
Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cyclooxygenase Protein Expression in Glioblastoma Cells Lines and Surgically Resected GBM Tissue
2.2. Effect of Exogenous PGE2 on GBM Cell Counts
2.3. Effect of the Non-Specific Cyclooxygenase Inhibitor, Ibuprofen, on GBM Cell Counts
2.4. Effect of the Specific Cyclooxygenase Inhibitors, SC560 (COX1) and NS398 (COX2), on GBM Cell Counts
2.5. Cyclooxygenase Inhibition Alters Cell Cycle in GBM Cell Lines
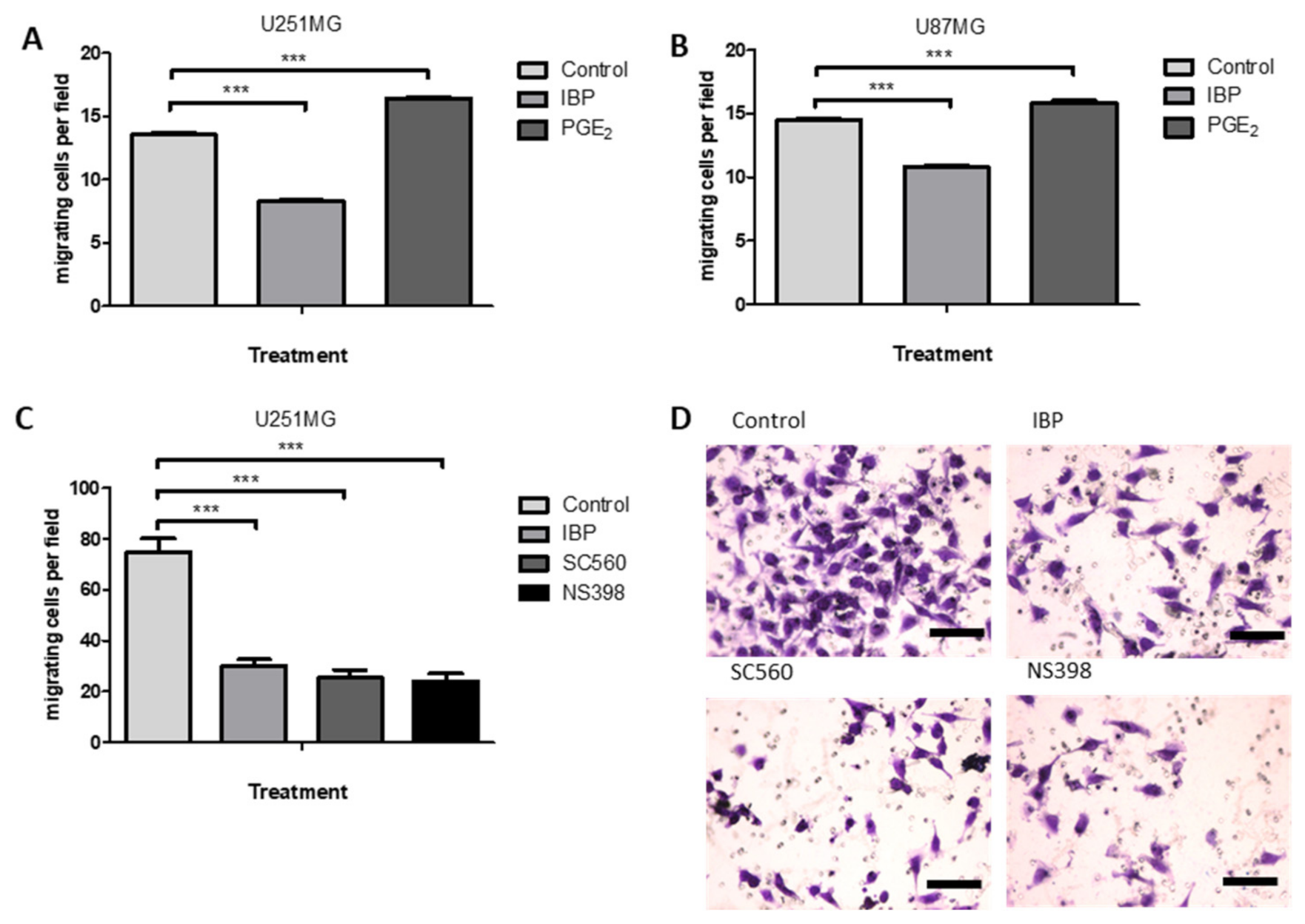
2.6. Cyclooxygenase Inhibition Alters Cell Migration in GBM Cell Lines
2.7. Inhibition of Prostaglandin E2 Receptors EP2 and EP4 Decrease Cell Counts in GBM Cell Lines
2.8. Inhibition of EP2 and EP4 Receptors Alters Cell Cycle in GBM Cell Lines
2.9. Effects of EP Receptor Inhibition on Cell Migration in GBM Cell Lines
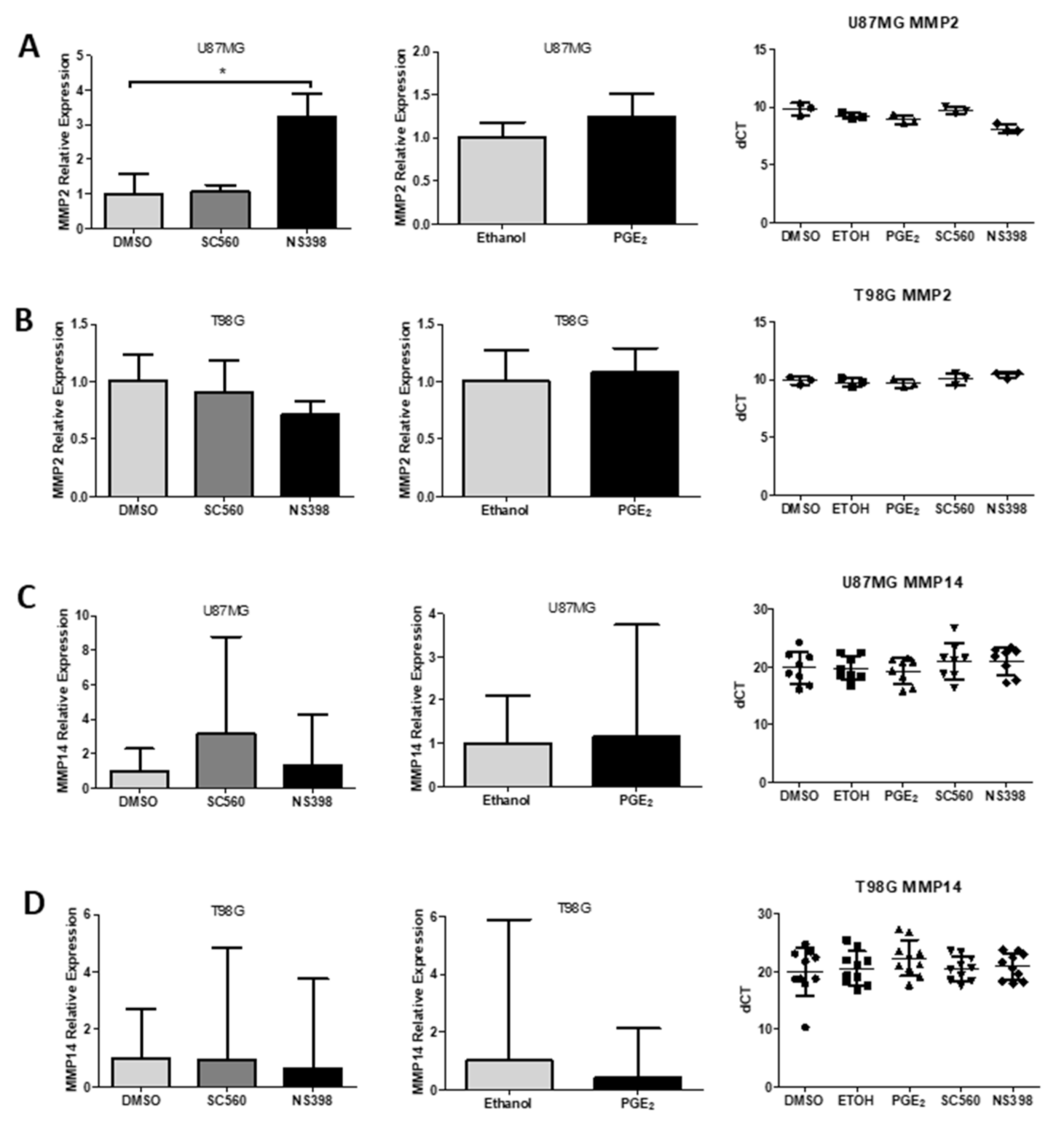
2.10. MMP2 and MMP14 Expression in GBM
2.11. MMP mRNA, Protein Expression and Activity
2.12. Influence of COX Inhibitors and PGE2 on MMPs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Total RNA Extraction/cDNA Synthesis
4.3. RT-PCR and RT-qPCR
4.4. Cell Counts Assay
4.5. Cell Cycle Assay—Propidium Iodide Fluorescence
4.6. Transwell Migration Assay
4.7. Protein Sample Preparation
4.8. Western Blot
4.9. Zymogram Assay
4.10. Immunohistochemical (IHC) Analysis by Light Microscopy
4.11. Immunocytochemical (ICC) Analysis by Light Microscopy
4.12. Patient Selection and Ethical Considerations
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AH6809 | An EP and DP receptor antagonist |
| BrdU | Bromodeoxyuridine |
| CCGA | Chinese glioma genome atlas |
| CNS | Central nervous system |
| COX1 | Cyclooxygenase 1 |
| COX2 | Cyclooxygenase 2 |
| DP | Prostaglandin D series receptor |
| EP | Prostaglandin E series receptor |
| GBM | Glioblastoma |
| IBP | Ibuprofen |
| L-161,982 | A selective EP4 antagonist |
| MMP2 | Matrix metalloproteinase 2 |
| MMP14 | Matrix metalloproteinase 14 |
| NS398 | A selective COX2 inhibitor |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| PGD1 | Prostaglandin D1 |
| PGD2 | Prostaglandin D2 |
| PGE1 | Prostaglandin E1 |
| PGE2 | Prostaglandin E2 |
| PTGS1 | Prostaglandin H synthase 1 |
| PTGS2 | Prostaglandin H synthase 2 |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| RT-qPCR | Real time quantitative polymerase chain reaction |
| SC560 | A selective COX1 inhibitor |
| TCGA | The cancer genome atlas |
