
Biomarkers in Glycogen Storage Diseases: An Update

 ,
,
Abstract
1. Background
- Glycogen storage disease type I (GSDI, Von Gierke disease) is characterized by the accumulation of glycogen and fat in the kidneys and liver, resulting in renomegaly and hepatomegaly, which may give rise to a hepatocellular adenoma (HCA) or even to become malignant as hepatocellular carcinoma (HCC) [3]. The overall incidence of GSDI is 1:100,000. Affected neonates who are not medically treated have severe hypoglycaemia. Other children not treated present with hepatomegaly, lactic acidosis, hyperuricemia, hyperlipidaemia, hypertriglyceridemia, or hypoglycaemic seizures, usually from four months after birth. Another issue to consider is impaired platelet function, which can lead to bleeding and epistaxis. The variant GSDIb shows an impaired neutrophil and monocyte function and chronic neutropenia, in addition to complications mentioned before. This leads to recurrent bacterial infections and oral and intestinal mucosal ulcers. In general, untreated GSDI quickly leads to multiple complications. Most affected individuals live to adulthood. GSDI is diagnosed by identifying biallelic pathogenic variants:
- GSDIa, caused by a deficiency of glucose-6-phosphatase (G6Pase or G6PC1) catalytic activity;
- GSDIb, GSDIc and GSDId, caused by different defects in the glucose-6-phosphate transporter (SLC37A4).
- Glycogen storage disease type II (GSDII, Pompe disease), with an incidence variability depending on ethnicity and geographic region, from 1:14,000 in African Americans to 1:100,000 in individuals of European descent, is classified by age of onset, organ involvement, severity, and rate of progression [4]:
- Infantile-onset Pompe disease (IOPD); in children younger than 12 months, the clinical manifestations include cardiomyopathy, which may already be evident in the uterus. However, the most typical onset of the disease occurs at around 4 months. In this case, patients most frequently present with hypotonia, a muscle weakness that occurs in a generalized manner, and hypertrophic cardiomyopathy. These patients usually have serious difficulties feeding and breathing. If not treated with enzyme replacement therapy (ERT), patients with IOPD usually die after two years from progressive left ventricular outflow obstruction and respiratory insufficiency.
- Late-onset Pompe disease (LOPD; including (a) individuals with onset before the age of 12 months without cardiomyopathy and (b) all individuals with onset after the age of 12 months) is characterized by proximal muscle weakness and respiratory insufficiency. Clinically significant cardiac involvement is uncommon.
- 3.
- Glycogen storage disease type III (GSDIII, Cori-Forbes disease), with an incidence of 1:100,000 too, is characterized by variable liver, cardiac muscle, and skeletal muscle involvement. GSDIIIa is the most common subtype, present in about 85% of affected individuals [5]. It manifests with liver and muscle involvement. GSDIIIb, with liver involvement only, comprises about 15% of all GSDIII. The clinical significance of GSDIII ranges from almost no symptoms to severe cardiac dysfunction, congestive heart failure, and, rarely, sudden death. Skeletal myopathy manifests as muscle weakness but is rarely evident in childhood because it progresses slowly. This acquires greater relevance in the third to fourth decades of life. The diagnosis is made by identifying biallelic pathogenic variants in glycogen debranching enzyme, amylo-alpha-1,6-glucosidase, or 4-alpha-glucanotransferase (AGL).
- 4.
- Type V glycogen storage disease (GSDV, McArdle’s disease) is a metabolic myopathy whose main characteristic is exercise intolerance. Incidence roughly reaches 1:100,000 [6]. Symptomatologically, patients present with rapid onset fatigue, myalgia, and muscle cramps [7]. The symptomatology is more acute with exercise, and symptoms are usually isometric or sustained aerobic exercise. It is common for patients to experience greater exercise by exploiting the “second wind” phenomenon, which relieves myalgia and fatigue after resting for a few minutes. Normally GSDV symptoms appears in the first decade of life. Twenty-five percent of patients will present with weakness that mainly affects the proximal muscles. The diagnosis is made using PYGM molecular genetic tests (which encode glycogen phosphorylase, muscle form). This is the only known gene associated with GSDV.
- 5.
- Glycogen storage disease types VIII and IX (GSDVIII and GSDIX), results from a liver phosphorylase b kinase enzyme deficiency, which plays an important role in the decomposition of glycogen [8]. One type is hepatic PhK deficiency (GSDIX), characterized by the presence of early-onset hepatomegaly. There is also growth retardation and, often, fasting ketosis and hypoglycaemia. Another variant is muscle PhK deficiency (GSDVIII), much less common and related to physical exercise intolerance, myalgia, muscular cramps, myoglobinuria, and progressive muscular weakness. The PhK enzyme comprises four subunits (α, β, γ, and δ). Pathogenic variants of PHKA1, which encodes the α subunit, cause the rare muscular deficiency of the X-linked disorder. The pathogenic variants of PHKA2, which also codes for the α subunit, are responsible for the most frequent variant, hepatic PhK deficiency (X-linked hepatic glycogenosis). Incidence for GSD IXα is about 1:100,000 individuals [9]. Pathogenic variants of PHKB, which encodes the β subunit, cause PhK deficiency in both the liver and muscle. Those of PHKG2, which encodes the γ subunit, cause PhK deficiency in the liver. The diagnosis is enhanced by clinical findings from assessing the activity of PhK in erythrocytes or the liver or muscle tissues (depending on the variant to be treated), and definitive confirmation from molecular genetic testing.
2. Materials and Methods
3. Markers Found in Von Gierke Disease (GSDI)
3.1. Hepatic Steatosis, Hepatocellular Adenoma, and Carcinoma in GSDI
3.1.1. Human Studies
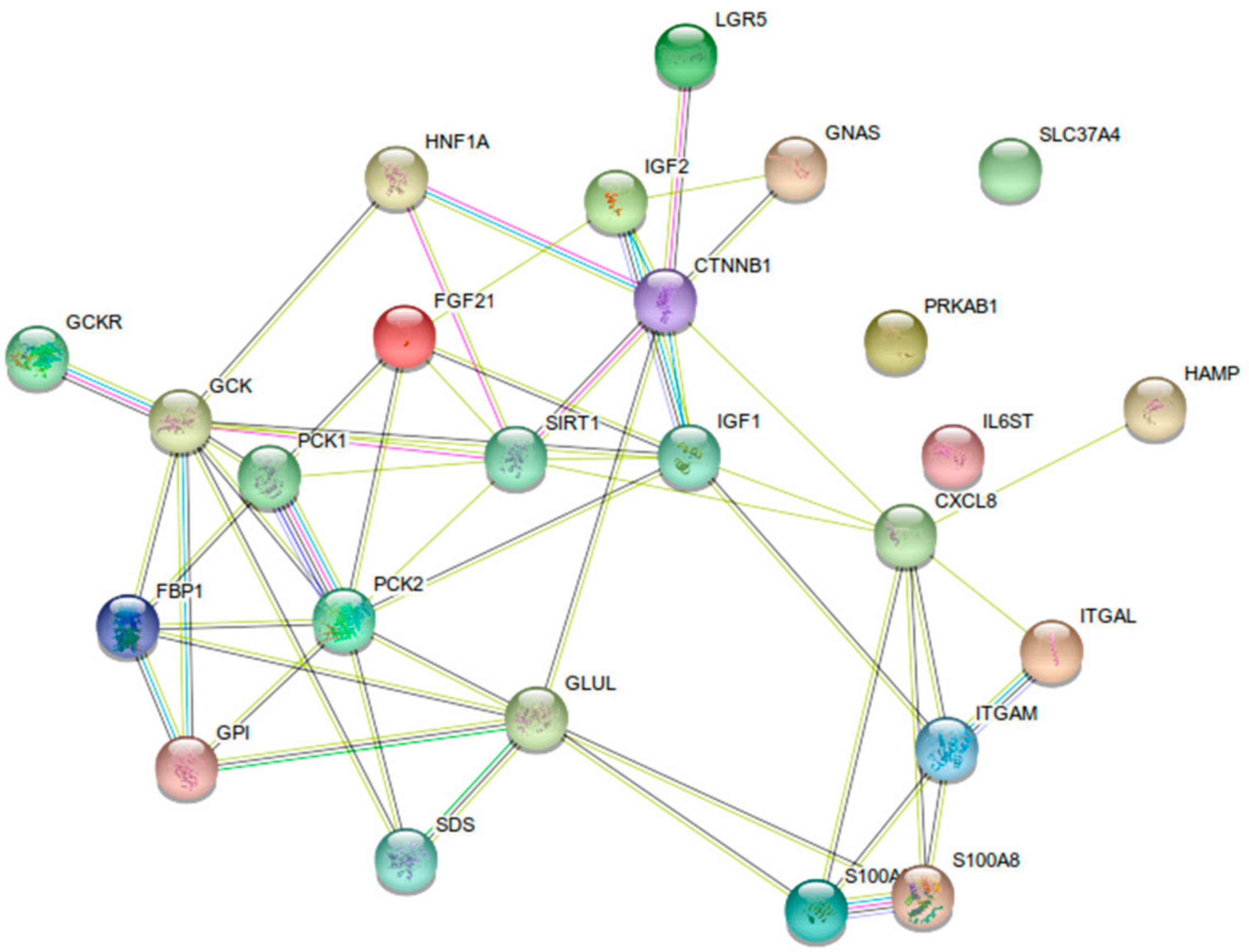
- Gluconeogenesis repression: decreased expression of phosphoenolpyruvate carboxykinase 1 and 2 (PCK1 and PCK2), glucose-6-phosphate transporter 1 (G6PT1), and fructose-1,6-bisphosphatase (FBP1);
- Glycolysis activation: increased expression of glucokinase (GCK) and glucose-6-phosphate isomerase (GPI), and decreased expression of glucokinase regulatory protein (GCKR);
- Fatty-acid synthesis activation: increased expression of acetyl-CoA carboxylase-α (ACACA), ATP citrate lyase (ACLY), fatty acid synthase (FASN), malic enzyme 1 (ME1), and fatty acid desaturase 1 and 2 (FADS1 and FADS2).
