
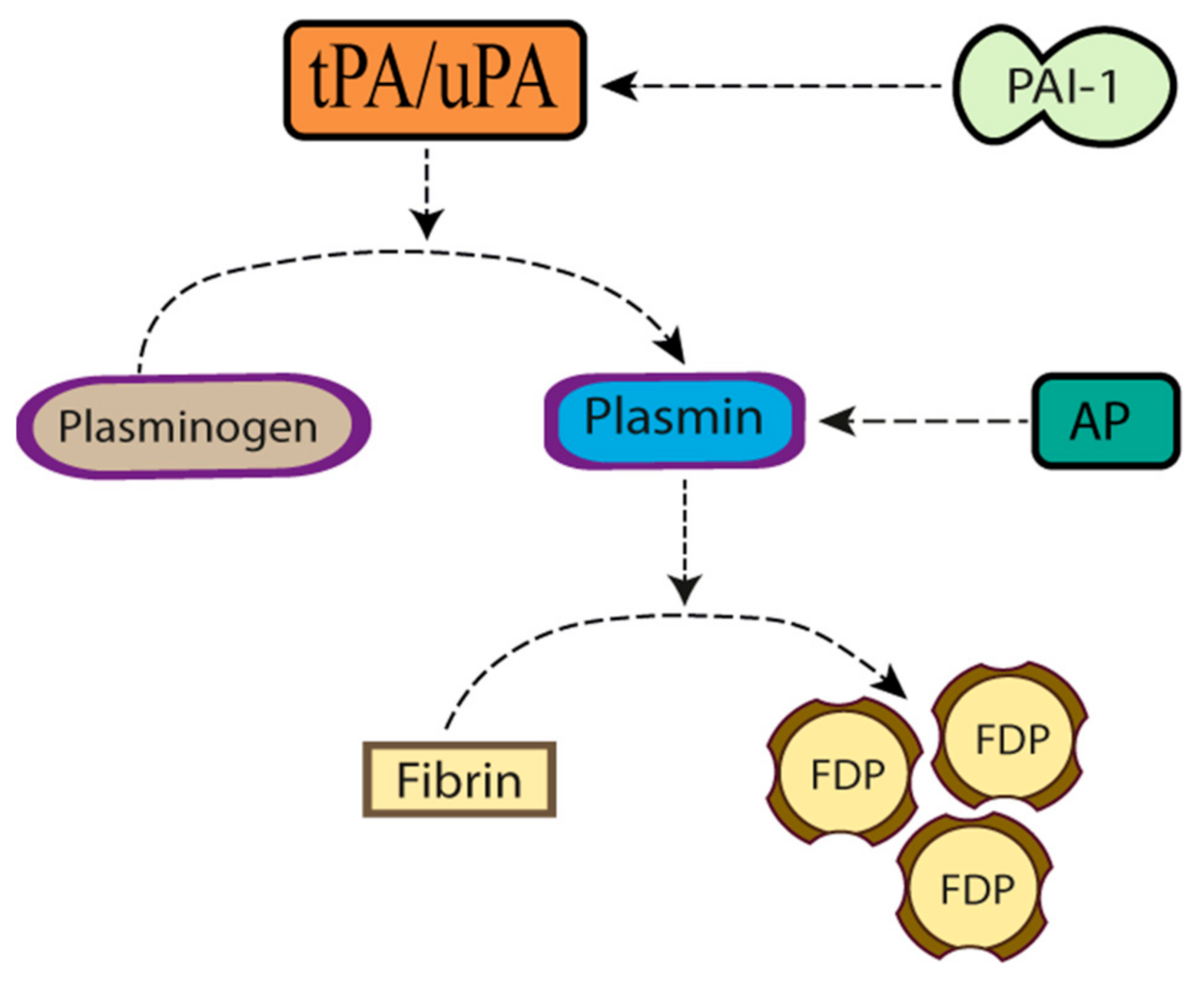
Plasminogen Activators in Neurovascular and Neurodegenerative Disorders



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Neurovascular Unit
3. Plasminogen Activators in the Neurovascular Unit under Physiological Conditions
3.1. Tissue-Type Plasminogen Activator
3.2. Tissue-Type Plasminogen Activator in the Neurovascular Unit
3.2.1. Cerebral Endothelial Cells
3.2.2. Pericytes
3.2.3. Astrocytes
3.2.4. tPA in Microglia
3.2.5. Neuronal tPA
3.3. Urokinase-Type Plasminogen Activator
3.4. Urokinase-Type Plasminogen Activator in the Neurovascular Unit (NVU)
3.4.1. Cerebral Endothelial Cells
3.4.2. Astrocytes and Microglia
3.4.3. Neurons
4. Plasminogen Activators in the Neurovascular Unit (NVU) under Ischemic Conditions
4.1. Endothelial Cells
4.1.1. Tissue-Type Plasminogen Activator
4.1.2. Urokinase-Type Plasminogen Activator
4.2. Astrocytes
4.2.1. Tissue-Type Plasminogen Activator
4.2.2. Urokinase-Type Plasminogen Activator
4.3. Microglia
4.3.1. Tissue-Type Plasminogen Activator
4.3.2. Urokinase-Type Plasminogen Activator
4.4. Neurons
4.4.1. Tissue-Type Plasminogen Activator
4.4.2. Urokinase-Type Plasminogen Activator
5. Plasminogen Activators in Neurodegenerative Disorders
5.1. Plasminogen Activators in Alzheimer’s Disease
5.2. Endothelial Cells
5.3. Astrocytes
5.4. Microglia
5.5. Neurons
5.5.1. Plasminogen Activators and the Formation of Aβ Deposits
5.5.2. Role of Plasminogen Activators in Synaptic Dysfunction in AD
5.5.3. Plasminogen Activators, Physical Activity and Alzheimer’s Disease
5.6. Plasminogen Activators in Parkinson’s Disease
5.7. Plasminogen Activators in Amyotrophic Lateral Sclerosis (ALS)
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Raum, D.; Marcus, D.; Alper, C.A.; Levey, R.; Taylor, P.D.; Starzl, T.E. Synthesis of Human Plasminogen by the Liver. Science 1980, 208, 1036–1037. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.E.; Martzen, M.R.; Ichinose, A.; Davie, E.W. Characterization of the Gene for Human Plasminogen, A Key Proenzyme in the Fibrinolytic System. J. Biol. Chem. 1990, 265, 6104–6111. [Google Scholar] [CrossRef]
- Das, R.; Pluskota, E.; Plow, E.F. Plasminogen and Its Receptors as Regulators of Cardiovascular Inflammatory Responses. Trends Cardiovasc. Med. 2010, 20, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Kim, S.-O.; Felez, J.; Grella, D.K.; Castellino, F.J.; Miles, L.A. Conversion of Glu-Plasminogen to Lys-Plasminogen Is Necessary for Optimal Stimulation of Plasminogen Activation on the Endothelial Cell Surface. J. Biol. Chem. 2001, 276, 19078–19083. [Google Scholar] [CrossRef]
- Lawrence, D.; Strandberg, L.; Grundström, T.; Ny, T. Purification of Active Human Plasminogen Activator Inhibitor 1 from Escherichia Coli. Comparison with Natural and Recombinant Forms Purified from Eucaryotic Cells. Eur. J. Biochem. 1989, 186, 523–533. [Google Scholar] [CrossRef]
- Draxler, D.F.; Sashindranath, M.; Medcalf, R.L. Plasmin: A Modulator of Immune Function. Semin. Thromb. Hemost. 2016, 43, 143–153. [Google Scholar] [CrossRef]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit—Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Pennica, D.; Holmes, W.E.; . Kohr, W.J.; Harkins, R.N.; Vehar, G.A.; Ward, C.A.; Bennett, W.F.; Yelverton, E.; Seeburg, P.H.; Heyneker, H.L.; et al. Cloning and Expression of Human Tissue-Type Plasminogen-Activator Cdna in Escherichia-Coli. Nature 1983, 301, 214–221. [Google Scholar] [CrossRef]
- Yepes, M.; Roussel, B.D.; Ali, C.; Vivien, D. Tissue-Type Plasminogen Activator in the Ischemic Brain: More than a Thrombolytic. Trends Neurosci. 2009, 32, 48–55. [Google Scholar] [CrossRef]
- Kim, J.A.; Tran, N.D.; Wang, S.-J.; Fisher, M.J. Astrocyte Regulation of Human Brain Capillary Endothelial Fibrinolysis. Thromb. Res. 2003, 112, 159–165. [Google Scholar] [CrossRef]
- Huber, D.; Cramer, E.M.; Kaufmann, J.E.; Meda, P.; Massé, J.-M.; Kruithof, E.K.O.; Vischer, U.M. Tissue-Type Plasminogen Activator (t-PA) is Stored in Weibel-Palade Bodies in Human Endothelial Cells Both in Vitro and in Vivo. Blood 2002, 99, 3637–3645. [Google Scholar] [CrossRef]
- Hanss, M.; Collen, D. Secretion of Tissue-Type Plasminogen Activator and Plasminogen Activator Inhibitor by Cultured Human Endothelial Cells: Modulation by Thrombin, Endotoxin, and Histamine. J. Lab. Clin. Med. 1987, 109, 97–104. [Google Scholar]
- Pepper, M.S.; Rosnoblet, C.; Di Sanza, C.; Kruithof, E.K. Synergistic Induction of t-PA by VASCULAR endothelial Growth Factor and Basic Fibroblast Growth Factor and Localization of t-PA to Weibel-Palade Bodies in Bovine Microvascular Endothelial Cells. Thromb. Haemost. 2001, 86, 702–709. [Google Scholar]
- Kawai, Y.; Matsumoto, Y.; Watanabe, K.; Yamamoto, H.; Satoh, K.; Murata, M.; Handa, M.; Ikeda, Y. Hemodynamic Forces Modulate the Effects of Cytokines on Fibrinolytic Activity of Endothelial Cells. Blood 1996, 87, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Chandler, W.L.; Levy, W.C.; Stratton, J.R. The Circulatory Regulation of TPA and UPA Secretion, Clearance, and Inhibition During Exercise and During the Infusion of Isoproterenol and Phenylephrine. Circulation 1995, 92, 2984–2994. [Google Scholar] [CrossRef]