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F.R. Cancer-related inflammation. Nat. Cell Biol. 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Gilroy, D.W.; Lawrence, T.; Perretti, M.; Rossi, A.G. Inflammatory Resolution: New opportunities for drug discovery. Nat. Rev. Drug Discov. 2004, 3, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Greene, E.R.; Huang, S.; Serhan, C.N.; Panigrahy, D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011, 96, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.M.; McReynolds, C.B.; Schmidt, W.K.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. [Google Scholar] [CrossRef]
- Gomes, R.N.; Da Costa, S.F.; Colquhoun, A. Eicosanoids and cancer. Clinics 2018, 73, e530. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-T.; Honn, K.V.; Nie, D. Cyclooxygenases, prostanoids, and tumor progression. Cancer Metastasis Rev. 2007, 26, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Hambek, M.; Baghi, M.; Wagenblast, J.; Schmitt, J.; Baumann, H.; Knecht, R. Inverse correlation between serum PGE2 and T classification in head and neck cancer. Head Neck 2007, 29, 244–248. [Google Scholar] [CrossRef]
- Panagopoulos, A.T.; Gomes, R.N.; Almeida, F.G.; Souza, F.D.C.; Veiga, J.C.E.; Nicolaou, A.; Colquhoun, A. The prostanoid pathway contains potential prognostic markers for glioblastoma. Prostaglandins Other Lipid Mediat. 2018, 137, 52–62. [Google Scholar] [CrossRef]
- Mattila, S.; Tuominen, H.; Koivukangas, J.; Stenbäck, F. The terminal prostaglandin synthases mPGES-1, mPGES-2, and cPGES are all overexpressed in human gliomas. Neuropathology 2009, 29, 156–165. [Google Scholar] [CrossRef]
- Panagopoulos, A.T.; Lancellotti, C.L.P.; Veiga, J.C.E.; De Aguiar, P.H.P.; Colquhoun, A. Expression of cell adhesion proteins and proteins related to angiogenesis and fatty acid metabolism in benign, atypical, and anaplastic meningiomas. J. Neuro-Oncol. 2008, 89, 73–87. [Google Scholar] [CrossRef]
- Colquhoun, A. Cell biology-metabolic crosstalk in glioma. Int. J. Biochem. Cell Biol. 2017, 89, 171–181. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Pan, P.-H.; Li, J.-R.; Ou, Y.-C.; Wang, J.-D.; Liao, S.-L.; Chen, W.-Y.; Wang, W.-Y.; Chen, C.-J. Aspirin Induced Glioma Apoptosis through Noxa Upregulation. Int. J. Mol. Sci. 2020, 21, 4219. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.N.; Colquhoun, A. E series prostaglandins alter the proliferative, apoptotic and migratory properties of T98G human glioma cells in vitro. Lipids Health Dis. 2012, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Qiu, J.; Li, Q.; Shi, Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends Cancer 2017, 3, 75–78. [Google Scholar] [CrossRef]