3.1.2. Animal Studies
3.2. Observed Predictor of Metabolic Control in GSDI
Human Studies
3.3. Inflammatory Bowel Disease in GSDI
Human Studies
3.4. Neutrophil Impairment in GSDIb
3.4.1. Human Studies
3.4.2. Animal Studies
3.5. Osteopenia and Osteoporosis in GSDI
3.5.1. Human Studies
3.5.2. Animal Studies
3.6. Potential Diagnostic Marker in GSDI
Human Studies
3.7. Unreliable Markers
Human Studies
- The enzyme biotinidase (BTD) catalyzes the hydrolysis of biocytin to biotin and is mostly produced in the liver. Its serum levels in patients with GSDI were evaluated according to Paesold-Burda et al. [65]. The normal levels of biotinidase in healthy people fall between 7.0 and 10.6 mU/mL.
- All GSDI patients analyzed showed elevations of these levels. In the case of GSD type I non-a, the values were increased 2.4 times. The sensitivity was 100% for GSDI patients. No specificity data was found in this study. In addition, no correlation was observed between the activity of BTD and age or dietary control. The enzyme’s activity was also independent of the liver glycogen content. However, another study by Angaroni et al. [66] revealed that not all GSDIa are linked to increased BTD activity, in contrast to previous studies [65,67,68,69]. The authors observed oscillating enzymatic values in GSDIa patients. In addition, they suggested that the presence of a functional polymorphism could influence the expression of this biomarker. The intra- and inter-individual variability of the BTD activity in the GSD individuals suggested that it had little value as a potential biomarker.
- One study on the importance of bone mineral density turnover in patients with hepatic GSDs concluded that some patients with GSDI had increased bone resorption, implied by increased urinary free pyridinoline (fPYD)/creatinine and free deoxypyridinoline (fDPD)/creatinine ratios and serum concentration of the carboxy-terminal telopeptide of type I collagen (ICTP). However, the authors found no correlation between any markers of bone destruction and total body z-score [70].
- After observing a sample of 8 patients (6 males, 2 females) with hepatocellular carcinoma (HCC) and GSDIa, one group suggested that α-fetoprotein (AFP) and carcinoembryonic antigen (CEA) do not appear to be reliable indicators of the presence of hepatic malignancy in patients with GSDI [71].
4. Putative Markers Found in Other Hepatic GSDs
4.1. Urinary Marker
Human Studies
4.2. Cardiomyopathy in GSDIII
Human Studies
4.3. Osteopenia and Osteoporosis in GSDIII
Human Studies
4.4. Unreliable Markers
Human Studies
5. Markers in Pompe Disease (GSDII)
5.1. Malfunction of Skeletal Muscle in GSDII
5.1.1. Human Studies
- Immature or regenerating muscle cells (TNNT2; MYH3; myosin VA, MYO5A; and RUNX related transcription factor 1, RUNX1),
- Inflammatory processes (chitinase 3 like 1, CHI3L1; macrophage expressed 1, MPEG1; TIMP metallopeptidase inhibitor 1, TIMP1; and lipopolysaccharide binding protein, LBP) and
- Apoptosis (cyclin-dependent kinase inhibitor 1A, CDKN1A; PERP TP53 apoptosis effector; and prune homolog 2, PRUNE2).
5.1.2. Animal Studies
- Lysosomal exocytosis was observed in the myotubes in which TFEB was administered. This causes a significant decrease in the amount of accumulated storage material (in particular, glycogen).
- Confocal microscopy techniques showed that overexpression of TFEB did not damage muscle tissue and that muscle fibers treated with TFEB showed increased fusion of lysosomes and autophagosomes. If the autophagic process in the cells was suppressed, the effect of TFEB on lysosomal clearance was blocked. This indicates that the autophagic process is necessary for the TFEB-mediated lysosomal clearance.
- The levels of the transcription factor TFEB were about 10 times higher in mice treated with TFEB, and accumulated glycogen was almost completely cleared. In addition, the muscle ultrastructure underwent improvement due to a reduction in the size and number of lysosomes containing glycogen.
5.2. Urinary Biomarker in GSDII
Human Studies
- The first trial showed that urinary and plasma Glc4 levels correlated with the motor response to treatment over 52 weeks for 11 infantile patients. Glc4 was measured by ultra-performance liquid chromatography-UV (UPLC-UV), to determine its levels in the urine of normal individuals (control group) and that of those with IOPD. This study found that changes in urinary Glc4 were related to the clinical response to ERT treatment. The urinary Glc4 levels were increased prior to treatment and decreased during treatment, normalizing or falling below the control range limits. After 2 months of treatment, they decreased and remained low during the first year of treatment. Therefore, the urinary levels of Glc4 correlated well with the clinical response to ERT.
- The second clinical trial was carried out with a different cohort of IOPD patients (n = 18) and described the correlation of urinary Glc4 with muscle glycogen content and motor function response to ERT over 2 to 3 years. The patients were classified into three groups. Group 1 (7 of 18 patients) had both the best motor function response in the initial phase and sustained clinical improvements by the end of the extension phase of the trial. Group 2 (6 of 18) showed measurable motor gains after 52 weeks of ERT but no further gains or suffered a clinical decline in the extension phase. Group 3 (5 of 18) failed to gain motor milestones at 52 weeks or by the end of the extension phase of the study and became invasive-ventilator dependent between 12 and 60 weeks of ERT. The results from this phase II/III clinical trial showed a strong correlation between urinary Glc4 and skeletal muscle glycogen at three different time points (0, 12, and 52 weeks). This latter trial also indicated that urinary Glc4 may have predictive value as a biomarker for IOPD patients. The value of Glc4 as a biomarker for monitoring the efficacy of treatment with serum enzyme markers of muscle damage is remarkable.
- In the study conducted by Sluiter et al. [105], the direct separation of underivatized Glc4 from its tetrasaccharide M4 isomer via rapid ultraperformance liquid chromatography–tandem mass spectrometry assay (UPLC-MS/MS) exhibited a very high diagnostic specificity of about 92%. This suggested the urinary measurement of Glc4 to be a suitable biomarker for the early diagnosis of PD and for the clinical course of the disease after the administration of ERT. This study was performed with urine obtained from adults, children, and infants from 65 GSDII patients (not only IOPD), eight with GSDIII, two with GSDIV, and six with GSDIX. The authors proposed UPLC-MS/MS as a convenient technique for detecting and quantifying Glc4 as a urine biomarker in GSDII patients. The results also support the suitability of Glc4 as a marker for GSDIII [106,107] in pediatric patients. This method is fast and analytical, has good diagnostic sensitivity, and is safe within acceptable limits.
- Huang and colleagues [108] concluded that urinary Glc4 is very helpful for distinguishing between newborns with pseudo-deficiency of lysosomal acid alpha-glucosidase and those with IOPD. They observed that the level of urinary Glc4 decreased rapidly if the newborn received ERT within 4 h of admission. Unexpectedly low results were previously observed, despite stability studies indicating that urinary Glc4 should be stable for at least 7 days at room temperature (most likely due to degradation by bacterial action or excessive urinary enzyme accumulation secondary to ERT). However, salivary Glc4 can be analyzed, and researchers are looking to assess its efficacy as a potential biomarker for Pompe patients [109].
- Bobillo Lobato [110] analyzed concentrations of Glc4 using HPLC (high performance liquid chromatography) in urine samples from 35 patients affected by PD (9 with infantile presentation and 26 with adult presentation) by comparing them to 40 control patients. The concentrations of Glc4 were normalized to those of urinary creatinine. It was observed that the Glc4 values decreased with age in both the affected and control groups. A ROC curve was developed, exhibiting an area of 0.980 for the infantile form of PD. A value of 4.925 mmol/mol of creatinine was set as the cut-off point, with a sensitivity and specificity of 85.7%. For the adult form of the disease, the area under the curve was 0.949, and 1.025 mmol/mol of creatinine was set as the cut-off point, with a sensitivity of 89.3% and a specificity of 88.5%. The values of Glc4 were elevated in PD, especially in patients with infantile presentation.
- In a retrospective review published in 2012, Young et al. [111] determined that urinary Glc4 had high sensitivity (95%) and specificity (84%) as a diagnostic biomarker in a group of pediatric and adult patients evaluated for PD between 2006 and 2011. Glc4 can be elevated in other conditions, so the authors recommended measuring Glc4 in conjunction with GAA activity for diagnostic purposes. They also concluded that Glc4 was useful for monitoring the response to ERT in these patients, especially for patients with infantile PD on ERT. A reduction into or close to the control range during the first few weeks of treatment was considered a good prognostic indicator by the authors. As a complement to Glc4 and other biomarkers, magnetic resonance spectroscopy and imaging techniques are also recommended for assessing the distribution of affected tissues and the degree of PD severity.