- Stein, C.; Brown, N.; E Vaughan, D.; Lang, C.C.; Wood, A.J. Regulation of Local Tissue-Type Plasminogen Activator Release by Endothelium-Dependent and Endothelium-Independent Agonists in Human Vasculature. J. Am. Coll. Cardiol. 1998, 32, 117–122. [Google Scholar] [CrossRef]
- Levin, E.G.; Del Zoppo, G.J. Localization of Tissue Plasminogen Activator in the Endothelium of a Limited Number of Vessels. Am. J. Pathol. 1994, 144, 855–861. [Google Scholar]
- Yang, F.; Liu, S.; Wang, S.-J.; Yu, C.; Paganini-Hill, A.; Fisher, M.J. Tissue Plasminogen Activator Expression and Barrier Properties of Human Brain Microvascular Endothelial Cells. Cell Physiol. Biochem. 2011, 28, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.-K.; Sun, E.-S.; Kim, Y.-H. Zinc-Triggered Induction of Tissue Plasminogen Activator and Plasminogen in Endothelial Cells and Pericytes. Exp. Neurobiol. 2013, 22, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Tran, N.D.; Li, Z.; Yang, F.; Zhou, W.; Fisher, M.J. Brain Endothelial Hemostasis Regulation by Pericytes. J. Cereb. Blood Flow Metab. 2006, 26, 209–217. [Google Scholar] [CrossRef]
- Polavarapu, R.; Gongora, M.C.; Yi, H.; Ranganthan, S.; Lawrence, D.A.; Strickland, D.; Yepes, M. Tissue-Type Plasminogen Activator–Mediated Shedding of Astrocytic Low-Density Lipoprotein Receptor–Related Protein Increases the Permeability of the Neurovascular Unit. Blood 2006, 109, 3270–3278. [Google Scholar] [CrossRef]
- Tang, H.; Fu, W.Y.; Ip, N.Y. Altered Expression of Tissue-Type Plasminogen Activator and Type 1 Inhibitor in Astrocytes of Mouse Cortex Following Scratch Injury in Culture. Neurosci. Lett. 2000, 285, 143–146. [Google Scholar] [CrossRef]
- Zhang, C.; An, J.; Haile, W.B.; Echeverry, R.; Strickland, D.K.; Yepes, M. Microglial Low-Density Lipoprotein Receptor-Related Protein 1 Mediates the Effect of Tissue-Type Plasminogen Activator on Matrix Metalloproteinase-9 Activity in the Ischemic Brain. J. Cereb. Blood Flow Metab. 2009, 29, 1946–1954. [Google Scholar] [CrossRef]
- Hu, K.; Yang, J.; Tanaka, S.; Gonias, S.L.; Mars, W.M.; Liu, Y. Tissue-Type Plasminogen Activator Acts as a Cytokine That Triggers Intracellular Signal Transduction and Induces Matrix Metalloproteinase-9 Gene Expression. J. Biol. Chem. 2006, 281, 2120–2127. [Google Scholar] [CrossRef]
- Polavarapu, R.; Gongora, M.C.; Winkles, J.A.; Yepes, M. Tumor Necrosis Factor-Like Weak Inducer of Apoptosis Increases the Permeability of the Neurovascular Unit through Nuclear Factor-Kappa B Pathway Activation. J. Neurosci. 2005, 25, 10094–10100. [Google Scholar] [CrossRef]
- Lin, L.; Wu, C.; Hu, K. Tissue Plasminogen Activator Activates NF-kappaB through a Pathway Involving Annexin A2/CD11b and Integrin-Linked Kinase. J. Am. Soc. Nephrol. 2012, 23, 1329–1338. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Shen, L.H.; Liu, X.; Hozeska-Solgot, A.; Zhang, R.L.; Zhang, Z.G.; Chopp, M. Multipotent Mesenchymal Stromal Cells Increase tPA Expression and Concomitantly Decrease PAI-1 Expression in Astrocytes through the Sonic Hedgehog Signaling Pathway after Stroke (In Vitro Study). J. Cereb. Blood Flow Metab. 2011, 31, 2181–2188. [Google Scholar] [CrossRef]
- Cassé, F.; Bardou, I.; Danglot, L.; Briens, A.; Montagne, A.; Parcq, J.; Alahari, A.; Galli, T.; Vivien, D.; Docagne, F. Glutamate Controls tPA Recycling by Astrocytes, Which in Turn Influences Glutamatergic Signals. J. Neurosci. 2012, 32, 5186–5199. [Google Scholar] [CrossRef]
- Vincent, V.A.; Löwik, C.W.; Verheijen, J.H.; De Bart, A.C.; Tilders, F.J.; Van Dam, A. Role of Astrocyte-Derived Tissue-Type Plasminogen Activator in the Regulation of Endotoxin-Stimulated Nitric Oxide Production by Microglial Cells. Glia 1998, 22, 130–137. [Google Scholar] [CrossRef]
- Siao, C.H.-J.; Susana, R.F.; Stella, E.T. Cell Type-Specific Roles for Tissue Plasminogen Activator Released by Neurons or Microglia after Excitotoxic Injury. J. Neurosci. 2003, 23, 3224–3242. [Google Scholar] [CrossRef]
- Siao, C.J.; Tsirka, S.E. Tissue Plasminogen Activator Mediates Microglial Activation via its Finger Domain through Annexin II. J. Neurosci. 2002, 22, 3352–3358. [Google Scholar] [CrossRef]
- Rogove, A.D.; Siao, C.; Keyt, B.; Strickland, S.; E Tsirka, S. Activation of Microglia Reveals a Non-Proteolytic Cytokine Function for Tissue Plasminogen Activator in the Central Nervous System. J. Cell Sci. 1999, 112, 4007–4016. [Google Scholar]
- Flavin, M.P.; Zhao, G. Tissue Plasminogen Activator Protects Hippocampal Neurons from Oxygen-Glucose Deprivation Injury. J. Neurosci. Res. 2001, 63, 388–394. [Google Scholar] [CrossRef]
- Krystosek, A.; Seeds, N.W. Plasminogen Activator Release at the Neuronal Growth Cone. Science 1981, 213, 1532–1534. [Google Scholar] [CrossRef]
- Seeds, N.W.; Basham, M.E.; Haffke, S.P. Neuronal Migration is Retarded in Mice Lacking the Tissue Plasminogen Activator Gene. Proc. Natl. Acad. Sci. USA 1999, 96, 14118–14123. [Google Scholar] [CrossRef]
- Lee, S.H.; Ko, H.M.; Kwon, K.J.; Lee, J.; Han, S.H.; Han, D.W.; Cheong, J.H.; Ryu, J.H.; Shin, C.Y. tPA Regulates Neurite Outgrowth by Phosphorylation of LRP5/6 in Neural Progenitor Cells. Mol. Neurobiol. 2014, 49, 199–215. [Google Scholar] [CrossRef]
