ligand Docking Simulation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgement
Conflicts of Interest
References
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Guertler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related protozoan pathogens, different diseases. J. Clin. Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, I.; Hodoki, Y.; Okazaki, Y.; Fujinaga, S.; Ohbayashi, K.; Nakano, S.-I. Widespread Dominance of Kinetoplastids and Unexpected Presence of Diplonemids in Deep Freshwater Lakes. Front. Microbiol. 2019, 10, 2375. [Google Scholar] [CrossRef]
- Sunyoto, T.; Boelaert, M.; Meheus, F. Understanding the economic impact of leishmaniasis on households in endemic countries: A systematic review. Expert Rev. Anti Infect. Ther. 2018, 17, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.S. The practical implications of comparative kinetoplastid genomics. In Comparative Genomics and Proteomics in Drug Discovery; CRC Press: Boca Raton, FL, USA, 2018; Volume 58, pp. 25–45. [Google Scholar]
- Escobar, P.; Matu, S.; Marques, C.; Croft, S.L. Sensitivities of Leishmania species to hexadecylphosphocholine (miltefosine), ET-18-OCH3 (edelfosine) and amphotericin B. Acta Trop. 2002, 81, 151–157. [Google Scholar] [CrossRef]
- Brun, R.; Schumacher, R.; Schmid, C.; Kunz, C.; Burri, C. The phenomenon of treatment failures in Human African Trypanosomiasis. Trop. Med. Int. Health 2001, 6, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; de Toranzo-Diaz, E. Toxic effects of nifurtimox and benznidazole, two drugs used against American trypanosomiasis (Chagas’ disease). Biomed. Environ. Sci. 1988, 1, 19–33. [Google Scholar]
- Crespillo-Andújar, C.; Chamorro-Tojeiro, S.; Norman, F.; Monge-Maillo, B.; López-Vélez, R.; Pérez-Molina, J. Toxicity of nifurtimox as second-line treatment after benznidazole intolerance in patients with chronic Chagas disease: When available options fail. Clin. Microbiol. Infect. 2018, 24, 1344.e1–1344.e4. [Google Scholar] [CrossRef]
- Atkins, J.F.; Gesteland, R.F.; Cech, T.R. RNA Worlds: From Life’s Origins to Diversity in Gene Regulation; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2010. [Google Scholar]
- Leontis, N.; Westhof, E. RNA 3D Structure Analysis and Prediction; Springer Science & Business Media: Berlin, Germany, 2012; Volume 27. [Google Scholar]
- AngelBello, A.J.; Rzuczek, S.G.; McKee, K.K.; Chen, J.L.; Olafson, H.; Cameron, M.D.; Moss, W.N.; Wang, E.T.; Disney, M.D. Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 7799–7804. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Wu, M.; Pushechnikov, A.; Aminova, O.; Disney, M.D. A Small Molecule Microarray Platform To Select RNA Internal Loop−Ligand Interactions. ACS Chem. Biol. 2007, 2, 745–754. [Google Scholar] [CrossRef]
- Disney, M.D. Targeting RNA with Small Molecules To Capture Opportunities at the Intersection of Chemistry, Biology, and Medicine. J. Am. Chem. Soc. 2019, 141, 6776–6790. [Google Scholar] [CrossRef]
- Steger, G.; Giegerich, R. RNA structure prediction. In RNA Structure and Folding; Klostermeier, D., Hammann, C., Eds.; De Gruyter: Berlin, Boston, 2013. [Google Scholar]