5.3. Cardiomyopathy in GSDII
Human Studies
5.4. Skin Fibroblasts as Another Assessment Tissue of GSDII Progression
Human Studies
6. Putative Markers in McArdle (GSDV)
6.1. Malfunction of Skeletal Muscle in GSDV
6.1.1. Human Studies
6.1.2. Animal Studies
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Complications/Risks Derived from Clinical Interventions
Appendix A.2. Hepatocellular Biopsy
- Uncommon: severe hemorrhage, including the pleural or peritoneal cavity, large hematoma, pneumothorax, subcutaneous emphysema, haemobilia, sepsis and septic shock, puncture of neighboring organs (kidney, pancreas, colon, extrahepatic bile ducts or gallbladder), peritonitis (including biliary), subdiaphragmatic abscess, intrahepatic arteriovenous fistula, rupture of the biopsy needle [139,140].
Appendix A.3. Skeletal Muscle Biopsy
Appendix A.4. Myocardial Biopsy
- Blood clots
- Bleeding from the biopsy site
- Cardiac arrhythmias
- Infection
- Recurrent laryngeal nerve injury
- Injury to the vein or artery
- Pneumothorax
- Rupture of the heart (very rare)
- Tricuspid regurgitation
References
- Ozen, H. Glycogen Storage Diseases: New Perspectives. World J. Gastroenterol. WJG 2007, 13, 2541–2553. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Labrune, P.A.; Laforet, P. The Glycogen Storage Diseases and Related Disorders. In Inborn Metabolic Diseases; Saudubray, J.-M., Baumgartner, M.R., Walter, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 121–137. ISBN 978-3-662-49771-5. [Google Scholar]
- Bali, D.S.; Chen, Y.-T.; Austin, S.; Goldstein, J.L. Glycogen Storage Disease Type I. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Leslie, N.; Bailey, L. Pompe Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Dagli, A.; Sentner, C.P.; Weinstein, D.A. Glycogen Storage Disease Type III. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- McMillan, B.M.; Hirshberg, J.S.; Cosgrove, S.C. McArdle disease causing rhabdomyolysis following vaginal delivery. Anaesth. Rep. 2019, 7, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.A.; Lucía, A.; Arenas, J.; Andreu, A.L. Glycogen Storage Disease Type V. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Goldstein, J.; Austin, S.; Kishnani, P.; Bali, D. Phosphorylase Kinase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Zhu, Q.; Wen, X.Y.; Zhang, M.Y.; Jin, Q.L.; Niu, J.Q. Mutation in PHKA2 leading to childhood glycogen storage disease type IXa: A case report and literature review. Medicine 2019, 98, e17775. [Google Scholar] [CrossRef] [PubMed]
- Winkel, L.P.F.; Hagemans, M.L.C.; van Doorn, P.A.; Loonen, M.C.B.; Hop, W.J.C.; Reuser, A.J.J.; Van der Ploeg, A.T. The Natural Course of Non-Classic Pompe’s Disease; a Review of 225 Published Cases. J. Neurol. 2005, 252, 875–884. [Google Scholar] [CrossRef]
- Müller-Felber, W.; Horvath, R.; Gempel, K.; Podskarbi, T.; Shin, Y.; Pongratz, D.; Walter, M.C.; Baethmann, M.; Schlotter-Weigel, B.; Lochmüller, H.; et al. Late Onset Pompe Disease: Clinical and Neurophysiological Spectrum of 38 Patients Including Long-Term Follow-up in 18 Patients. Neuromuscul. Disord. NMD 2007, 17, 698–706. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, P. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Brit. Med. J. 2009, 339, 1–8. [Google Scholar] [CrossRef]
- Calderaro, J.; Labrune, P.; Morcrette, G.; Rebouissou, S.; Franco, D.; Prévot, S.; Quaglia, A.; Bedossa, P.; Libbrecht, L.; Terracciano, L.; et al. Molecular Characterization of Hepatocellular Adenomas Developed in Patients with Glycogen Storage Disease Type I. J. Hepatol. 2013, 58, 350–357. [Google Scholar] [CrossRef]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-Frame Somatic Deletions Activate Gp130 in Inflammatory Hepatocellular Tumours. Nature 2009, 457, 200–204. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated Analysis of Somatic Mutations and Focal Copy-Number Changes Identifies Key Genes and Pathways in Hepatocellular Carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Bioulac-Sage, P.; Rebouissou, S.; Thomas, C.; Blanc, J.-F.; Saric, J.; Sa Cunha, A.; Rullier, A.; Cubel, G.; Couchy, G.; Imbeaud, S.; et al. Hepatocellular Adenoma Subtype Classification Using Molecular Markers and Immunohistochemistry. Hepatology 2007, 46, 740–748. [Google Scholar] [CrossRef]
- Rebouissou, S.; Imbeaud, S.; Balabaud, C.; Boulanger, V.; Bertrand-Michel, J.; Tercé, F.; Auffray, C.; Bioulac-Sage, P.; Zucman-Rossi, J. HNF1alpha Inactivation Promotes Lipogenesis in Human Hepatocellular Adenoma Independently of SREBP-1 and Carbohydrate-Response Element-Binding Protein (ChREBP) Activation. J. Biol. Chem. 2007, 282, 14437–14446. [Google Scholar] [CrossRef]
- Kim, S.Y.; Weinstein, D.A.; Starost, M.F.; Mansfield, B.C.; Chou, J.Y. Necrotic Foci, Elevated Chemokines and Infiltrating Neutrophils in the Liver of Glycogen Storage Disease Type Ia. J. Hepatol. 2008, 48, 479–485. [Google Scholar] [CrossRef]
- Huang, Y.S.; Chan, C.Y.; Wu, J.C.; Pai, C.H.; Chao, Y.; Lee, S.D. Serum Levels of Interleukin-8 in Alcoholic Liver Disease: Relationship with Disease Stage, Biochemical Parameters and Survival. J. Hepatol. 1996, 24, 377–384. [Google Scholar] [CrossRef]
- Swiatkowska-Stodulska, R.; Bakowska, A.; Drobińska-Jurowiecka, A. Interleukin-8 in the Blood Serum of Patients with Alcoholic Liver Disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2006, 12, CR215–CR220. [Google Scholar]
- Grefhorst, A.; Schreurs, M.; Oosterveer, M.H.; Cortés, V.A.; Havinga, R.; Herling, A.W.; Reijngoud, D.-J.; Groen, A.K.; Kuipers, F. Carbohydrate-Response-Element-Binding Protein (ChREBP) and Not the Liver X Receptor α (LXRα) Mediates Elevated Hepatic Lipogenic Gene Expression in a Mouse Model of Glycogen Storage Disease Type 1. Biochem. J. 2010, 432, 249–254. [Google Scholar] [CrossRef]
- Farah, B.L.; Landau, D.J.; Sinha, R.A.; Brooks, E.D.; Wu, Y.; Fung, S.Y.S.; Tanaka, T.; Hirayama, M.; Bay, B.-H.; Koeberl, D.D.; et al. Induction of Autophagy Improves Hepatic Lipid Metabolism in Glucose-6-Phosphatase Deficiency. J. Hepatol. 2016, 64, 370–379. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Oosterveer, M.H.; Mithieux, G.; Rajas, F. Mechanisms by Which Metabolic Reprogramming in GSD1 Liver Generates a Favorable Tumorigenic Environment. J. Inborn Errors Metab. Screen. 2016, 4, 2326409816679429. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Chuang, T.-P.; Bali, D.; Koeberl, D.; Austin, S.; Weinstein, D.A.; Murphy, E.; Chen, Y.-T.; Boyette, K.; Liu, C.-H.; et al. Chromosomal and Genetic Alterations in Human Hepatocellular Adenomas Associated with Type Ia Glycogen Storage Disease. Hum. Mol. Genet. 2009, 18, 4781–4790. [Google Scholar] [CrossRef]
- Chiu, L.-Y.; Kishnani, P.S.; Chuang, T.-P.; Tang, C.-Y.; Liu, C.-Y.; Bali, D.; Koeberl, D.; Austin, S.; Boyette, K.; Weinstein, D.A.; et al. Identification of Differentially Expressed microRNAs in Human Hepatocellular Adenoma Associated with Type I Glycogen Storage Disease: A Potential Utility as Biomarkers. J. Gastroenterol. 2014, 49, 1274–1284. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Roy, C.N.; Fleming, M.D.; Loda, M.F.; Wolfsdorf, J.I.; Andrews, N.C. Inappropriate expression of hepcidin is associated with iron refractory anemia: Implications for the anemia of chronic disease. Blood 2002, 100, 3776–3781. [Google Scholar] [CrossRef]