- Muller, C.M.; Griesinger, C.B. Tissue Plasminogen Activator Mediates Reverse Occlusion Plasticity in Visual Cortex. Nat. Neurosci. 1998, 1, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.H.; Xin, H.; Li, Y.; Zhang, R.L.; Cui, Y.; Zhang, L.; Lü, M.; Zhang, Z.G.; Chopp, M. Endogenous Tissue Plasminogen Activator Mediates Bone Marrow Stromal Cell-Induced Neurite Remodeling after Stroke in Mice. Stroke 2011, 42, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Sappino, A.P.; Madani, R.; Huarte, J.; Belin, D.; Kiss, J.Z.; Wohlwend, A.; Vassalli, J.D. Extracellular Proteolysis in the Adult Murine Brain. J. Clin. Investig. 1993, 92, 679–685. [Google Scholar] [CrossRef]
- Nicole, O.; Docagne, F.; Ali, C.; Margaill, I.; Carmeliet, P.; MacKenzie, E.T.; Vivien, D.; Buisson, A. The Proteolytic Activity of Tissue-Plasminogen Activator Enhances NMDA Receptor-Mediated Signaling. Nat. Med. 2001, 7, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Medcalf, R.L. Tissue-Type Plasminogen Activator: A Multifaceted Modulator of Neurotransmission and Synaptic Plasticity. Neuron 2006, 50, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Gilbert, M.E.; Colicos, M.A.; Kandel, E.R.; Kuhl, D. Tissue-Plasminogen Activator is Induced as an Immediate-Early Gene during Seizure, Kindling and Long-Term Potentiation. Nature 1993, 361, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Seeds, N.W.; Basham, M.E.; Ferguson, J.E. Absence of Tissue Plasminogen Activator Gene or Activity Impairs Mouse Cerebellar Motor Learning. J. Neurosci. 2003, 23, 7368–7375. [Google Scholar] [CrossRef] [PubMed]
- Seeds, N.W.; Williams, B.L.; Bickford, P.C. Tissue Plasminogen Activator Induction in Purkinje Neurons after Cerebellar Motor Learning. Science 1995, 270, 1992–1994. [Google Scholar] [CrossRef]
- Pawlak, R.; Magarinos, A.M.; Melchor, J.; McEwen, B.; Strickland, S. Tissue Plasminogen Activator in the Amygdala is Critical for Stress-Induced Anxiety-Like Behavior. Nat. Neurosci. 2003, 6, 168–174. [Google Scholar] [CrossRef]
- Stepanova, V.V.; Tkachuk, V.A. Urokinase as a Multidomain Protein and Polyfunctional Cell Regulator. Biochemistry 2002, 67, 109–118. [Google Scholar]
- Danø, K.; Andreasen, P.; Grøndahl-Hansen, J.; Kristensen, P.; Nielsen, L.; Skriver, L. Plasminogen Activators, Tissue Degradation, and Cancer. Adv. Cancer Res. 1985, 44, 139–266. [Google Scholar]
- Stoppelli, M.; Tacchetti, C.; Cubellis, M.; Corti, A.; Hearings, V.J.; Cassani, G.; Appella, E.; Blasi, F. Autocrine Saturation of Pro-urokinase Receptors on Human A431 Cells. Cell 1986, 45, 675–684. [Google Scholar] [CrossRef]
- Cubellis, M.V.; Wun, T.C.; Blasi, F. Receptor-Mediated Internalization and Degradation of Urokinase is Caused by Its Specific Inhibitor PAI-1. EMBO J. 1990, 9, 1079–1085. [Google Scholar] [CrossRef]
- Cao, D.J.; Guo, Y.L.; Colman, R.W. Urokinase-Type Plasminogen Activator Receptor is Involved in Mediating the Apoptotic Effect of Cleaved High Molecular Weight Kininogen in Human Endothelial Cells. Circ. Res. 2004, 94, 1227–1234. [Google Scholar] [CrossRef]
- Breuss, J.M.; Uhrin, P. VEGF-Initiated Angiogenesis and the uPA/uPAR System. Cell Adh. Migr. 2012, 6, 535–615. [Google Scholar] [CrossRef]
- Prager, G.W.; Breuss, J.M.; Steurer, S.; Mihaly, J.; Binder, B.R. Vascular Endothelial Growth Factor (VEGF) Induces Rapid Prourokinase (pro-uPA) Activation on the Surface of Endothelial Cells. Blood 2004, 103, 955–962. [Google Scholar] [CrossRef]
- Pepper, M.S.; Vassalli, J.D.; Montesano, R.; Orci, L. Urokinase-Type Plasminogen Activator is Induced in Migrating Capillary Endothelial Cells. J. Cell Biol. 1987, 105, 2535–2541. [Google Scholar] [CrossRef]
- Stie, J.; Fox, D. Induction of Brain Microvascular Endothelial Cell Urokinase Expression by Cryptococcus Neoformans Facilitates Blood-Brain Barrier Invasion. PLoS ONE 2012, 7, e49402. [Google Scholar] [CrossRef]
- Benton, R.L.; Maddie, M.A.; Dincman, T.A.; Hagg, T.; Whittemore, S.R. Transcriptional Activation of Endothelial Cells by TGFbeta Coincides with Acute Microvascular Plasticity Following Focal Spinal Cord Ischaemia/Reperfusion Injury. ASN Neuro. 2009, 1, AN20090008. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sawaya, R.; Mohanam, S.; Bindal, A.K.; Bruner, J.M.; Oka, K.; Rao, V.H.; Tomonaga, M.; Nicolson, G.L.; Rao, J.S. Expression and Localization of Urokinase-Type Plasminogen Activator in Human Astrocytomas in Vivo. Cancer Res. 1994, 54, 3656–3661. [Google Scholar]
- Diaz, A.; Merino, P.; Manrique, L.G.; Ospina, J.P.; Cheng, L.; Wu, F.; Jeanneret, V.; Yepes, M. A Cross-talk Between Neuronal Urokinase-type Plasminogen Activator (uPA) and Astrocytic uPA Receptor (uPAR) Promotes Astrocytic Activation and Synaptic Recovery in the Ischemic Brain. J. Neurosci. 2017, 37, 10310–10322. [Google Scholar] [CrossRef]
- Diaz, A.; Merino, P.; Manrique, L.G.; Cheng, L.; Yepes, M. Urokinase-Type Plasminogen Activator (uPA) Protects the Tripartite Synapse in the Ischemic Brain via Ezrin-Mediated Formation of Peripheral Astrocytic Processes. J. Cereb. Blood Flow Metab. 2018, 39, 2157–2171. [Google Scholar] [CrossRef]
- Rudlin, C.R. A Three-Dimensional Representation of Linear Growth and Skeletal Maturation. Acta. Paediatr. Scand. Suppl. 1989, 356, 46–50. [Google Scholar] [CrossRef]