- Piatkowski, P.; Kasprzak, J.M.; Kumar, D.; Magnus, M.; Chojnowski, G.; Bujnicki, J.M. RNA 3D structure modeling by combination of template-based method ModeRNA, template-free folding with SimRNA, and refinement with QRNAS. In RNA Structure Determination; Humana Press: New York, NY, USA, 2016; pp. 217–235. [Google Scholar]
- Tahi, F.; Tran, v.T.; Boucheham, A. In Silico Prediction of RNA Secondary Structure. In Promoter Associated RNA: Methods and Protocols; Springer: New York, NY, USA, 2017; pp. 145–168. [Google Scholar]
- Gutell, R.R. Comparative Analysis of the Higher-Order Structure of RNA. In Biophysics of RNA Folding; Russell, R., Ed.; Springer New York: New York, NY, USA, 2013; pp. 11–22. [Google Scholar]
- Mwangi, H.N.; Wagacha, P.; Mathenge, P.; Sijenyi, F.; Mulaa, F. Structure of the 40S ribosomal subunit of Plasmodium falciparum by homology and de novo modeling. Acta Pharm. Sin. B 2017, 7, 97–105. [Google Scholar] [CrossRef]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009, 25, 1974. [Google Scholar] [CrossRef]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Zok, T.; Antczak, M.; Zurkowski, M.; Popenda, M.; Blazewicz, J.; Adamiak, R.W.; Szachniuk, M. RNApdbee 2.0: Multifunctional tool for RNA structure annotation. Nucleic Acids Res. 2018, 46, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Martinez, H.M.; Maizel, J.V.; Shapiro, B.A. RNA2D3D: A program for Generating, Viewing, and Comparing 3-Dimensional Models of RNA. J. Biomol. Struct. Dyn. 2008, 25, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M.; et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002, 3, 1–31. [Google Scholar]
- Weissman, K.J.; Müller, R. Myxobacterial secondary metabolites: Bioactivities and modes-of-action. Nat. Prod. Rep. 2010, 27, 1276–1295. [Google Scholar] [CrossRef]
- Bader, C.D.; Panter, F.; Müller, R. In depth natural product discovery - Myxobacterial strains that provided multiple secondary metabolites. Biotechnol. Adv. 2020, 39, 107480. [Google Scholar] [CrossRef]
- Whitworth, D.E. Genome-wide analysis of myxobacterial two-component systems: Genome relatedness and evolutionary changes. BMC Genom. 2015, 16, 780. [Google Scholar] [CrossRef] [PubMed]
- Dehhaghi, M.; Mohammadipanah, F.; Guillemin, G.J. Myxobacterial natural products: An under-valued source of products for drug discovery for neurological disorders. NeuroToxicology 2018, 66, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Mueller, R. The biosynthetic potential of myxobacteria and their impact in drug discovery. Curr. Opin. Drug Discov. Dev. 2009, 12, 220. [Google Scholar]
- Hoffmann, T.; Krug, D.; Bozkurt, N.; Duddela, S.; Jansen, R.; Garcia, R.; Gerth, K.; Steinmetz, H.; Müller, R. Correlating chemical diversity with taxonomic distance for discovery of natural products in myxobacteria. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Garcia, R.O.; Krug, D.; Müller, R. Discovering natural products from myxobacteria with emphasis on rare producer strains in combination with improved analytical methods. Methods Enzymol. 2009, 458, 59–91. [Google Scholar] [PubMed]
- Nett, M.; König, G.M. The chemistry of gliding bacteria. Nat. Prod. Rep. 2007, 24, 1245–1261. [Google Scholar] [CrossRef]