- Kim, G.-Y.; Kwon, J.H.; Cho, J.-H.; Zhang, L.; Mansfield, B.C.; Chou, J.Y. Downregulation of Pathways Implicated in Liver Inflammation and Tumorigenesis of Glycogen Storage Disease Type Ia Mice Receiving Gene Therapy. Hum. Mol. Genet. 2017, 26, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-Y.; Lee, Y.M.; Cho, J.-H.; Pan, C.-J.; Jun, H.S.; Springer, D.A.; Mansfield, B.C.; Chou, J.Y. Mice Expressing Reduced Levels of Hepatic Glucose-6-Phosphatase-α Activity Do Not Develop Age-Related Insulin Resistance or Obesity. Hum. Mol. Genet. 2015, 24, 5115–5125. [Google Scholar] [CrossRef] [PubMed]
- Nerstedt, A.; Cansby, E.; Amrutkar, M.; Smith, U.; Mahlapuu, M. Pharmacological Activation of AMPK Suppresses Inflammatory Response Evoked by IL-6 Signalling in Mouse Liver and in Human Hepatocytes. Mol. Cell. Endocrinol. 2013, 375, 68–78. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-κB and STAT3—Key Players in Liver Inflammation and Cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-Dependent Transcription and Cell Survival by the SIRT1 Deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, L.; Chen, X.; Lu, Q.; Yang, Y.; Liu, J.; Ma, X. SIRT1 Counteracted the Activation of STAT3 and NF-κB to Repress the Gastric Cancer Growth. Int. J. Clin. Exp. Med. 2014, 7, 5050–5058. [Google Scholar]
- Chen, I.-C.; Chiang, W.-F.; Huang, H.-H.; Chen, P.-F.; Shen, Y.-Y.; Chiang, H.-C. Role of SIRT1 in Regulation of Epithelial-to-Mesenchymal Transition in Oral Squamous Cell Carcinoma Metastasis. Mol. Cancer 2014, 13, 254. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Ye, X.; Guo, Y.; Zhang, Q.; Chen, W.; Hua, X.; Liu, W.; Yang, Y.; Chen, G. βKlotho Suppresses Tumor Growth in Hepatocellular Carcinoma by Regulating Akt/GSK-3β/Cyclin D1 Signaling Pathway. PLoS ONE 2013, 8, e55615. [Google Scholar] [CrossRef]
- Gertz, M.; Fischer, F.; Nguyen, G.T.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 Inhibits Sirtuins by Exploiting Their Unique NAD+-Dependent Deacetylation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef]
- Cho, J.-H.; Kim, G.-Y.; Pan, C.-J.; Anduaga, J.; Choi, E.-J.; Mansfield, B.C.; Chou, J.Y. Downregulation of SIRT1 Signaling Underlies Hepatic Autophagy Impairment in Glycogen Storage Disease Type Ia. PLoS Genet. 2017, 13, e1006819. [Google Scholar] [CrossRef]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel Insights into ChREBP Regulation and Function. Trends Endocrinol. Metab. TEM 2013, 24, 257–268. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Melis, D.; Pivonello, R.; Parenti, G.; Della Casa, R.; Salerno, M.; Balivo, F.; Piccolo, P.; Di Somma, C.; Colao, A.; Andria, G. The Growth Hormone-Insulin-Like Growth Factor Axis in Glycogen Storage Disease Type 1: Evidence of Different Growth Patterns and Insulin-Like Growth Factor Levels in Patients with Glycogen Storage Disease Type 1a and 1b. J. Pediatr. 2010, 156, 663–670.e1. [Google Scholar] [CrossRef]
- Lawrence, N.T.; Chengsupanimit, T.; Brown, L.M.; Derks, T.G.J.; Smit, G.P.A.; Weinstein, D.A. Inflammatory Bowel Disease in Glycogen Storage Disease Type Ia: A Case Series. J. Pediatr. Gastroenterol. Nutr. 2014. [Google Scholar] [CrossRef]
- Burri, E.; Beglinger, C. Faecal Calprotectin in the Diagnosis of Inflammatory Bowel Disease. Biochem. Med. 2011, 21, 245–253. [Google Scholar] [CrossRef]
- Foell, D.; Frosch, M.; Sorg, C.; Roth, J. Phagocyte-Specific Calcium-Binding S100 Proteins as Clinical Laboratory Markers of Inflammation. Clin. Chim. Acta Int. J. Clin. Chem. 2004, 344, 37–51. [Google Scholar] [CrossRef]
- Hsu, K.; Champaiboon, C.; Guenther, B.D.; Sorenson, B.S.; Khammanivong, A.; Ross, K.F.; Geczy, C.L.; Herzberg, M.C. Anti-Infective Protective Properties of S100 Calgranulins. Anti Inflamm. Anti Allergy Agents Med. Chem. 2009, 8, 290–305. [Google Scholar] [CrossRef]
- Manolakis, A.C.; Kapsoritakis, A.N.; Tiaka, E.K.; Potamianos, S.P. Calprotectin, Calgranulin C, and Other Members of the S100 Protein Family in Inflammatory Bowel Disease. Dig. Dis. Sci. 2011, 56, 1601–1611. [Google Scholar] [CrossRef]
- Konikoff, M.R.; Denson, L.A. Role of fecal calprotectin as a biomarker of intestinal inflammation in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 524–534. [Google Scholar] [CrossRef]
- Jin Jeong, S. The role of fecal calprotectin in pediatric disease. Korean J. Pediatr. 2019, 62, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.; Burch, N.; Morgan, J.; Harrison, B.; Pandey, S.; Pagnamenta, A.T.; Arancibia, C.; Bailey, A.; Barnes, E.; Bird-Lieberman, B.; et al. Remission of inflammatory bowel disease in glucose-6-phosphatase 3 deficiency by allogeneic haematopoietic stem cell transplantation. J. Crohns Colitis 2020, 14, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Veiga-da-Cunha, M.; Chevalier, N.; Stephenne, X.; Defour, J.P.; Paczia, N.; Ferster, A.; Achouri, Y.; Dewulf, J.P.; Linster, C.L.; Bommer, G.T.; et al. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with G6PT and G6PC3 deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.C.; Elling, R.; Maag, B.; Wortmann, S.B.; Derks, T.G.J.; Hannibal, L.; Schumann, A.; Rosenbaum-Fabian, S.; Spiekerkoetter, U. Improved inflammatory bowel disease, wound healing and normal oxidative burst under treatment with empagliflozin in glycogen storage disease type Ib. Orphanet J. Rare Dis. 2020, 15, 8–15. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Van Hove, J.L.K.; Derks, T.G.J.; Chevalier, N.; Knight, V.; Koller, A.; Oussoren, E.; Mayr, J.A.; van Spronsen, F.J.; Lagler, F.B.; et al. Treating neutropenia and neutrophil dysfunction in glycogen storage disease type Ib with an SGLT2 inhibitor. Blood 2020, 136, 1033–1043. [Google Scholar] [CrossRef]
- Jun, H.S.; Weinstein, D.A.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Molecular Mechanisms of Neutrophil Dysfunction in Glycogen Storage Disease Type Ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef]
- Kim, G.-Y.; Lee, Y.M.; Kwon, J.H.; Jun, H.S.; Chou, J. Glycogen Storage Disease Type Ib Neutrophils Exhibit Impaired Cell Adhesion and Migration. Biochem. Biophys. Res. Commun. 2017, 482, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Chihara, K.; Sugimoto, T. The Action of GH/IGF-I/IGFBP in Osteoblasts and Osteoclasts. Horm. Res. 1997, 48 (Suppl. 5), 45–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-M.; Zhao, H.-Y.; Ning, G.; Chen, Y.; Zhang, L.-Z.; Sun, L.-H.; Zhao, Y.-J.; Xu, M.-Y.; Chen, J.-L. IGF-1 as an Early Marker for Low Bone Mass or Osteoporosis in Premenopausal and Postmenopausal Women. J. Bone Miner. Metab. 2008, 26, 159–164. [Google Scholar] [CrossRef]
- Frystyk, J. Free Insulin-Like Growth Factors—Measurements and Relationships to Growth Hormone Secretion and Glucose Homeostasis. Growth. Horm. IGF Res. 2004, 14, 337–375. [Google Scholar] [CrossRef]
- Bach, L.A.; Headey, S.J.; Norton, R.S. IGF-Binding Proteins–the Pieces Are Falling into Place. Trends Endocrinol. Metab. Tem 2005, 16, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.; Højlund, K.; Beck-Nielsen, H.; Sandahl Christiansen, J.; Orskov, H.; Frystyk, J. Free Rather Than Total Circulating Insulin-Like Growth Factor-I Determines the Feedback on Growth Hormone Release in Normal Subjects. J. Clin. Endocrinol. Metab. 2005, 90, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Maïmoun, L.; Coste, O.; Galtier, F.; Mura, T.; Mariano-Goulart, D.; Paris, F.; Sultan, C. Bone Mineral Density Acquisition in Peripubertal Female Rhythmic Gymnasts Is Directly Associated with Plasma IGF1/IGF-Binding Protein 3 Ratio. Eur. J. Endocrinol. 2010, 163, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.-W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-aguirre, J.; Wan, J.; et al. Low Protein Intake Is Associated with a Major Reduction in IGF-1, Cancer, and Overall Mortality in the 65 and Younger but Not Older Population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef]