- Ribeiro, G.; Benadiba, M.; Colquhoun, A.; Silva, D.D.O. Diruthenium(II,III) complexes of ibuprofen, aspirin, naproxen and indomethacin non-steroidal anti-inflammatory drugs: Synthesis, characterization and their effects on tumor-cell proliferation. Polyhedron 2008, 27, 1131–1137. [Google Scholar] [CrossRef]
- Benadiba, M.; Costa, I.D.M.; Santos, R.L.S.R.; Serachi, F.O.; Silva, D.D.O.; Colquhoun, A. Growth inhibitory effects of the Diruthenium-Ibuprofen compound, [Ru2Cl(Ibp)4], in human glioma cells in vitro and in the rat C6 orthotopic glioma in vivo. JBIC J. Biol. Inorg. Chem. 2014, 19, 1025–1035. [Google Scholar] [CrossRef]
- Rehman, H.U.; Freitas, T.E.; Gomes, R.N.; Colquhoun, A.; Silva, D.D.O. Axially-modified paddlewheel diruthenium(II,III)-ibuprofenato metallodrugs and the influence of the structural modification on U87MG and A172 human glioma cell proliferation, apoptosis, mitosis and migration. J. Inorg. Biochem. 2016, 165, 181–191. [Google Scholar] [CrossRef]
- Jackson, C.; Ruzevick, J.; Phallen, J.; Belcaid, Z.; Lim, M. Challenges in Immunotherapy Presented by the Glioblastoma Multiforme Microenvironment. Clin. Dev. Immunol. 2011, 2011, 1–20. [Google Scholar] [CrossRef]
- Vempati, P.; Karagiannis, E.D.; Popel, A.S. A Biochemical Model of Matrix Metalloproteinase 9 Activation and Inhibition. J. Biol. Chem. 2007, 282, 37585–37596. [Google Scholar] [CrossRef]
- Yong, V.W. Metalloproteinases: Mediators of Pathology and Regeneration in the CNS. Nat. Rev. Neurosci. 2005, 6, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Molecular Determinants of Malignant Brain Cancers: From Intracellular Alterations to Invasion Mediated by Extracellular Vesicles. Int. J. Mol. Sci. 2017, 18, 2774. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, C. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World, J. Clin. Oncol. 2012, 3, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44–46, 207–223. [Google Scholar] [CrossRef]
- Jana, S.; Chatterjee, K.; Ray, A.K.; DasMahapatra, P.; Swarnakar, S. Regulation of Matrix Metalloproteinase-2 Activity by COX-2-PGE2-pAKT Axis Promotes Angiogenesis in Endometriosis. PLoS ONE 2016, 11, e0163540. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.T.; Gomes, R.N.; Panagopoulos, A.T.; De Almeida, F.G.; Veiga, J.C.E.; Colquhoun, A. Opposing roles of PGD2 in GBM. Prostaglandins Other Lipid Mediat. 2018, 134, 66–76. [Google Scholar] [CrossRef]