- Mwangi, H.N.; Gitonga, P.M.; Wagacha, P.W.; Sijenyi, F.; Mulaa, F. Integrating mechanism-based screening paradigm into homology and de novo modeling exemplified by Mycobacterium Tuberculosis 30S ribosomal structure and its potential application as a screening target. Int. J. Sci. Eng. Res. 2018, 9, 305–317. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Berman, H.M.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of Ensemble-Average Pairwise Root Mean-Square Deviation from Experimental B-Factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Studio, D. Discovery Studio; Accelrys: San Diego, CA, USA, 2008. [Google Scholar]
- Bhardwaj, P.; Biswas, G.P.; Bhunia, B. Docking-based inverse virtual screening strategy for identification of novel protein targets for triclosan. Chemosphere 2019, 235, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R.; Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L. Schrodinger Suite 2012 Induced Fit. Docking Protocol; Glide Version 5.8; Schrodinger, LLC: New York, NY, USA, 2012. [Google Scholar]
- Schrödinger Glide; LLC: New York, NY, USA, 2017.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Row # | Organism (2) | L(3) | RT(4) | RC | Size | Cmp | Acc | Common Name | Phylogeny[M] (1) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Trypanosoma brucei | N | R | 16S | 2251 | 100 | M12676 | kinetoplasts | cellular organisms …» |
| 2 | Trypanosoma cruzi | N | R | 16S | 2315 | 100 | AF245382 | kinetoplasts | cellular organisms …» |
| 3 | Leishmania major | N | R | 16S | 2203 | 100 | AC005806 | kinetoplasts | cellular organisms …» |
| Species | Name | 18SrRNA.std.egy | 18SrRNA.opt.egy |
|---|---|---|---|
| Leishmania major | Total Inter energy | 908487.3182 | −83582.63882 |
| Total intra energy (-Gamma en | −17864.6686 | −17828.1188 | |
| Total Gamma Terms Energy | 1746.8654 | 1743.13568 | |
| Total Gap Geometry Penalty | 3108.37922 | 2746.02559 | |
| Total Restraint Energy | 0 | 3550.87301 | |
| TOTAL STRUCTURE ENERGY | 895477.8943 | −96921.59635 | |
| Trypanosoma brucei | Total Inter energy | 2021190.532 | −102281.5112 |
| Total intra energy (-Gamma en | 71145.86365 | 10625.04139 | |
| Total Gamma Terms Energy | 2357.05629 | 2277.69707 | |
| Total Gap Geometry Penalty | 24166.41849 | 9500.81691 | |
| Total Restraint Energy | 0 | 8078.41829 | |
| TOTAL STRUCTURE ENERGY | 2118859.871 | −79877.95586 | |
| Trypanosoma cruzi | Total Inter energy | 7208497.219 | −98209.94034 |
| Total intra energy (-Gamma en | 208432.083 | −7806.99519 | |
| Total Gamma Terms Energy | 2458.82781 | 2440.13835 | |
| Total Gap Geometry Penalty | 35870.86855 | 12018.7965 | |
| Total Restraint Energy | 0 | 10017.37844 | |
| TOTAL STRUCTURE ENERGY | 7455258.998 | −91558.00067 |
| Compound Name | Compounds with Activity on All More Negative Kinetoplastids, ACE −400 | ||
|---|---|---|---|
| T. Brucei | T. Cruzi | L. Major | |
| Angiolam A | −491.7 | −673.71 | −550.93 |
| Apicularen B | −549.58 | −529.41 | −585.93 |
| Archazolid A | −516.32 | −470.74 | −413.53 |
| Cittilin A | −495.42 | −529.71 | −520.78 |
| Epothilone B | −573.04 | −513.65 | −346.85 |
| Leupyrin | −598.53 | −648.66 | −393.82 |
| Myxothiazol | −595.18 | −573.36 | −449.9 |
| Sorangicin A | −466.93 | −466.93 | −456.49 |
| Spirangien B | −503.52 | −576.45 | −516.62 |
| Sulfangolid A | −613.53 | −613.53 | −643.25 |
| Compound Name | Compounds with Activity on All More Negative Kinetoplastids ACE −400 | |||||