- Fontana, L.; Weiss, E.P.; Dennis, T.; Klein, S.; Holloszy, J.O.; Sanitá, S.; Elena, R. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell 2008, 7, 681–687. [Google Scholar] [CrossRef]
- Wang, Y.; Nishida, S.; Boudignon, B.M.; Burghardt, A.; Elalieh, H.Z.; Hamilton, M.M.; Majumdar, S.; Halloran, B.P.; Clemens, T.L.; Bikle, D.D. IGF-I Receptor Is Required for the Anabolic Actions of Parathyroid Hormone on Bone. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2007, 22, 1329–1337. [Google Scholar] [CrossRef]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 Maintains Bone Mass by Activation of mTOR in Mesenchymal Stem Cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef]
- Kurbatova, O.; Surkov, A.; Polyakova, S.; Miroskhina, L.; Semenova, G.; Samokhina, I.; Izmailova, T.; Kapustina, E.; Dukhova, Z.; Zakirov, R.; et al. Lymphocytes Intracellular Enzymes Activity in Children with Hepatic Form of Glycogen Storage Disease. J. Inherit. Metab. Dis. 2013, 36 (Suppl. 2), S226. [Google Scholar] [CrossRef]
- Paesold-Burda, P.; Baumgartner, M.R.; Santer, R.; Bosshard, N.U.; Steinmann, B. Elevated Serum Biotinidase Activity in Hepatic Glycogen Storage Disorders—A Convenient Biomarker. J. Inherit. Metab. Dis. 2007, 30, 896–902. [Google Scholar] [CrossRef]
- Angaroni, C.J.; Giner-Ayala, A.N.; Hill, L.P.; Guelbert, N.B.; Paschini-Capra, A.E.; Dodelson de Kremer, R. Evaluation of the Biotinidase Activity in Hepatic Glycogen Storage Disease Patients. Undescribed Genetic Finding Associated with Atypical Enzymatic Behavior: An Outlook. J. Inherit. Metab. Dis. 2010, 33, S289–S294. [Google Scholar] [CrossRef]
- Hug, G.; Chuck, G.; Tsoras, M. Increased Serum Biotinidase Activity in Glycogen-Storage-Disease Type-Ia. In Proceedings of the Pediatric Research; Williams & Wilkins: Baltimore, MD, USA, 1994; Volume 35, p. A203. [Google Scholar]
- Burlina, A.B.; Dermikol, M.; Mantau, A.; Piovan, S.; Grazian, L.; Zacchello, F.; Shin, Y. Increased Plasma Biotinidase Activity in Patients with Glycogen Storage Disease Type Ia: Effect of Biotin Supplementation. J. Inherit. Metab. Dis. 1996, 19, 209–212. [Google Scholar] [CrossRef]
- Wolf, B.; Freehauf, C.L.; Thomas, J.A.; Gordon, P.L.; Greene, C.L.; Ward, J.C. Markedly Elevated Serum Biotinidase Activity May Indicate Glycogen Storage Disease Type Ia. J. Inherit. Metab. Dis. 2003, 26, 805–809. [Google Scholar] [CrossRef]
- Cabrera-Abreu, J.; Crabtree, N.J.; Elias, E.; Fraser, W.; Cramb, R.; Alger, S. Bone Mineral Density and Markers of Bone Turnover in Patients with Glycogen Storage Disease Types I, III and IX. J. Inherit. Metab. Dis. 2004, 27, 1–9. [Google Scholar] [CrossRef]
- Franco, L.M.; Krishnamurthy, V.; Bali, D.; Weinstein, D.A.; Arn, P.; Clary, B.; Boney, A.; Sullivan, J.; Frush, D.P.; Chen, Y.-T.; et al. Hepatocellular Carcinoma in Glycogen Storage Disease Type Ia: A Case Series. J. Inherit. Metab. Dis. 2005, 28, 153–162. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein-Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Melis, D.; Rossi, A.; Pivonello, R.; Del Puente, A.; Pivonello, C.; Cangemi, G.; Negri, M.; Colao, A.; Andria, G.; Parenti, G. Reduced Bone Mineral Density in Glycogen Storage Disease Type III: Evidence for a Possible Connection Between Metabolic Imbalance and Bone Homeostasis. Bone 2016, 86, 79–85. [Google Scholar] [CrossRef]
- Ugorski, M.; Seder, A.; Lundblad, A.; Zopf, D. Studies on the Metabolic Origin of a Glucose-Containing Tetrasaccharide in Human Urine. J. Exp. Pathol. 1983, 1, 27–38. [Google Scholar]
- Walker, G.J.; Whelan, W.J. The Mechanism of Carbohydrase Action. 7. Stages in the Salivary Alpha-Amylolysis of Amylose, Amvlopectin and Glycogen. Biochem. J. 1960, 76, 257–263. [Google Scholar] [CrossRef]
- Piraud, M.; Pettazzoni, M.; de Antonio, M.; Vianey-Saban, C.; Froissart, R.; Chabrol, B.; Young, S.; Laforêt, P. Urine glucose tetrasaccharide: A good biomarker for glycogenoses type II and III? A study of the French cohort. Mol. Genet. Metab. Rep. 2020, 23, 100583. [Google Scholar] [CrossRef]
- Sidbury, J.B.; Cornblath, M.; Fisher, J.; House, E. GLYCOGEN in Erythrocytes of Patients with Glycogen Storage Disease. Pediatrics 1961, 27, 103–111. [Google Scholar]
- An, Y.; Young, S.P.; Hillman, S.L.; Van Hove, J.L.K.; Chen, Y.T.; Millington, D.S. Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of pompe disease. Anal. Biochem. 2000, 287, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; Wortmann, S.B.; van Essen, H.Z.; van Sambeek, R.L.; Wevers, R.; van Diggelen, O.P. Biochemical characteristics and increased tetraglucoside excretion in patients with phosphorylase kinase deficiency. J. Inherit. Metab. Dis. 2005, 28, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Halaby, C.A.; Young, S.P.; Austin, S.; Stefanescu, E.; Bali, D.; Clinton, L.K.; Smith, B.; Pendyal, S.; Upadia, J.; Schooler, G.R.; et al. Liver fibrosis during clinical ascertainment of glycogen storage disease type III: A need for improved and systematic monitoring. Genet. Med. 2019, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Heiner-Fokkema, M.R.; van der Krogt, J.; de Boer, F.; Fokkert-Wilts, M.J.; Maatman, R.G.H.J.; Hoogeveen, I.J.; Derks, T.G.J. The multiple faces of urinary glucose tetrasaccharide as biomarker for patients with hepatic glycogen storage diseases. Genet. Med. 2020, 22, 1915–1916. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.D.; Yi, H.; Austin, S.L.; Thurberg, B.L.; Young, S.P.; Fyfe, J.C.; Kishnani, P.S.; Sun, B. Natural Progression of Canine Glycogen Storage Disease Type IIIa. Comp. Med. 2016, 66, 41–51. [Google Scholar]
- Alpert, J.S.; Thygesen, K.; Antman, E.; Bassand, J.P. Myocardial Infarction Redefined–a Consensus Document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J. Am. Coll. Cardiol. 2000, 36, 959–969. [Google Scholar]
- Takahashi, M.; Lee, L.; Shi, Q.; Gawad, Y.; Jackowski, G. Use of Enzyme Immunoassay for Measurement of Skeletal Troponin-I Utilizing Isoform-Specific Monoclonal Antibodies. Clin. Biochem. 1996, 29, 301–308. [Google Scholar] [CrossRef]
- Wilkinson, J.M.; Grand, R.J. Comparison of Amino Acid Sequence of Troponin I from Different Striated Muscles. Nature 1978, 271, 31–35. [Google Scholar] [CrossRef]
- Cummins, P.; Perry, S.V. Troponin I from Human Skeletal and Cardiac Muscles. Biochem. J. 1978, 171, 251–259. [Google Scholar] [CrossRef]
- Cummins, P.; Young, A.; Auckland, M.L.; Michie, C.A.; Stone, P.C.; Shepstone, B.J. Comparison of Serum Cardiac Specific Troponin-I with Creatine Kinase, Creatine Kinase-MB Isoenzyme, Tropomyosin, Myoglobin and C-Reactive Protein Release in Marathon Runners: Cardiac or Skeletal Muscle Trauma? Eur. J. Clin. Investig. 1987, 17, 317–324. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, Y.; Zhang, L.; Xu, W.; Zhou, X. Diagnostic and prognostic value of biomarkers in acute myocardial infarction. Postgrad. Med. J. 2019, 95, 210–216. [Google Scholar] [CrossRef]