- Casibang, M.; Moody, T.W. AH6809 antagonizes non-small cell lung cancer prostaglandin receptors. Lung Cancer 2002, 36, 33–42. [Google Scholar] [CrossRef]
- Cherukuri, D.P.; Chen, X.B.; Goulet, A.-C.; Young, R.N.; Han, Y.; Heimark, R.L.; Regan, J.W.; Meuillet, E.; Nelson, M.A. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp. Cell Res. 2007, 313, 2969–2979. [Google Scholar] [CrossRef]
- Souza, F.C.; Ferreira, M.T.; Colquhoun, A. Influence of Lipoxygenase Inhibition on Glioblastoma Cell Biology. Int. J. Mol. Sci. 2020, 21, 8395. [Google Scholar] [CrossRef]
- Chopra, S.; Overall, C.M.; Dufour, A. Matrix metalloproteinases in the CNS: Interferons get nervous. Cell. Mol. Life Sci. 2019, 76, 3083–3095. [Google Scholar] [CrossRef]
- Itoh, Y.; Binner, S.; Nagase, H. Steps involved in activation of the complex of pro-matrix metalloproteinase 2 (progelatinase A) and tissue inhibitor of metalloproteinases (TIMP)-2 by 4-aminophenylmercuric acetate. Biochem. J. 1995, 308, 645–651. [Google Scholar] [CrossRef]
- Koo, B.-H.; Kim, Y.H.; Han, J.H.; Kim, D.-S. Dimerization of Matrix Metalloproteinase-2 (MMP-2). J. Biol. Chem. 2012, 287, 22643–22653. [Google Scholar] [CrossRef]
- Pahwa, S.; Stawikowski, M.J.; Fields, G.B. Monitoring and Inhibiting MT1-MMP during Cancer Initiation and Progression. Cancers 2014, 6, 416–435. [Google Scholar] [CrossRef]
- Koul, D.; Parthasarathy, R.; Shen, R.A.; Davies, M.A.; Jasser, S.; Chintala, S.K.; Rao, J.S.; Sun, Y.; Benvenisite, E.N.; Liu, T.-J.; et al. Suppression of matrix metalloproteinase-2 gene expression and invasion in human glioma cells by MMAC/PTEN. Oncogene 2001, 20, 6669–6678. [Google Scholar] [CrossRef]
- Cesarini, V.; Martini, M.; Vitiani, L.R.; Gravina, G.L.; Di Agostino, S.; Graziani, G.; D’Alessandris, Q.G.; Pallini, R.; LaRocca, L.M.; Rossi, P.; et al. Type 5 phosphodiesterase regulates glioblastoma multiforme aggressiveness and clinical outcome. Oncotarget 2017, 8, 13223–13239. [Google Scholar] [CrossRef]
- Toth, M.; Sohail, A.; Fridman, R. Assessment of Gelatinases (MMP-2 and MMP-9) by Gelatin Zymography. In Metastasis Research Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 163–174. [Google Scholar]
- Roomi, M.W.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Modulation of MMP-2 and MMP-9 secretion by cytokines, inducers and inhibitors in human glioblastoma T-98G cells. Oncol. Rep. 2017, 37, 1907–1913. [Google Scholar] [CrossRef]