- Merino, P.; Diaz, A.; Jeanneret, V.; Wu, F.; Torre, E.; Cheng, L.; Yepes, M. Urokinase-Type Plasminogen Activator (uPA) Binding to the uPA Receptor (uPAR) Promotes Axonal Regeneration in the Central Nervous System. J. Biol. Chem. 2017, 292, 2741–2753. [Google Scholar] [CrossRef]
- Wu, F.; Catano, M.; Echeverry, R.; Torre, E.; Haile, W.B.; An, J.; Chen, C.; Cheng, L.; Nicholson, A.; Tong, F.C.; et al. Urokinase-Type Plasminogen Activator Promotes Dendritic Spine Recovery and Improves Neurological Outcome Following Ischemic Stroke. J. Neurosci. 2014, 34, 14219–14232. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Lee, K.J.; Seo, J.-W.; Byun, C.J.; Chung, S.-J.; Suh, D.C.; Carmeliet, P.; Koh, J.-Y.; Kim, J.S.; Lee, J.-Y. Neuroprotection by Urokinase Plasminogen Activator in the Hippocampus. Neurobiol. Dis. 2012, 46, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lino, N.; Fiore, L.; Rapacioli, M.; Teruel, L.; Flores, V.; Scicolone, G.; Sánchez, V. uPA-uPAR Molecular Complex is Involved in Cell Signaling during Neuronal Migration and Neuritogenesis. Dev. Dyn. 2014, 243, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Farias-Eisner, R.; Vician, L.; Silver, A.; Reddy, S.; Rabbani, S.A.; Herschman, H.R. The Urokinase Plasminogen Activator Receptor (UPAR) is Preferentially Induced by Nerve Growth Factor in PC12 Pheochromocytoma Cells and is Required for NGF-Driven Differentiation. J. Neurosci. 2000, 20, 230–239. [Google Scholar] [CrossRef]
- Gilder, A.S.; Jones, K.A.; Hu, J.; Wang, L.; Chen, C.C.; Carter, B.S.; Gonias, S.L. Soluble Urokinase Receptor Is Released Selectively by Glioblastoma Cells That Express Epidermal Growth Factor Receptor Variant III and Promotes Tumor Cell Migration and Invasion. J. Biol. Chem. 2015, 2902, 14798–14809. [Google Scholar] [CrossRef]
- Semina, E.; Rubina, K.; Sysoeva, V.; Rysenkova, K.; Klimovich, P.; Plekhanova, O.; Tkachuk, V. Urokinase and Urokinase Receptor Participate in Regulation of Neuronal Migration, Axon Growth and Branching. Eur. J. Cell Biol. 2016, 95, 295–310. [Google Scholar] [CrossRef]
- Rivellini, C.; Dina, G.; Porrello, E.; Cerri, F.; Scarlato, M.; Domi, T.; Ungaro, D.; Del Carro, U.; Bolino, A.; Quattrini, A.; et al. Urokinase Plasminogen Receptor and the Fibrinolytic Complex Play a Role in Nerve Repair after Nerve Crush in Mice, and in Human Neuropathies. PLoS ONE 2012, 7, e32059. [Google Scholar] [CrossRef]
- Diaz, A.; Merino, P.; Guo, J.D.; Yepes, M.A.; McCann, P.; Katta, T.; Tong, E.M.; Torre, E.; Rangaraju, S.; Yepes, M. Urokinase-Type Plasminogen Activator Protects Cerebral Cortical Neurons from Soluble Abeta-Induced Synaptic Damage. J. Neurosci. 2020, 40, 4251–4263. [Google Scholar] [CrossRef]
- Feigin, V.L.; Norrving, B.; Mensah, G.A. Global Burden of Stroke. Circ. Res. 2017, 120, 439–448. [Google Scholar] [CrossRef]
- Yepes, M.; Sandkvist, M.; Wong, M.K.; Coleman, T.A.; Smith, E.; Cohan, S.L.; Lawrence, D.A. Neuroserpin Reduces Cerebral Infarct Volume and Protects Neurons from Ischemia-Induced Apoptosis. Blood 2000, 96, 569–576. [Google Scholar] [CrossRef]
- Rossetti, L.; Hu, M. Skeletal Muscle Glycogenolysis is More Sensitive to Insulin than is Glucose Transport/Phosphorylation. Relation to the Insulin-Mediated Inhibition of Hepatic Glucose Production. J. Clin. Investig. 1993, 92, 2963–2974. [Google Scholar] [CrossRef]
- Gris, J.; Schved, J.; Brun, S.; Brunschwig, C.; Petris, I.; Lassonnery, M.; Martinez, P.; Sarlat, C. Venous Occlusion and Chronic Cigarette Smoking: Dose-Dependent Decrease in the Measurable Release of Tissue-Type Plasminogen Activator and Von Willebrand Factor. Atherosclerosis 1991, 91, 247–255. [Google Scholar] [CrossRef]
- Gong, P.; Li, M.; Zou, C.; Tian, Q.; Xu, Z. Tissue Plasminogen Activator Causes Brain Microvascular Endothelial Cell Injury After Oxygen Glucose Deprivation by Inhibiting Sonic Hedgehog Signaling. Neurochem. Res. 2019, 44, 441–449. [Google Scholar] [CrossRef]
- Lindgren, A.; Lindoff, C.; Norrving, B.; Åstedt, B.; Johansson, B.B. Tissue Plasminogen Activator and Plasminogen Activator Inhibitor-1 in Stroke Patients. Stroke 1996, 27, 1066–1071. [Google Scholar] [CrossRef]
- The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Stroke rt. Tissue Plasminogen Activator for Acute Ischemic Stroke. N. Engl. J. Med. 1995, 333, 1581–1587. [Google Scholar] [CrossRef]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with Alteplase 3 to 4.5 Hours after Acute Ischemic Stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef]
- Van Geffen, M.; Cugno, M.; Lap, P.; Loof, A.; Cicardi, M.; Van Heerde, W. Alterations of Coagulation and Fibrinolysis in Patients with Angioedema Due to C1-Inhibitor Deficiency. Clin. Exp. Immunol. 2012, 167, 472–478. [Google Scholar] [CrossRef]
- Baker, S.K.; Chen, Z.-L.; Norris, E.H.; Revenko, A.S.; MacLeod, A.R.; Strickland, S. Blood-Derived Plasminogen Drives Brain Inflammation and Plaque Deposition in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, 9687–9696. [Google Scholar] [CrossRef]
- Chen, Z.-L.; Revenko, A.S.; Singh, P.; MacLeod, A.R.; Norris, E.H.; Strickland, S. Depletion of Coagulation Factor XII Ameliorates Brain Pathology and Cognitive Impairment in Alzheimer Disease Mice. Blood 2017, 129, 2547–2556. [Google Scholar] [CrossRef]