- Austin, S.L.; Proia, A.D.; Spencer-Manzon, M.J.; Butany, J.; Wechsler, S.B.; Kishnani, P.S. Cardiac Pathology in Glycogen Storage Disease Type III. JIMD Rep. 2012, 6, 65–72. [Google Scholar] [CrossRef]
- Cochrane, A.B.; Fedson, S.E.; Cronin, D.C. Nesiritide as Bridge to Multi-Organ Transplantation: A Case Report. Transplant. Proc. 2007, 39, 308–310. [Google Scholar] [CrossRef]
- Palermo, A.T.; Palmer, R.E.; So, K.S.; Oba-Shinjo, S.M.; Zhang, M.; Richards, B.; Madhiwalla, S.T.; Finn, P.F.; Hasegawa, A.; Ciociola, K.M.; et al. Transcriptional Response to GAA Deficiency (Pompe Disease) in Infantile-Onset Patients. Mol. Genet. Metab. 2012, 106, 287–300. [Google Scholar] [CrossRef]
- Batzios, S.P.; Zafeiriou, D.I.; Vargiami, E.; Karakiulakis, G.; Papakonstantinou, E. Altered Expression of MMP-2 and MMP-9 in the Serum of Patients with Lysosomal Storage Disorders. J. Inherit. Metab. Dis. 2013, 36 (Suppl. 2), S288. [Google Scholar] [CrossRef]
- Hůlková, H.; Ledvinová, J.; Asfaw, B.; Koubek, K.; Kopriva, K.; Elleder, M. Lactosylceramide in Lysosomal Storage Disorders: A Comparative Immunohistochemical and Biochemical Study. Virchows Arch. Int. J. Pathol. 2005, 447, 31–44. [Google Scholar] [CrossRef]
- Pascual, J.M.; Roe, C.R. Systemic Metabolic Abnormalities in Adult-Onset Acid Maltase Deficiency: Beyond Muscle Glycogen Accumulation. JAMA Neurol. 2013, 70, 756–763. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baligand, C.; Todd, A.G.; Lee-McMullen, B.; Vohra, R.S.; Byrne, B.J.; Falk, D.J.; Walter, G.A. 13C/31P MRS Metabolic Biomarkers of Disease Progression and Response to AAV Delivery of hGAA in a Mouse Model of Pompe Disease. Mol. Ther. Methods Clin. Dev. 2017, 7, 42–49. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. TFEB Regulates Autophagy: An Integrated Coordination of Cellular Degradation and Recycling Processes. Autophagy 2011, 7, 1379–1381. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.-A.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription Factor EB (TFEB) Is a New Therapeutic Target for Pompe Disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Giacinti, C.; Nardis, C.; Borsellino, G.; Rizzuto, E.; Nicoletti, C.; Wannenes, F.; Battistini, L.; Rosenthal, N.; Molinaro, M.; et al. Local Expression of IGF-1 Accelerates Muscle Regeneration by Rapidly Modulating Inflammatory Cytokines and Chemokines. FASEB J. 2007, 21, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of Skeletal Muscle Mass in Mice by a New TGF-Beta Superfamily Member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.; Han, D.-S.; Hwu, W.-L.; Thurberg, B.L.; Yang, W.-S. Myostatin and Insulin-Like Growth Factor I: Potential Therapeutic Biomarkers for Pompe Disease. PLoS ONE 2013, 8, e71900. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Young, S.P.; Kishnani, P.S.; Millington, D.S.; Amalfitano, A.; Corz, D.; Chen, Y.-T. Glucose Tetrasaccharide as a Biomarker for Monitoring the Therapeutic Response to Enzyme Replacement Therapy for Pompe Disease. Mol. Genet. Metab. 2005, 85, 247–254. [Google Scholar] [CrossRef]
- Young, S.P.; Zhang, H.; Corzo, D.; Thurberg, B.L.; Bali, D.; Kishnani, P.S.; Millington, D.S. Long-Term Monitoring of Patients with Infantile-Onset Pompe Disease on Enzyme Replacement Therapy Using a Urinary Glucose Tetrasaccharide Biomarker. Genet. Med. Off. J. Am. Coll. Med. Genet. 2009, 11, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, W.; van den Bosch, J.C.; Goudriaan, D.A.; van Gelder, C.M.; de Vries, J.M.; Huijmans, J.G.M.; Reuser, A.J.J.; van der Ploeg, A.T.; Ruijter, G.J.G. Rapid Ultraperformance Liquid Chromatography–Tandem Mass Spectrometry Assay for a Characteristic Glycogen-Derived Tetrasaccharide in Pompe Disease and Other Glycogen Storage Diseases. Clin. Chem. 2012, 58, 1139–1147. [Google Scholar] [CrossRef]
- Chester, M.A.; Lundblad, A.; Häger, A.; Sjöblad, S.; Loonen, C.; Tager, J.M.; Zopf, D. Increased Urinary Excretion of a Glycogen-Derived Tetrasaccharide in Heterozygotes with Glycogen Storage Diseases Type II and III. Lancet 1983, 1, 994–995. [Google Scholar] [CrossRef]
- Oberholzer, K.; Sewell, A.C. Unique Oligosaccharide (Apparently Glucotetrasaccharide) in Urine of Patients with Glycogen Storage Diseases. Clin. Chem. 1990, 36, 1381. [Google Scholar] [CrossRef]
- Huang, C.-K.; Liao, H.-C.; Hsieh, Y.-P.; Chen, Y.-C.; Yang, C.-F.; Niu, D.-M. AB067. Glucose Tetrasaccharide (Glc4) Level in Urine Sample as a Biomarker for Pompe Patients. Ann. Transl. Med. 2015, 3. [Google Scholar] [CrossRef]
- Prunty, H.; Broomfield, A.; Vellodi, A.; Cleary, M.; Harvey, G.; Burke, D.; Lukovic, B.; Heales, E. Salivary Glucose Tetrasaccharide (GLC4) as a Potential Biomarker in Infantile Pompe Disease. J. Inherit. Metab. Dis. 2013, 36 (Suppl. 2), S292. [Google Scholar] [CrossRef]
- Bobillo Lobato, J.; Durán Parejo, P.; Tejero Díez, P.; Jiménez Jiménez, L.M. Tetra-Saccharide Glucose as a Diagnostic Biomarker for Pompe Disease: A Study with 35 Patients. Med. Clin. 2013, 141, 106–110. [Google Scholar] [CrossRef]
- Young, S.P.; Piraud, M.; Goldstein, J.L.; Zhang, H.; Rehder, C.; Laforet, P.; Kishnani, P.S.; Millington, D.S.; Bashir, M.R.; Bali, D.S. Assessing Disease Severity in Pompe Disease: The Roles of a Urinary Glucose Tetrasaccharide Biomarker and Imaging Techniques. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160C, 50–58. [Google Scholar] [CrossRef]
- Young, S.P.; Stevens, R.D.; An, Y.; Chen, Y.T.; Millington, D.S. Analysis of a glucose tetrasaccharide elevated in Pompe disease by stable isotope dilution-electrospray ionization tandem mass spectrometry. Anal. Biochem. 2003, 316, 175–180. [Google Scholar] [CrossRef]
- Cox, T.M. Biomarkers in lysosomal storage diseases. In Fabry Disease: Perspectives from 5 Years of Fos; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; pp. 1–16. [Google Scholar]
- Gaze, D.C.; Lawson, G.J.; Harris, A.; Collinson, P.O. Evidence of Cardiomyocyte Necrosis in Glycogen Storage Disease Type II. Ann. Clin. Biochem. 2007, 44, 86–88. [Google Scholar] [CrossRef]
- Sawada, R.; Jardine, K.A.; Fukuda, M. The Genes of Major Lysosomal Membrane Glycoproteins, Lamp-1 and Lamp-2. 5′-Flanking Sequence of Lamp-2 Gene and Comparison of Exon Organization in Two Genes. J. Biol. Chem. 1993, 268, 9014–9022. [Google Scholar] [CrossRef]
- Carlsson, S.R.; Fukuda, M. The Lysosomal Membrane Glycoprotein Lamp-1 Is Transported to Lysosomes by Two Alternative Pathways. Arch. Biochem. Biophys. 1992, 296, 630–639. [Google Scholar] [CrossRef]
- Karageorgos, L.E.; Isaac, E.L.; Brooks, D.A.; Ravenscroft, E.M.; Davey, R.; Hopwood, J.J.; Meikle, P.J. Lysosomal Biogenesis in Lysosomal Storage Disorders. Exp. Cell Res. 1997, 234, 85–97. [Google Scholar] [CrossRef]
- Meikle, P.J.; Yan, M.; Ravenscroft, E.M.; Isaac, E.L.; Hopwood, J.J.; Brooks, D.A. Altered Trafficking and Turnover of LAMP-1 in Pompe Disease-Affected Cells. Mol. Genet. Metab. 1999, 66, 179–188. [Google Scholar] [CrossRef]
- Santacatterina, F.; Chamorro, M.; de Arenas, C.N.; Navarro, C.; Martín, M.A.; Cuezva, J.M.; Sánchez-Aragó, M. Quantitative Analysis of Proteins of Metabolism by Reverse Phase Protein Microarrays Identifies Potential Biomarkers of Rare Neuromuscular Diseases. J. Transl. Med. 2015, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Liotta, L.A.; Espina, V. Reverse Phase Protein Microarrays Advance to Use in Clinical Trials. Mol. Oncol. 2010, 4, 461–481. [Google Scholar] [CrossRef] [PubMed]