- Hagemann, C.; Anacker, J.; Haas, S.; Riesner, D.; Schömig, B.; Ernestus, R.-I.; Vince, G.H. Comparative expression pattern of Matrix-Metalloproteinases in human glioblastoma cell-lines and primary cultures. BMC Res. Notes 2010, 3, 293. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Cao, L.; Chen, X.-Y.; Zhao, J.; Gao, L.; Li, S.-Z.; Fei, Z. High expression of MMP9 in glioma affects cell proliferation and is associated with patient survival rates. Oncol. Lett. 2017, 13, 1325–1330. [Google Scholar] [CrossRef]
- Kassem, K.M.; Clevenger, M.H.; Szandzik, D.L.; Peterson, E.; Harding, P. PGE2 Reduces MMP-14 and Increases Plasminogen Activator Inhibitor-1 in Cardiac Fibroblasts. Prostaglandins Other Lipid Mediat. 2014, 113–115, 62–68. [Google Scholar] [CrossRef][Green Version]
- Annabi, B.; Laflamme, C.; Sina, A.; Lachambre, M.-P.; Béliveau, R. A MT1-MMP/NF-κB signaling axis as a checkpoint controller of COX-2 expression in CD133(+) U87 glioblastoma cells. J. Neuroinflam. 2009, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Marshall, O.J. PerlPrimer: Cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 2004, 20, 2471–2472. [Google Scholar] [CrossRef]
- Auld, D.S. [14] Removal and replacement of metal ions in metallopeptidases. Methods Enzymol. 1995, 248, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, M.T.; Miyake, J.A.; Gomes, R.N.; Feitoza, F.; Stevannato, P.B.; da Cunha, A.S.; Serachi, F.d.O.; Panagopoulos, A.T.; Colquhoun, A. Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro. Int. J. Mol. Sci. 2021, 22, 4297. https://doi.org/10.3390/ijms22094297
Ferreira MT, Miyake JA, Gomes RN, Feitoza F, Stevannato PB, da Cunha AS, Serachi FdO, Panagopoulos AT, Colquhoun A. Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro. International Journal of Molecular Sciences. 2021; 22(9):4297. https://doi.org/10.3390/ijms22094297
Chicago/Turabian StyleFerreira, Matthew Thomas, Juliano Andreoli Miyake, Renata Nascimento Gomes, Fábio Feitoza, Pollyana Bulgarelli Stevannato, Andrew Silva da Cunha, Fernanda de Oliveira Serachi, Alexandros Theodoros Panagopoulos, and Alison Colquhoun. 2021. "Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro" International Journal of Molecular Sciences 22, no. 9: 4297. https://doi.org/10.3390/ijms22094297
APA StyleFerreira, M. T., Miyake, J. A., Gomes, R. N., Feitoza, F., Stevannato, P. B., da Cunha, A. S., Serachi, F. d. O., Panagopoulos, A. T., & Colquhoun, A. (2021). Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro. International Journal of Molecular Sciences, 22(9), 4297. https://doi.org/10.3390/ijms22094297