- Draxler, D.F.; Lee, F.; Ho, H.; Keragala, C.B.; Medcalf, R.L.; Niego, B. t-PA Suppresses the Immune Response and Aggravates Neurological Deficit in a Murine Model of Ischemic Stroke. Front Immunol. 2019, 10, 591. [Google Scholar] [CrossRef]
- Graham, C.H.; Fitzpatrick, T.E.; McCrae, K.R. Hypoxia Stimulates Urokinase Receptor Expression through a Heme Protein-Dependent Pathway. Blood 1998, 91, 3300–3307. [Google Scholar] [CrossRef] [PubMed]
- E Kroon, M.; Koolwijk, P.; Van Der Vecht, B.; Van Hinsbergh, V.W. Urokinase Receptor Expression on Human Microvascular Endothelial Cells is Increased by Hypoxia: Implications for Capillary-Like Tube Formation in a Fibrin Matrix. Blood 2000, 96, 2775–2783. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- del Zoppo, G.J.; Higashida, R.T.; Furlan, A.J.; Pessin, M.S.; Rowley, H.A.; Gent, M. PROACT: A Phase II Randomized Trial of Recombinant Pro-Urokinase by Direct Arterial Delivery in Acute Middle Cerebral Artery Stroke. PROACT Investigators. Prolyse in Acute Cerebral Thromboembolism. Stroke 1998, 29, 4–11. [Google Scholar] [CrossRef]
- Wardlaw, J.M. Overview of Cochrane Thrombolysis Meta-Analysis. Neurology 2001, 57, 69–76. [Google Scholar] [CrossRef]
- Zhang, X.; Polavarapu, R.; She, H.; Mao, Z.; Yepes, M. Tissue-Type Plasminogen Activator and the Low-Density Lipoprotein Receptor-Related Protein Mediate Cerebral Ischemia-Induced Nuclear Factor-kappaB Pathway Activation. Am. J. Pathol. 2007, 171, 1281–1290. [Google Scholar] [CrossRef]
- Yepes, M.; Sandkvist, M.; Moore, E.G.; Bugge, T.H.; Strickland, D.K.; Lawrence, D.A. Tissue-Type Plasminogen Activator Induces Opening of the Blood-Brain Barrier via the LDL Receptor-Related Protein. J. Clin. Investig. 2003, 112, 1533–1540. [Google Scholar] [CrossRef]
- Kidwell, C.S.; Latour, L.; Saver, J.L.; Alger, J.R.; Starkman, S.; Duckwiler, G.; Jahan, R.; Viñuela, F.; Kang, D.-W.; Warach, S. Thrombolytic Toxicity: Blood Brain Barrier Disruption in Human Ischemic Stroke. Cerebrovasc. Dis. 2008, 25, 338–343. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Zhang, C.; Chopp, M.; Gosiewska, A.; Hong, K. Delayed Administration of Human Umbilical Tissue-Derived Cells Improved Neurological Functional Recovery in a Rodent Model of Focal Ischemia. Stroke 2011, 42, 1437–1444. [Google Scholar] [CrossRef]
- Bretscher, A.; A Edwards, K.; Fehon, R.G. ERM Proteins and Merlin: Integrators at the Cell Cortex. Nat. Rev. Mol. Cell Biol. 2002, 3, 586–599. [Google Scholar] [CrossRef]
- Tsukita, S.; Yonemura, S. Cortical Actin Organization: Lessons from ERM (Ezrin/Radixin/Moesin) Proteins. J. Biol. Chem. 1999, 274, 34507–34510. [Google Scholar] [CrossRef]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the Cell Cortex: The Role of ERM Proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef]
- Reichenbach, A.; Derouiche, A.; Kirchhoff, F. Morphology and Dynamics of Perisynaptic Glia. Brain Res. Rev. 2010, 63, 11–25. [Google Scholar] [CrossRef]
- Lavialle, M.; Aumann, G.; Anlauf, E.; Pröls, F.; Arpin, M.; Derouiche, A. Structural Plasticity of Perisynaptic Astrocyte Processes Involves Ezrin and Metabotropic Glutamate Receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 12915–12919. [Google Scholar] [CrossRef]
- Taylor, R.A.; Sansing, L.H. Microglial Responses after Ischemic Stroke and Intracerebral Hemorrhage. Clin. Dev. Immunol. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Gravanis, I.; Tsirka, S.E. Tissue Plasminogen Activator and Glial Function. Glia 2004, 49, 177–183. [Google Scholar] [CrossRef]
- Zhang, C.; An, J.; Strickland, D.K.; Yepes, M. The Low-Density Lipoprotein Receptor-Related Protein 1 Mediates Tissue-Type Plasminogen Activator-Induced Microglial Activation in the Ischemic Brain. Am. J. Pathol. 2009, 174, 586–594. [Google Scholar] [CrossRef]
- Su, E.J.; Fredriksson, L.; Geyer, M.; Folestad, E.; Cale, J.; Andrae, J.; Gao, Y.; Pietras, K.; Mann, K.; Yepes, M.; et al. Activation of PDGF-CC by Tissue Plasminogen Activator Impairs Blood-Brain Barrier Integrity during Ischemic Stroke. Nat. Med. 2008, 14, 731–737. [Google Scholar] [CrossRef]
- Kuo, P.C.; Weng, W.T.; Scofield, B.A.; Furnas, D.; Paraiso, H.C.; Intriago, A.J.; Bosi, K.D.; Yu, I.C.; Yen, J.H. Interferon-Beta Alleviates Delayed tPA-Induced Adverse Effects via Modulation of MMP3/9 Production in Ischemic Stroke. Blood Adv. 2020, 4, 4366–4381. [Google Scholar] [CrossRef]
- Washington, R.A.; Becher, B.; Balabanov, R.; Antel, J.; Dore-Duffy, P. Expression of the Activation Marker Urokinase Plasminogen-Activator Receptor in Cultured Human Central Nervous System Microglia. J. Neurosci. Res. 1996, 45, 392–399. [Google Scholar] [CrossRef]