- Nogales-Gadea, G.; Consuegra-García, I.; Rubio, J.C.; Arenas, J.; Cuadros, M.; Camara, Y.; Torres-Torronteras, J.; Fiuza-Luces, C.; Lucia, A.; Martín, M.A.; et al. A Transcriptomic Approach to Search for Novel Phenotypic Regulators in McArdle Disease. PLoS ONE 2012, 7, e31718. [Google Scholar] [CrossRef] [PubMed]
- Féasson, L.; Stockholm, D.; Freyssenet, D.; Richard, I.; Duguez, S.; Beckmann, J.S.; Denis, C. Molecular Adaptations of Neuromuscular Disease-Associated Proteins in Response to Eccentric Exercise in Human Skeletal Muscle. J. Physiol. 2002, 543, 297–306. [Google Scholar] [CrossRef]
- Brancaccio, P.; Maffulli, N.; Limongelli, F.M. Creatine Kinase Monitoring in Sport Medicine. Br. Med. Bull. 2007, 81–82, 209–230. [Google Scholar] [CrossRef]
- Olerud, J.E.; Homer, L.D.; Carroll, H.W. Serum Myoglobin Levels Associated with Severe Exercise. Mil. Med. 1981, 146, 274–276. [Google Scholar] [CrossRef]
- Valle, G.; Faulkner, G.; De Antoni, A.; Pacchioni, B.; Pallavicini, A.; Pandolfo, D.; Tiso, N.; Toppo, S.; Trevisan, S.; Lanfranchi, G. Telethonin, a Novel Sarcomeric Protein of Heart and Skeletal Muscle. FEBS Lett. 1997, 415, 163–168. [Google Scholar] [CrossRef]
- Kollár, V.; Szatmári, D.; Grama, L.; Kellermayer, M.S.Z. Dynamic Strength of Titin’s Z-Disk End. J. Biomed. Biotechnol. 2010, 2010, 838530. [Google Scholar] [CrossRef][Green Version]
- Dahlqvist, J.R.; Voss, L.G.; Lauridsen, T.; Krag, T.O.; Vissing, J. A Pilot Study of Muscle Plasma Protein Changes After Exercise. Muscle Nerve 2014, 49, 261–266. [Google Scholar] [CrossRef]
- Scalco, R.S.; Gardiner, A.R.; Pitceathly, R.D.S.; Hilton-Jones, D.; Schapira, A.H.; Turner, C.; Parton, M.; Desikan, M.; Barresi, R.; Marsh, J.; et al. CAV3 Mutations Causing Exercise Intolerance, Myalgia and Rhabdomyolysis: Expanding the Phenotypic Spectrum of Caveolinopathies. Neuromuscul. Disord. NMD 2016, 26, 504–510. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Santos-Lozano, A.; Llavero, F.; Campo, R.; Nogales-Gadea, G.; Díez-Bermejo, J.; Baladrón, C.; González-Murillo, Á.; Arenas, J.; Martín, M.A.; et al. Muscle Molecular Adaptations to Endurance Exercise Training Are Conditioned by Glycogen Availability: A Proteomics-Based Analysis in the McArdle Mouse Model. J. Physiol. 2018, 596, 1035–1061. [Google Scholar] [CrossRef]
- Bassel-Duby, R.; Olson, E.N. Signaling Pathways in Skeletal Muscle Remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef]
- Koulmann, N.; Bigard, A.-X. Interaction between Signalling Pathways Involved in Skeletal Muscle Responses to Endurance Exercise. Pflug. Arch. Eur. J. Physiol. 2006, 452, 125–139. [Google Scholar] [CrossRef]
- Favier, F.B.; Benoit, H.; Freyssenet, D. Cellular and Molecular Events Controlling Skeletal Muscle Mass in Response to Altered Use. Pflug. Arch. Eur. J. Physiol. 2008, 456, 587–600. [Google Scholar] [CrossRef]
- Russell, A.P. Molecular Regulation of Skeletal Muscle Mass. Clin. Exp. Pharmacol. Physiol. 2010, 37, 378–384. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Zhen, Y.-Y.; Chien, K.-Y.; Lee, I.-C.; Lin, W.-C.; Chen, M.-Y.; Pai, L.-M. LIMCH1 Regulates Nonmuscle Myosin-II Activity and Suppresses Cell Migration. Mol. Biol. Cell 2017, 28, 1054–1065. [Google Scholar] [CrossRef]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-Wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef]
- Ha, H.C.; Snyder, S.H. Poly(ADP-Ribose) Polymerase Is a Mediator of Necrotic Cell Death by ATP Depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. P38 MAP-Kinases Pathway Regulation, Function and Role in Human Diseases. Biochim. Et Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Ho, R.C.; Alcazar, O.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. P38gamma MAPK Regulation of Glucose Transporter Expression and Glucose Uptake in L6 Myotubes and Mouse Skeletal Muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R342–R349. [Google Scholar] [CrossRef]
- Kleiner, D.E. Hepatocellular carcinoma: Liver biopsy in the balance. Hepatology 2018, 68, 13–15. [Google Scholar] [CrossRef]
- Tommaso, L.D.; Spadaccini, M.; Donadon, M.; Personeni, N.; Elamin, A.; Aghemo, A.; Lleo, A. Role of liver biopsy in hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 6041–6052. [Google Scholar] [CrossRef]
- Gallo, A.; Abraham, A.; Katzberg, H.D.; Ilaalagan, S.; Bril, V.; Breiner, A. Muscle biopsy technical safety and quality using a self-contained, vacuum-assisted biopsy technique. Neuromuscul. Disord. 2018, 28, 450–453. [Google Scholar] [CrossRef]
- Aburahma, S.K.; Wicklund, M.P.; Quan, D. Take two: Utility of the repeat skeletal muscle biopsy. Muscle Nerve 2019, 60, 41–46. [Google Scholar] [CrossRef]
- Nishikawa, T.; Sekiguchi, M.; Ishibashi-Ueda, H. More than 50 Years after Konno’s Development of the Endomyocardial Biopsy. Int. Heart J. 2017, 58, 840–846. [Google Scholar] [CrossRef]
- Veinot, J.P. Endomyocardial biopsy–when and how? Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2011, 20, 291–296. [Google Scholar] [CrossRef]
- Grankvist, R.; Chireh, A.; Sandell, M.; Mukarram, A.K.; Jaff, N.; Berggren, I.; Persson, H.; Linde, C.; Arnberg, F.; Lundberg, J.; et al. Myocardial micro-biopsy procedure for molecular characterization with increased precision and reduced trauma. Sci. Rep. 2020, 10, 8029. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Biomarkers | Sample | Species | Comments | Risks-Complications | Status | References |
|---|---|---|---|---|---|---|---|
| Diagnosis | HNF1A | Liver | Human | Inactivation in GSDI-related HCA | High risk * | Lack of clinical validation in GSDs | [13,24] |
| IL6ST, GNAS | Liver | Human | Mutated in IHCA | High risk * | Lack of clinical validation in GSDs | [13] | |
| CTNNB1, IL6ST | Liver | Human | Mutated in bHCA | High risk * | Lack of clinical validation in GSDs | [13,15,24] | |
| IL6ST | Liver | Human | Activation in IHCA | High risk * | Lack of clinical validation in GSDs | [13,14] | |
| LGR5 and/or GLUL | Liver | Human | High expression in bHCA | High risk * | Lack of clinical validation in GSDs | [13,16] | |
| PCK1, PCK2, G6PT1, FBP1, GCKR | Liver | Human | Decreased expression in GSD non-tumor livers | High risk * | Lack of clinical validation in GSDs | [13] | |
| GCK, GPI | Liver | Human | Increased expression in GSD non-tumor livers | High risk * | Lack of clinical validation in GSDs | [13] | |
| IGF2 | Liver | Human | Higher levels from HCA to HCC | High risk * | Lack of clinical validation in GSDs | [24] | |
| Calprotectin | Faeces | Human | High concentrations in GSDIa-associated IBD | Non-invasive, No risk | Tested in GSDs clinical trials | [41,42,43,44,45] | |
| SDH, GPDH | Blood (lymphocytes) | Human | Decreased in mitochondrial enzyme activity, resulting in loss of glycolysis | Low risk | Lack of clinical validation in GSDs | [64] | |
| NADH-H | Blood (lymphocytes) | Human | Increased in mitochondrial enzyme activity, resulting in loss of glycolysis | Low risk | Lack of clinical validation in GSDs | [64] | |
| HEPC | Liver | Human | Increased in some HCA, mediator in the anemia of GSDIa | High risk * | Lack of clinical validation in GSDs | [26] | |
| CD11b and CD11a | Blood (neutrophils) | Mouse | Impaired neutrophil migration and adhesion with decreased expression | - | Not yet tested in humans with GSD | [53] | |
| Prognosis | IGF2 | Blood | Human | predictor of metabolic control | High risk * | Lack of clinical validation in GSDs | [40] |