|---|---|---|---|---|---|---|
| T. Brucei | T. Cruzi | L. Major | ||||
| Angiolam A | −491.7 | G92,G93,A434,A450,G470,G473,G495,U496,U510 | −673.71 | A55,U56,G92,G93,A434,A450,G473,G495,U496,U510 | −550.93 | U1259,G1261,A1262,C1543,G1544,C1545,A1546,C1547,U1548,A1549,C1550,A1551,G1662 |
| Apicularen B | −549.58 | G1253,A1254,C1255,A1257,U1258,G1260,U2230,G2231 | −529.41 | G1109,U1110,A1134,C1135,U1150,G1151,U1152,C1153 | −585.93 | U27,A28,A40,G41,G407,A421,U422,U423,A813 |
| Archazolid A | −516.32 | G690,U691,U692,A693,G1281,A1282,C1283,A1284,G1460,A1461,A1470,G1471,G1472,U1473,G1478 | −470.74 | C94,U427,A472,C474,A475,G476,G477,C478,A485 | −413.53 | C164,G165,U445,C448,U449,A450,G465,G466, |
| Cittilin A | −495.42 | A43,A47,G48,C94,U95,C492,A493,G494,C496TTTT | −529.71 | U716,G719,G738,U740,G741,A742,C1051,U1052 | −520.78 | A26,U27,A40,C419,G420,A421,U422,U423, |
| Epothilone B | −573.04 | A55,U56,A90,U91,G92,A468,G513,U514,C515,A527,U528,A530 | −513.65 | U56,A90,G92,A450,U496,C497,A512 | −346.85 | G42,C50,A471,G472,G473,C474,A481 |
| Leupyrin | −598.53 | G1532,C1533,A1534,U1663,U1683,G1686,A1690,U1691,A2092,U2093 | −648.66 | C1804,A1807,U1809,A1810,A1813,U1884,U1887 | −393.82 | A103,C105,G107,A108,A347,C349,U350 |
| Myxothiazol | −595.18 | A55,U56,G92,U467,A468,C489,G513,U514,C515,A527,U528,A530 | −573.36 | C49,C50,A434,G435,U449,A450,G470,C471,A472,G473, | −449.9 | C203,A205,G206,C218,U224,C225,U227,G228, |
| Sorangicin A | −466.93 | A1240,A1241,G1253,C1255,C1256,A1257,U1258,U1259,U2220 | −466.93 | −456.49 | C235,C236,A237,A304,U306, | |
| Spirangien B | −503.52 | C59,U60,A64,G79,G80,A520,A521,C525,G526 | −576.45 | A100,G106,G407,G409,C423,G424,A864,A902 | −516.62 | A1294,C1535,C1536,A1538,A1549,A1642,U1644,U1645,A1689 |
| Sulfangolid A | −613.53 | A105,G373,C374,G428,A885,U919,A920,C921,A922 | −613.53 | −643.25 | A26,U27,G41,G407,G408,U422,U423,A813 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwangi, H.N.; Muge, E.K.; Wagacha, P.W.; Ndakala, A.; Mulaa, F.J. Methods for Identifying Microbial Natural Product Compounds that Target Kinetoplastid RNA Structural Motifs by Homology and De Novo Modeled 18S rRNA. Int. J. Mol. Sci. 2021, 22, 4493. https://doi.org/10.3390/ijms22094493
Mwangi HN, Muge EK, Wagacha PW, Ndakala A, Mulaa FJ. Methods for Identifying Microbial Natural Product Compounds that Target Kinetoplastid RNA Structural Motifs by Homology and De Novo Modeled 18S rRNA. International Journal of Molecular Sciences. 2021; 22(9):4493. https://doi.org/10.3390/ijms22094493
Chicago/Turabian StyleMwangi, Harrison Ndung’u, Edward Kirwa Muge, Peter Waiganjo Wagacha, Albert Ndakala, and Francis Jackim Mulaa. 2021. "Methods for Identifying Microbial Natural Product Compounds that Target Kinetoplastid RNA Structural Motifs by Homology and De Novo Modeled 18S rRNA" International Journal of Molecular Sciences 22, no. 9: 4493. https://doi.org/10.3390/ijms22094493
APA StyleMwangi, H. N., Muge, E. K., Wagacha, P. W., Ndakala, A., & Mulaa, F. J. (2021). Methods for Identifying Microbial Natural Product Compounds that Target Kinetoplastid RNA Structural Motifs by Homology and De Novo Modeled 18S rRNA. International Journal of Molecular Sciences, 22(9), 4493. https://doi.org/10.3390/ijms22094493