- Cunningham, O.; Campion, S.; Perry, V.H.; Murray, C.; Sidenius, N.; Docagne, F.; Cunningham, C. Microglia and the urokinase plasminogen activator receptor/uPA system in innate brain inflammation. Glia 2009, 57, 1802–1814. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.M.; Cho, K.S.; Choi, M.S.; Lee, S.H.; Han, S.-H.; Kang, Y.-S.; Kim, H.J.; Cheong, J.H.; Shin, C.Y.; Ko, K.H. Urokinase-Type Plasminogen Activator Induces BV-2 Microglial Cell Migration Through Activation of Matrix Metalloproteinase-9. Neurochem. Res. 2010, 35, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Tsirka, S.E.; Strickland, S.; Stieg, P.E.; Soriano, S.G.; Lipton, S.A. Tissue plasminogen activator (tPA) increase neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat. Med. 1998, 4, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, R.; Wu, J.; Haile, W.B.; Guzman, J.; Yepes, M. Tissue-type plasminogen activator is a neuroprotectant in the mouse hippocampus. J. Clin. Investig. 2010, 120, 2194–2205. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; De Mol, M.; Lijnen, H.R.; Carmeliet, P.; Collen, D. Role of Plasminogen System Components in Focal Cerebral Ischemic Infarction: A Gene Targeting and Gene Transfer Study in Mice. Circulation 1999, 99, 2440–2444. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, P.; Wang, L.; Seeds, N.; McComb, J.G.; Yamada, S.; Griffin, J.H.; Carmeliet, P.; Weiss, M.H.; Zlokovic, B.V. Tissue Plasminogen Activator (tPA) Deficiency Exacerbates Cerebrovascular Fibrin Deposition and Brain Injury in a Murine Stroke Model: Studies in tPA-Deficient Mice and Wild-Type Mice on a Matched Genetic Background. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2801–2806. [Google Scholar] [CrossRef]
- Wu, F.; Wu, J.; Nicholson, A.D.; Echeverry, R.; Haile, W.B.; Catano, M.; An, J.; Lee, A.K.; Duong, D.; Dammer, E.B.; et al. Tissue-Type Plasminogen Activator Regulates the Neuronal Uptake of Glucose in the Ischemic Brain. J. Neurosci. 2012, 32, 9848–9858. [Google Scholar] [CrossRef]
- Klein, G.M.; Li, H.; Sun, P.; Buchan, A.M. Tissue Plasminogen Activator does not Increase Neuronal Damage in Rat Models of Global and Focal Ischemia. Neurology 1999, 52, 1381–1384. [Google Scholar] [CrossRef]
- Hacke, W.; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of Outcome with Early Stroke Treatment: Pooled Analysis of ATLANTIS, ECASS, and NINDS rt-PA Stroke Trials. Lancet 2004, 363, 768–774. [Google Scholar]
- Adams, H.P., Jr.; Del Zoppo, G.; Alberts, M.J.; Bhatt, D.L.; Brass, L.; Furlan, A.; Grubb, R.L.; Higashida, R.T.; Jauch, E.C.; Kidwell, C.; et al. Guidelines for the Early Management of Adults with Ischemic Stroke: A Guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Circulation 2007, 115, 478–534. [Google Scholar]
- Haile, W.B.; Wu, J.; Echeverry, R.; Wu, F.; An, J.; Yepes, M. Tissue-Type Plasminogen Activator has a Neuroprotective Effect in the Ischemic Brain Mediated by Neuronal TNF-α. J. Cereb. Blood Flow Metab. 2012, 32, 57–69. [Google Scholar] [CrossRef]
- Tsirka, S.E.; Gualandris, A.; Amaral, D.G.; Strickland, S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nat. Cell Biol. 1995, 377, 340–344. [Google Scholar] [CrossRef]
- Chen, Z.-L.; Strickland, S. Neuronal Death in the Hippocampus Is Promoted by Plasmin-Catalyzed Degradation of Laminin. Cell 1997, 91, 917–925. [Google Scholar] [CrossRef]
- Reddrop, C.; Moldrich, R.X.; Beart, P.M.; Farso, M.; Liberatore, G.T.; Howells, D.W.; Petersen, K.-U.; Schleuning, W.-D.; Medcalf, R.L. Vampire Bat Salivary Plasminogen Activator (Desmoteplase) Inhibits Tissue-Type Plasminogen Activator-Induced Potentiation of Excitotoxic Injury. Stroke 2005, 36, 1241–1246. [Google Scholar] [CrossRef]
- Wu, F.; Echeverry, R.; Wu, J.; An, J.; Haile, W.B.; Cooper, D.S.; Catano, M.; Yepes, M. Tissue-Type Plasminogen Activator Protects Neurons from Excitotoxin-Induced Cell Death via Activation of the ERK1/2-CREB-ATF3 Signaling Pathway. Mol. Cell Neurosci. 2013, 52, 9–19. [Google Scholar] [CrossRef]
- Salles, F.J.; Strickland, S. Localization and Regulation of the Tissue Plasminogen Activator-Plasmin System in the Hippocampus. J. Neurosci. 2002, 22, 2125–2134. [Google Scholar] [CrossRef]
- Kim, Y.H. Nonproteolytic Neuroprotection by Human Recombinant Tissue Plasminogen Activator. Science 1999, 284, 647–650. [Google Scholar] [CrossRef]
- Parcq, J.; Bertrand, T.; Montagne, A.; Baron, A.F.; Macrez, R.; Billard, J.M.; Briens, A.; Hommet, Y.; Wu, J.; Yepes, M.; et al. Unveiling an Exceptional Zymogen: The Single-Chain Form of tPA is a Selective Activator of NMDA Receptor-Dependent Signaling and Neurotoxicity. Cell Death Differ. 2012, 19, 1983–1991. [Google Scholar] [CrossRef]
- Anfray, A.; Brodin, C.; Drieu, A.; Potzeha, F.; Dalarun, B.; Agin, V.; Vivien, D.; Orset, C. Single- and Two- Chain Tissue Type Plasminogen Activator Treatments Differentially Influence Cerebral Recovery after Stroke. Exp. Neurol. 2021, 338, 113606. [Google Scholar] [CrossRef]
- Nagai, N.; Okada, K.; Kawao, N.; Ishida, C.; Ueshima, S.; Collen, D.; Matsuo, O. Urokinase-Type Plasminogen Activator receptor (uPAR) Augments Brain Damage in a Murine Model of Ischemic Stroke. Neurosci. Lett. 2008, 432, 46–49. [Google Scholar] [CrossRef]
- Morales, D.; McIntosh, T.; Conte, V.; Fujimoto, S.; Graham, D.; Grady, M.S.; Stein, S.C. Impaired Fibrinolysis and Traumatic Brain Injury in Mice. J. Neurotrauma 2006, 23, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The Public Health Impact of Alzheimer’s Disease, 2000–2050: Potential Implication of Treatment Advances. Annu. Rev. Public Health 2002, 23, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J.; Soiza, R.L. Evidence of Endothelial Dysfunction in the Development of Alzheimer’s Disease: Is Alzheimer’s a Vascular Disorder? Am. J. Cardiovasc. Dis. 2013, 3, 197–226. [Google Scholar] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral Blood Flow Regulation and Neurovascular Dysfunction in Alzheimer Disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Park, L.; Zhou, J.; Koizumi, K.; Wang, G.; Anfray, A.; Ahn, S.J.; Seo, J.; Zhou, P.; Zhao, L.; Paul, S.; et al. tPA Deficiency Underlies Neurovascular Coupling Dysfunction by Amyloid-beta. J. Neurosci. 2020, 40, 8160–8173. [Google Scholar] [CrossRef]