| miR-32, miR-199a-5p, miR-199b-5p, miR-214, miR-582-5p, miR-130b | Liver | Human | Differential expression in GSDIa HCA | High risk * | Lack of clinical validation in GSDs | [25] | |
| IL8 | Blood | Human | Stimulated by lipids accumulation | High risk * | Lack of clinical validation in GSDs | [18] | |
| IGF1 | Bone | Mouse | Expression promotes osteoblastic activity on bones | - | Lack of clinical validation in GSDs | [54,55,56,57,58,59,62,63,73] | |
| AMPK, SIRT1, FGF21, β-klotho | Liver | Mouse | Activated in the AAV-NT individuals | - | Not yet tested in humans with GSD | [27,28,37] | |
| Therapeutic | IGF2 | Liver | Human | A therapy targeting IGF2 could prevent malignancy | High risk * | Lack of clinical validation in GSDs | [24] |
| 1,5AG | Blood (neutrophils) | Human | Inhibits the first step of glycolysis and reduces neutrophils survival | Low risk | Tested in GSDs clinical trials | [49] | |
| miR-224, miR-452 | Liver | Human | High overexpression in GSDIa HCA | High risk * | Lack of clinical validation in GSDs | [25] |
| Type | Biomarkers | Sample | Species | Comments | Risks-Complications | Status | References |
|---|---|---|---|---|---|---|---|
| Diagnosis | Tn | Cardiac tissue | Human | Levels significantly elevated in a GSDIII patient with cardiomyopathy | High risk * | Lack of clinical validation in GSDs | [89] |
| Glc4 | Urine, Blood (erythrocytes) | Human | Found in GSDIII and GSDIX patients | Non-invasive, no risk/Low risk | Lack of clinical validation in GSDs | [76,77,78,79] | |
| Prognosis | Glc4 | Urine, Blood (erythrocytes) | Human | Decreased Glc4 excretions reflected improve in fasting tolerance for GSDIII pediatric patients | Non-invasive, no risk/Low risk | Lack of clinical validation in GSDs | [81] |
| Glc4 | Urine, Blood (erythrocytes) | Human | Decrease may be secondary to fibrosis progression leading to cirrhosis | Non-invasive, no risk/Low risk | Lack of clinical validation in GSDs | [80] | |
| IGF1 | Blood | Human | Reduced levels may contribute to reduced BMD in GSDIII individuals | Middle risk | Lack of clinical validation in GSDs | [54,55,56,57,58,59,62,63,73] |
| Type | Biomarkers | Sample | Species | Comments | Risks-Complications | Status | References |
|---|---|---|---|---|---|---|---|
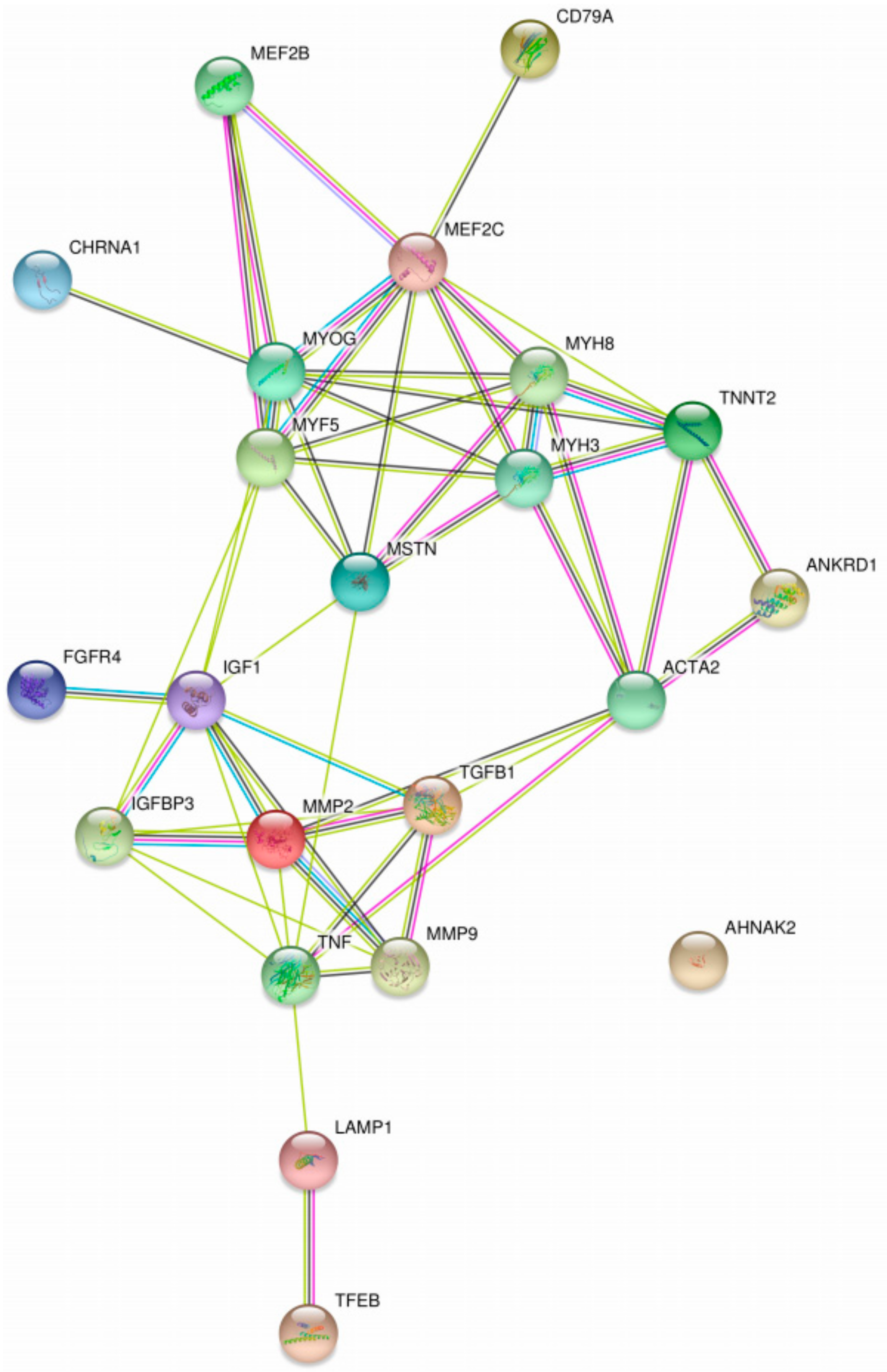
| Diagnosis | MYH3, MYH8, MYOG, MYF5, MEF2B, MEF2C, TNNT2, CHRNA1, ANKRD1, AHNAK2 | Skeletal muscle | Human | Increased in IOPD | High risk * | Lack of clinical validation in GSDs | [91] |
| TGFβ, TNFα, CD79A | Skeletal muscle | Human | Upregulated in IOPD patients | High risk * | Lack of clinical validation in GSDs | [91] | |
| ACTA2, FGFR4 | Skeletal muscle | Human | Increased/unchanged in IOPD biceps, while decreased in IOPD quadriceps | High risk * | Limited value due to variability | [91] | |
| LacCer | Liver, heart, and skeletal muscle | Human | Increased storage in patients | High risk * | Lack of clinical validation in GSDs | [93] | |
| TNNT2 | Blood | Human | Presence in a clinical case was an indicator of necrosis | High risk * | Lack of clinical validation in GSDs | [114] | |
| IGF1, IGFBP3 | Blood | Human | Elevated in 26 patients | Low risk | Lack of clinical validation in GSDs | [94] | |
| Glc4 | Urine, Blood | Human | High sensitivity (95%) and specificity (84%); very helpful for distinguishing IOPD newborns | Non-invasive, no risk/Low risk | Validated by clinical trials | [82,108,111] | |
| MMP2, MMP9 | Blood | Human | Differential expression with other LSDs | Low risk | Lack of clinical validation in GSDs | [92] | |
| Prognosis | MMP2, MMP9 | Blood | Human | Altered protein expression after initiation of ERT in 2 patients | Low risk | Lack of clinical validation in GSDs | [92] |
| LAMP1 | Skin | Human | Extended trafficking time inside fibroblasts for patients | High risk * | Lack of clinical validation in GSDs | [117,118] | |
| 31P MRS PME | - | Mouse | Elevated in the skeletal muscle of GAA−/− mice | - | Not yet tested in humans with GSD | [95] | |
| TFEB | Skeletal muscle | Mouse | High expression gave rise to lysosomal clearing of glycogen excess, and improvement of muscle ultrastructure | - | Not yet tested in humans with GSD | [99] | |
| Therapeutic | IGF1, MSTN | Blood | Human | Lower levels in patients before ERT, after ERT is increased | High risk * | Lack of clinical validation in GSDs | [102] |
| Glc4 | Urine, Blood | Human | Levels correlated with motor response to ERT | Non-invasive, no risk/Low risk | Validated by clinical trials | [103,104,111] | |
| MMP2, MMP9 | Blood | Human | Altered protein expression after initiation of ERT in 2 patients | Low risk | Lack of clinical validation in GSDs | [92] | |
| 31P MRS PME | - | Mouse | High potential for observing metabolic changes after GAA gene therapy | - | Not yet tested in humans with GSD | [95] | |
| TFEB | Skeletal muscle | Mouse | Treated mice showed glycogen accumulation was almost completely cleared | - | Not yet tested in humans with GSD | [99] |
| Type | Biomarkers | Sample | Species | Comments | Risks-Complications | Status | References |
|---|---|---|---|---|---|---|---|
| Diagnosis | PDH E1α, COX1, LDH-A, ACACB, CAPN3, CADH15, CKM, GYS1 | Skeletal muscle | Human | Downregulated in patients | High risk * | Lack of clinical validation in GSDs | [119,120,121] |
| β-F1-ATPase/LDH-A | Skeletal muscle | Human | Upregulated in patients | High risk * | Lack of clinical validation in GSDs | [119] | |
| SERCA1 | Skeletal muscle | Human | Downregulated in patients | High risk * | Lack of clinical validation in GSDs | [121] | |
| CAV3 | Skeletal muscle | Human | Myalgia, exercise intolerance and recurrent rhabdomyolysis appeared in patients with CAV3 mutations | High risk * | Lack of clinical validation in GSDs | [128] | |
| Prognosis | TNNI2 | Blood | Human | Increased levels after induced muscle damage, only in patients | Low risk | Lack of clinical validation in GSDs | [127] |
| LIMCH1, TIGD4 | Skeletal muscle | Mouse | High expression after endurance exercise training in both McArdle and wild-type mice | - | Not yet tested in humans with GSD | [129] | |
| MAPK12, PARP1 | Skeletal muscle | Mouse | Strong expression in McArdle mice after endurance exercise | - | Not yet tested in humans with GSD | [129,137,138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molares-Vila, A.; Corbalán-Rivas, A.; Carnero-Gregorio, M.; González-Cespón, J.L.; Rodríguez-Cerdeira, C. Biomarkers in Glycogen Storage Diseases: An Update. Int. J. Mol. Sci. 2021, 22, 4381. https://doi.org/10.3390/ijms22094381
Molares-Vila A, Corbalán-Rivas A, Carnero-Gregorio M, González-Cespón JL, Rodríguez-Cerdeira C. Biomarkers in Glycogen Storage Diseases: An Update. International Journal of Molecular Sciences. 2021; 22(9):4381. https://doi.org/10.3390/ijms22094381
Chicago/Turabian StyleMolares-Vila, Alberto, Alberte Corbalán-Rivas, Miguel Carnero-Gregorio, José Luís González-Cespón, and Carmen Rodríguez-Cerdeira. 2021. "Biomarkers in Glycogen Storage Diseases: An Update" International Journal of Molecular Sciences 22, no. 9: 4381. https://doi.org/10.3390/ijms22094381
APA StyleMolares-Vila, A., Corbalán-Rivas, A., Carnero-Gregorio, M., González-Cespón, J. L., & Rodríguez-Cerdeira, C. (2021). Biomarkers in Glycogen Storage Diseases: An Update. International Journal of Molecular Sciences, 22(9), 4381. https://doi.org/10.3390/ijms22094381