- Davis, J.; Wagner, M.R.; Zhang, W.; Xu, F.; Van Nostrand, W.E. Amyloid Beta-Protein Stimulates the Expression of Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR) in Human Cerebrovascular Smooth Muscle Cells. J. Biol. Chem. 2003, 278, 19054–19061. [Google Scholar] [CrossRef]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 Transports Amyloid-Beta(1-42) Across the Blood-Brain Barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef]
- Lillis, A.P.; Mikhailenko, I.; Strickland, D.K. Beyond Endocytosis: LRP Function in Cell Migration, Proliferation and Vascular Permeability. J. Thromb. Haemost. 2005, 3, 1884–1893. [Google Scholar] [CrossRef]
- Rodríguez-Arellano, J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in Physiological Aging and Alzheimer’s Disease. Neurosci. 2016, 323, 170–182. [Google Scholar] [CrossRef]
- A Bates, K.; Fonte, J.; A Robertson, T.; Martins, R.N.; Harvey, A.R. Chronic Gliosis Triggers Alzheimer’s Disease-Like Processing of Amyloid Precursor Protein. Neurosci. 2002, 113, 785–796. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-Associated Astrocytes in Alzheimer’s Disease and Aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The Blood-Brain Barrier: An Overview: Structure, Regulation, and Clinical Implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L.; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Mehra, A.; Ali, C.; Parcq, J.; Vivien, D.; Docagne, F. The Plasminogen Activation System in Neuroinflammation. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 395–402. [Google Scholar] [CrossRef]
- ElAli, A.; Bordeleau, M.; Thériault, P.; Filali, M.; Lampron, A.; Rivest, S. Tissue-Plasminogen Activator Attenuates Alzheimer’s Disease-Related Pathology Development in APPswe/PS1 Mice. Neuropsychopharmacology 2016, 41, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.F.; Beach, T.G. Increased Expression of the Urokinase Plasminogen-Activator Receptor in Amyloid Beta Peptide-Treated Human Brain Microglia and in AD Brains. Brain Res. 2002, 926, 69–79. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Mohammad, D.; O Trépanier, M.; Giuliano, V.; Bazinet, R.P. Markers of Microglia in Post-Mortem Brain Samples from Patients with Alzheimer’s Disease: A Systematic Review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Muller, U.C.; Deller, T.; Korte, M. Not Just Amyloid: Physiological Functions of the Amyloid Precursor Protein Family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Selkoe, D.J. Toward a Comprehensive Theory for Alzheimer’s Disease. Hypothesis: Alzheimer’s Disease is Caused by the Cerebral Accumulation and Cytotoxicity of Amyloid Beta-Protein. Ann. NY Acad. Sci. 2000, 924, 17–25. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Yamazaki, T.; Citron, M.; Podlisny, M.B.; Koo, E.H.; Teplow, D.B.; Haass, C. The Role of APP Processing and Trafficking Pathways in the Formation of Amyloid Beta-Protein. Ann. NY Acad. Sci. 1996, 777, 57–64. [Google Scholar] [CrossRef]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s Disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.D. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-Amyloid Accumulation in APP Mutant Neurons Reduces PSD-95 and GluR1 in Synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Aizenstein, H.J.; Nebes, R.D.; Saxton, J.A.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Ziolko, S.K.; James, J.A.; Snitz, B.E.; Houck, P.R.; et al. Frequent Amyloid Deposition Without Significant Cognitive Impairment Among the Elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.L.; Willis, B.A.; Hawdon, A.; Natanegara, F.; Chua, L.; Foster, J.; Shcherbinin, S.; Ardayfio, P.; Sims, J.R. Donanemab (LY3002813) Dose-Escalation Study in Alzheimer’s Disease. Alzheimers Dement (NY) 2021, 7, e12112. [Google Scholar]
- Ledesma, M.D.; Da Silva, J.S.; Crassaerts, K.; Delacourte, A.; De Strooper, B.; Dotti, C.G. Brain Plasmin Enhances APP Alpha-Cleavage and Abeta Degradation and is Reduced in Alzheimer’s Disease Brains. EMBO Rep. 2000, 1, 530–535. [Google Scholar] [CrossRef]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The Plasmin System is Induced by and Degrades Amyloid-Beta Aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef]
- Tucker, H.M.; Kihiko-Ehmann, M.; Wright, S.; Rydel, R.E.; Estus, S. Tissue Plasminogen Activator Requires Plasminogen to Modulate Amyloid-Beta Neurotoxicity and Deposition. J. Neurochem. 2000, 75, 2172–2177. [Google Scholar] [CrossRef]
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315. [Google Scholar] [CrossRef]
- Barker, R.; Love, S.; Kehoe, P.G. Plasminogen and Plasmin in Alzheimer’s Disease. Brain Res. 2010, 1355, 7–15. [Google Scholar] [CrossRef]
- Dotti, C.G.; Galvan, C.; Ledesma, M.D. Plasmin Deficiency in Alzheimer’s Disease Brains: Causal or Casual? Neurodegener. Dis. 2004, 1, 205–212. [Google Scholar] [CrossRef]
- Kingston, I.B.; Castro, M.J.; Anderson, S. In Vitro Stimulation of Tissue-Type Plasminogen Activator by Alzheimer Amyloid Beta-Peptide Analogues. Nat. Med. 1995, 1, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Melchor, J.P.; Pawlak, R.; Strickland, S. The Tissue Plasminogen Activator-Plasminogen Proteolytic Cascade Accelerates Amyloid-Beta (Abeta) Degradation and Inhibits Abeta-Induced Neurodegeneration. J. Neurosci. 2003, 23, 8867–8871. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.; Keohane, M.E.; Vandenberg, S.R.; Gonias, S.L. Plasminogen activator inhibitor-1 in the cerebrospinal fluid as an index of neurological disease. Blood Coagul. Fibrinolysis 1994, 5, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Van Groen, T.; Katre, A.; Cao, D.; Kadisha, I.; Ballinger, C.; Wang, L.; Carroll, S.L.; Li, L. Knockout of Plasminogen Activator Inhibitor 1 Gene Reduces Amyloid Beta Peptide Burden in a Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2011, 32, 1079–1089. [Google Scholar] [CrossRef]
- Medina, M.G.; Ledesma, M.D.; Domínguez, J.E.; Medina, M.; Zafra, D.; Alameda, F.; Dotti, C.G.; Navarro, P. Tissue Plasminogen Activator Mediates Amyloid-Induced Neurotoxicity via Erk1/2 Activation. EMBO J. 2005, 24, 1706–1716. [Google Scholar] [CrossRef]
- Tucker, H.M.; Kihiko-Ehmann, M.; Estus, S. Urokinase-Type Plasminogen Activator Inhibits Amyloid-Beta Neurotoxicity and Fibrillogenesis via Plasminogen. J. Neurosci. Res. 2002, 70, 249–255. [Google Scholar] [CrossRef]
- Price, J.L.; Morris, J.C. Tangles and Plaques in Nondemented Aging and "Preclinical" Alzheimer’s Disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease is A Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-Beta-Induced Neuronal Dysfunction in Alzheimer’s Disease: From Synapses toward Neural Networks. Nat Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR Removal Underlies Abeta-Induced Synaptic Depression and Dendritic Spine Loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble Oligomers of Amyloid Beta Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural Oligomers of the Amyloid-Beta Protein Specifically Disrupt Cognitive Function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Stratton, J.R.; Chandler, W.L.; Schwartz, R.S.; Cerqueira, M.D.; Levy, W.C.; E Kahn, S.; Larson, V.G.; Cain, K.C.; Beard, J.C.; Abrass, I.B. Effects of physical conditioning on fibrinolytic variables and fibrinogen in young and old healthy adults. Circulation 1991, 83, 1692–1697. [Google Scholar] [CrossRef]
- Yaffe, K.; Barnes, D.; Nevitt, M.; Lui, L.Y.; Covinsky, K. A Prospective Study of Physical Activity and Cognitive Decline in Elderly Women: Women Who Walk. Arch. Intern. Med. 2001, 161, 1703–1708. [Google Scholar] [CrossRef]
- Smith, P.J.; Blumenthal, J.A.; Hoffman, B.M.; Cooper, H.; Strauman, T.A.; Welsh-Bohmer, K.; Browndyke, J.N.; Sherwood, A. Aerobic Exercise and Neurocognitive Performance: A Meta-Analytic Review of Randomized Controlled Trials. Psychosom. Med. 2010, 72, 239–252. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kim, K.S.; Choi, Y.R.; Park, J.Y.; Lee, J.H.; Kim, D.K.; Lee, S.J.; Paik, S.R.; Jou, I.; Park, S.M. Proteolytic Cleavage of Extracellular Alpha-Synuclein by Plasmin: Implications for Parkinson Disease. J. Biol. Chem. 2012, 287, 24862–24872. [Google Scholar] [CrossRef]
- Reuland, C.J.; Church, F.C. Synergy between plasminogen activator inhibitor-1, α-synuclein, and neuroinflammation in Parkinson’s disease. Med. Hypotheses 2020, 138, 109602. [Google Scholar] [CrossRef]
- Pan, H.; Zhao, Y.; Zhai, Z.; Zheng, J.; Zhou, Y.; Zhai, Q.; Cao, X.; Tian, J.; Zhao, L. Role of Plasminogen Activator Inhibitor-1 in the Diagnosis and Prognosis of Patients with Parkinson’s Disease. Exp. Ther. Med. 2018, 15, 5517–5522. [Google Scholar] [CrossRef]
- Demestre, M.; Howard, R.S.; Orrell, R.W.; Pullen, A.H. Serine proteases purified from sera of patients with amyotrophic lateral sclerosis (ALS) induce contrasting cytopathology in murine motoneurones to IgG. Neuropathol. Appl. Neurobiol. 2006, 32, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Glas, M.; Popp, B.; Angele, B.; Koedel, U.; Chahli, C.; Schmalix, W.; Anneser, J.; Pfister, H.; Lorenzl, S. A role for the urokinase-type plasminogen activator system in amyotrophic lateral sclerosis. Exp. Neurol. 2007, 207, 350–356. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yepes, M.; Woo, Y.; Martin-Jimenez, C. Plasminogen Activators in Neurovascular and Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4380. https://doi.org/10.3390/ijms22094380
Yepes M, Woo Y, Martin-Jimenez C. Plasminogen Activators in Neurovascular and Neurodegenerative Disorders. International Journal of Molecular Sciences. 2021; 22(9):4380. https://doi.org/10.3390/ijms22094380
Chicago/Turabian StyleYepes, Manuel, Yena Woo, and Cynthia Martin-Jimenez. 2021. "Plasminogen Activators in Neurovascular and Neurodegenerative Disorders" International Journal of Molecular Sciences 22, no. 9: 4380. https://doi.org/10.3390/ijms22094380
APA StyleYepes, M., Woo, Y., & Martin-Jimenez, C. (2021). Plasminogen Activators in Neurovascular and Neurodegenerative Disorders. International Journal of Molecular Sciences, 22(9), 4380. https://doi.org/10.3390/ijms22094380